
The Role of PTEN in Epithelial–Mesenchymal Transition


Institute of Cytology RAS, 194064 St. Petersburg, Russia
*
Authors to whom correspondence should be addressed.
Cancers 2022, 14(15), 3786; https://doi.org/10.3390/cancers14153786
Submission received: 20 April 2022
/
Revised: 31 July 2022
/
Accepted: 2 August 2022
/
Published: 3 August 2022
(This article belongs to the Special Issue PTEN: Regulation, Signalling and Targeting in Cancer)
Abstract
:Simple Summary
The PTEN phosphatase is a ubiquitously expressed tumor suppressor, which inhibits the PI3K/AKT pathway in the cell. The PI3K/AKT pathway is considered to be one of the main signaling pathways that drives the proliferation of cancer cells. Furthermore, the same pathway controls the epithelial–mesenchymal transition (EMT). EMT is an evolutionarily conserved developmental program, which, upon aberrant reactivation, is also involved in the formation of cancer metastases. Importantly, metastasis is the leading cause of cancer-associated deaths. In this review, we discuss the literature data that highlight the role of PTEN in EMT. Based on this knowledge, we speculate about new possible strategies for cancer treatment.
Abstract
Phosphatase and Tensin Homolog deleted on Chromosome 10 (PTEN) is one of the critical tumor suppressor genes and the main negative regulator of the PI3K pathway. PTEN is frequently found to be inactivated, either partially or fully, in various malignancies. The PI3K/AKT pathway is considered to be one of the main signaling cues that drives the proliferation of cells. Perhaps it is not surprising, then, that this pathway is hyperactivated in highly proliferative tumors. Importantly, the PI3K/AKT pathway also coordinates the epithelial–mesenchymal transition (EMT), which is pivotal for the initiation of metastases and hence is regarded as an attractive target for the treatment of metastatic cancer. It was shown that PTEN suppresses EMT, although the exact mechanism of this effect is still not fully understood. This review is an attempt to systematize the published information on the role of PTEN in the development of malignant tumors, with a main focus on the regulation of the PI3K/AKT pathway in EMT.
1. Introduction
Metastasis is a biological event during which the cellular activities of migration, survival and proliferation are tightly coordinated to ensure that cancer cells leaving the primary tumor location are able to set up a new colony in another metastatic niche. The epithelial–mesenchymal transition (EMT) is a process by which epithelial cells lose their apicobasal polarity and intercellular contacts, transforming into mesenchymal cells, and thus acquiring the ability to migrate and invade. The EMT program is instrumental to such biological processes as embryonic development and metastasis [1]. The main events associated with EMT are the downregulation of E-cadherin and zona occludens 1 (ZO-1) as epithelial markers, and the induction of mesenchymal markers such as vimentin, N/P-cadherins, and β-catenin. These are achieved by a fine-tuned network of transcription factors and signaling pathways [1,2]. The core set of EMT master regulators includes zinc-finger E-box-binding homeobox 1 (ZEB1) and ZEB2, Snail (SNAI1), Slug (SNAI2), and Twist-related protein 1 (TWIST1) [3].
Upon the loss of E-cadherin, epithelial cells in the primary carcinoma lose their cell–cell adhesion properties and focal adhesion contacts with the extra cellular matrix (ECM), resulting in loss of epithelial polarity. However, it should be noted that the majority of cancer cells from the primary tumor acquire an intermediate state of EMT or pseudo-EMT that is characterized by both epithelial and mesenchymal phenotypes [4]. Due to the intrinsic heterogeneity of tumors, only several individual cells or groups of cells are able to detach from the primary tumor and intravasate into the bloodstream to subsequently form metastases. Tumor cells extravasate from blood vessels near specific metastatic niches and undergo the reverse process, called mesenchymal–epithelial transition (MET). This is an important step for the colonization of new tumor niches and, ultimately, for the formation of metastasis, since MET causes cells to proliferate. It is important to mention that EMT in cancer cells is not only reversible but can be stopped at any stage [5].
Recent studies have shown that EMT also plays an important role in the acquisition of drug resistance by cancer cells [6,7] as well as resistance to radiotherapy [8,9]. EMT-induced drug resistance is mediated by attenuating the proliferation rate, upregulating drug efflux, blocking the apoptotic mechanisms of cancer cells, and by increasing evasion from immune surveillance [10,11,12].
Furthermore, activation of the EMT program may also trigger the generation of cancer stem cells (CSCs), which are suggested to be another mechanism of drug resistance. CSCs, also known as tumor-initiating cells (TICs), comprise a class of pseudo-multipotent cells that are able to transdifferentiate into different subtypes within a tumor-initiating tissue [13,14].
PTEN is the main negative regulator of the PI3K/AKT pathway, and hence is considered to be one of the major tumor suppressors. Thus, it is not surprising that the loss of its expression or functions is observed in different cancers [15]. The PI3K/AKT signaling pathway plays an important role in carcinogenesis by orchestrating a number of cellular processes including proliferation, differentiation, metabolism, and migration. The PI3K/AKT pathway is intimately intertwined with EMT at multiple levels.
A large number of studies have shown that PTEN negatively affects EMT. However, the exact role of PTEN in metastasis remains unclear. In this review we discuss the role of PTEN in metastasis, including crosstalk between the signaling networks of PTEN and EMT, which ultimately affects the gene expression programs of cellular plasticity and metastasis.
2. PI3K Pathway and PTEN
2.1. The PI3K Pathway
PI3K pathway signaling is activated by cell surface located growth factor receptors (RTKs), G protein-coupled receptors (GPCRs), cytokine receptors, and integrins [16]. RTKs are a family of high-affinity cell surface receptors that sense growth factors, hormones, cytokines, and other signaling molecules, and convert these signals into the phosphorylation cascade inside the cell. Over 58 gene encoding RTKs have been identified in the human genome [17].
The key element of the PI3K/AKT signaling pathway, PI3K, is a member of the family of enzymes that phosphorylate phosphatidylinositol at the 3′-position of the inositol ring [18]. The PI3K family members can be divided into three classes depending on their structural organization and substrate specificity. The most important role in the formation of cancerous tumors is type I PI3K. They are represented by heterodimers consisting of the catalytic subunit p110 (p110α, p110β, p110γ, and p110δ) and one of the regulatory subunit p85α, p85β, p55α, p55γ, p50α, p101, and p87 [19]. Typically, the catalytic subunit (p110) binds to the regulatory subunit to stabilize the PI3K heterodimer, and thus inhibits PI3K activation. The p85 regulatory subunit of PI3K contains SH2-domains (Src homology-2) that mediate binding to adaptor proteins or tyrosine receptors to facilitate localization and activation of PI3K at the membrane [20]. There is less information about PI3K types II and III; however, it is known that PI3K II consists only of catalytic subunits PI3KC2α, PI3KC2β, or PI3KC2γ, in contrast to class III, which exists in a heterodimeric form and consists of the catalytic subunit Vps34 and the regulatory protein p150 [21].
Activation of type I PI3K occurs through distinct cellular responses downstream of various receptors, including receptor tyrosine kinases (RTKs), cytokine receptors, B-cell antigen receptors, some integrins, and a subset of G protein-coupled receptors. Activation of the receptors upon binding the ligand leads to the autophosphorylation of tyrosine residues within the RTK, and PI3K is then recruited to the membrane by direct binding to consensus phosphotyrosine residues located in the C-terminus of the receptor via one of the two SH2-domains in the PI3K regulatory subunit. This leads to the activation of the catalytic PI3K subunit, resulting in the formation of a secondary messenger phosphatidylinositol (3,4,5)-trisphosphate (PIP3) from the phosphatidylinositol (4,5)-bisphosphate (PIP2) substrate. PIP3 then recruits signaling proteins to the inner membrane that contain pleckstrin domains (PH), including 3′-phosphoinositide-dependent kinase 1 (PDK1) and protein kinase B (AKT). PH-domains are the conserved domains found in hundreds of mammalian proteins involved in intracellular signaling and are responsible for recruiting proteins to lipid membranes [22].
The most important mediator of the PI3K pathway is protein kinase AKT [23]. There are three isoforms of AKT: AKT1, AKT2, and AKT3 [24,25,26]. These isoforms have a common conserved domain structure, consisting of the N-terminal domain (pleckstrin homology; PH-domain), the kinase domain, and the C-terminal regulatory domain containing a hydrophobic motif [26]. It was shown that AKT1 is expressed ubiquitously, while AKT2 is expressed in insulin-sensitive tissues, and AKT3 is expressed in the brain and testes [27]. PIP3 can bind the PH-domain of AKT and induce the translocation of AKT to membranes, where AKT is phosphorylated at two sites: Thr308 and Ser473, which, in turn, leads to the activation of the enzyme [28,29,30]. PDK1 is responsible for the phosphorylation of Thr308 [31], and the phosphorylation of Ser473, which can be carried out by integrin-linked kinase (ILK) or the mTORC2 protein complex, PKCε [32,33,34]. According to Ebner et al. [35], active AKT is predominantly associated with cellular membranes and is critically dependent on the engagement with PIP3. Accordingly, the transforming mutation D323A, which uncouples kinase activation from PIP3 binding, leads to the accumulation of hyperactive phosphorylated AKT in the cytosol [35].
The activation and stabilization of AKT is regulated by phosphorylation not only at the two major sites (Thr308 and Ser473) but also at many additional sites. It was shown that AKT1 has 31 potential phosphorylation sites, while AKT2 possesses 22, and AKT3 has 18 phosphorylation sites [36]. For example, AKT can be phosphorylated by CK2 at Ser129, which augments its catalytic activity [37]. Liu et al. [38] found that phosphorylation at S477/T479 was essential for AKT activation.
The activated form of AKT promotes cell survival by suppressing the expression of genes responsible for cell death, or by activating the expression of genes that contribute to survival [39,40]. Thus, AKT has been shown to inhibit the activity of the following: Foxo family members FKHR (FoxO1), FHHRL1 (FoxO3), and AFX (FoxO4); Bad, which is a pro-apoptotic member of the Bcl-2 protein family; kinase ASK1, which activates JNK and p38 MAP kinases; and procaspase-9 which plays an important role in the signaling pathway of apoptosis.
The Forkhead (FoxO) family of transcription factors includes FKHR/FoxO1, FoxO2, FKHRL1/FoxO3, and AFX/FoxO4 [41]. Forkhead proteins are important downstream targets for AKT, and their phosphorylation by AKT can regulate cell survival by changing the expression of their target genes [42]. In addition, these transcription factors can inhibit cell proliferation by suppressing cyclin D, and can induce apoptosis by transactivating the Fas ligand [43,44].
PI3K/AKT/mTOR/p70S6K is one of the main signaling pathways that regulate autophagy under stress conditions, for example, during fasting, oxidative stress, or infection [45]. It has been shown that GSK3β is responsible for the induction of apoptosis upon inhibition of AKT. Suppression of GSK3β also decreases the level of basal apoptosis and induces proliferation [46].
The protein kinase mTOR (also called the mammalian target of rapamycin) is an evolutionarily conserved protein from yeast to humans. The mTOR activity is finely controlled both positively and negatively by upstream regulators. Positive regulators of mTOR signaling include growth factors and their receptors, such as insulin-like growth factor-1 (IGF-1) and its cognate receptor IFGR-1, members of the human epidermal growth factor receptor (HER) family and their associated ligands, as well as vascular endothelial growth factor receptors (VEGFR) and their ligands that signal mTOR via PI3K/AKT [47]. mTOR exists in the cell as a subunit of the intracellular multimolecular signaling complexes TORC1 and TORC2 [48]. The mTORC1 complex consists of mTOR, Raptor, mLST8, and PRAS40. Importantly, it was found to be sensitive to rapamycin. This discovery allowed to develop the first generation of mTOR inhibitors. The mTORC2 complex consists of mTOR, Rictor, Sin1, and mLST8. It is less sensitive to rapamycin, and its role in normal cell function and tumorigenesis is not fully understood [49]. It is important to underline that mTORC2 activates AKT, thereby promoting cell proliferation and survival. The canonical mTOR activation pathway depends on PI3K/AKT signaling, but alternative non-AKT-dependent activation pathways are also known, such as the Ras/MEK/ERK pathway [50] (Figure 1).
2.2. PTEN
Phosphatase and Tensin Homolog deleted on Chromosome 10 (PTEN) is a tumor suppressor that exhibits phosphatase activity preferentially against members of the PI3K oncogenic pathway. Thus, PTEN is considered to be a major negative regulator of the PI3K/AKT pathway. Oppositely, PTEN inactivation augments the activity of the PI3K/AKT pathway, resulting in the better survival, proliferation, differentiation, and migration of cancer cells.
Under normal conditions, only a small fraction of PTEN molecules bind to the plasma membrane and the rest mainly localize in the cytoplasm. To perform its membrane-bound functions, PTEN first needs to be activated through post-translational modifications and then recruited to the plasma membrane. PTEN acts as a lipid phosphatase that removes the phosphate group from the second messenger PIP3 (phosphatidylinositol (3,4,5)-trisphosphate) to generate PIP2 (phosphatidylinositol (4,5)-bisphosphate), thereby controlling cellular proliferation, survival, growth, and migration [51]. Notably, PTEN acts not only through the lipid but also via its protein phosphatase activity. PTEN is considered to be a dual protein and lipid phosphatase. Furthermore, PTEN can operate non-enzymatically [52].
The regulation of PTEN expression occurs at multiple levels, including epigenetic silencing, transcriptional repression, post-transcriptional regulation by miRNAs, and disruption of competitive endogenous RNA (ceRNA) networks. In addition, PTEN-interacting proteins also affect the activity and functions of PTEN [53,54].
Not surprisingly, PTEN itself, or its functions, are frequently lost either partially or fully in different types of cancer [55]. PTEN inactivation can occur through various genetic alterations such as epigenetic mechanisms (promoter hypermethylation), large chromosomal deletions, or point mutations [56].
PTEN activity can also be regulated by various signaling cues. For example, the sphingosine 1-phosphate2 receptor (S1P2R), which belongs to the S1PR family of G protein-coupled receptors (GPCR), has been shown to activate PTENs by coupling them to the Rho-dependent activation. As a result, the PI-3 kinase pathway is inhibited and cell migration is attenuated [57].
Lima-Fernandes et al. [58] used an elegant in cellulo approach to monitor the conformational state of PTEN in response to various extracellular signals. By employing the intramolecular bioluminescence resonance energy transfer (BRET)-based PTEN conformation-specific biosensor as a reporter, they uncovered that PTEN was activated by several G protein-coupled receptors, including the thromboxane A2 receptor (TPαR) and the muscarinic M1 receptor (M1MR), which were not previously recognized as PTEN regulators.
As mentioned above, PTEN localizes both to the cytoplasm and nucleus. At the plasma membrane, PTEN plays an important role in activating apoptosis by suppressing AKT and, as a result, activating caspase-8 and caspase-9, increasing expression of several pro-apoptotic genes [59].
Localization and activity of PTEN is regulated largely through post-translational modifications. PTEN monoubiquitination is implicated in the mediation of the nuclear accumulation of PTEN by several E3 ligases such as NEDD4, WWP2, XIAP, CHIP, and others, whereas, on the contrary, the polyubiquitination of PTEN induces its proteasome-mediated degradation [60,61,62,63,64,65]. SUMOylation of PTEN at lysine 254 was shown to be also involved in its nuclear translocation [66]. In this respect, it should be noted that Ca2+ mediates the interaction of PTEN with major vault protein (MVP) and regulates the localization of PTEN in the nucleus [67]. Furthermore, it was shown that PNUTS (protein phosphatase-1 nuclear targeting subunit, PPP1R10) interacts with PTEN, blocking the access of PTEN to membrane lipids and hence activating its translocation to the nucleus [68]. In turn, nuclear PTEN participates in DNA repair and replication, and gene expression, as well as controlling cell cycle progression [69]. Notably, members of the PI3K pathway, PI3K and AKT, are also found in the nucleus. Thus, PTEN can act as a lipid phosphatase both at the plasma and nuclear membranes [70,71].
Importantly, PTEN also regulates the stability and transcriptional activity of another tumor suppressor, p53 [72]. Moreover, PTEN was demonstrated to be a transcriptional target of p53 [73], thus forming a positive regulatory feedback loop. On the contrary, another transcriptional target of p53, E3-ubiquitin ligase MDM2, which is a negative regulator of p53, mediates its proteasome-mediated degradation [74]. Various inhibitors of the p53-Mdm2 interaction have been developed and are currently being investigated at different stages of clinical trials [75,76]. Interestingly, AKT can phosphorylate MDM2, which leads to the stabilization of the latter and thus keeps low levels of the tumor suppressor p53 [77]. In turn, inhibition of the PI3K/AKT pathway by PTEN mediates the accumulation of MDM2 in the cytoplasm [78] (Figure 2). Chung et al. [79] showed that, being localized in the nucleus, PTEN inhibits the expression of cyclin D1, thereby causing cell cycle arrest. Nuclear PTEN plays an important role in the maintenance of chromosomal integrity by association with the centromere specific binding protein C (CENP-C)—a key component of centromeres that is important for kinetochore attachment during mitosis [80,81]. Moreover, PTEN regulates the expression of and recruits Rad51 to chromatin favoring double strand break (DSB) repair [81]. Rad51 is one of the central figure factors of the DSB repair machinery that mediates homologous recombination repair [82]. In the nucleus, PTEN may act as regulator of DNA repair at different levels, and it is not surprising that cells lacking nuclear PTEN are sensitive to DNA damage. Importantly, the functioning of PTEN in the nucleus does not require its functional catalytic domain [66].
It is important to note that a number of mutations in the Tp53 gene convert p53 from a tumor suppressor to an oncogene. In this respect, Li et al. [83] showed that by increasing the level of mutant p53 (gain-of-function) via the inhibition of Mdm2-mediated degradation, PTEN can also be an oncogene. Thus, the mutational statuses of p53 and PTEN are important factors to be considered for understanding the aggressiveness of a tumor.
3. PTEN and EMT
3.1. PTEN and EMT Transcription Factors
The ZEB family of transcription factors consists of two members: ZEB1 (alternatively, TCF8 and δEF1) and ZEB2 (SIP1 and ZFXH1B). The members of this protein family interact with DNA through the binding of the two zinc-finger domains to E-boxes, and repress the expression of E-cadherin (Figure 3).
Liu et al. [84] showed that the repression of ZEB1 leads to the induction of PTEN. More detailed experiments revealed that ZEB1 affects the AKT pathway and inhibits the expression of PTEN in breast cancer cells. Moreover, ZEB1 binds to the PTEN promoter region in the MCF7 cell line. Using a luciferase reporter assay, Gu et al. [85] demonstrated that ZEB1 suppresses the transcription of the PTEN gene and as a result activates the AKT pathway.
ZEB2 is the transcription factor that, along with ZEB1, controls EMT. Interestingly, ZEB2 transcripts act as ceRNA towards miRNA targeting PTEN in melanoma cells. Moreover, the depletion of ZEB2 activates the AKT pathway and enhances oncogenic cell transformation of melanoma [86].
The SNAIL family of transcription repressors encompasses three members: SNAIL1 (SNAIL), SLUG (SNAIL2), and SNAIL3 (SMUC). Being important effectors of EMT, these proteins inhibit the expression of E-cadherin. In addition, the SNAIL family members were also shown to participate in PTEN regulation. It was demonstrated that SNAIL1 prevents p53 association with the PTEN promoter region after γ-radiation, thus downregulating the expression of PTEN [73,87]. Another member of this protein family, SLUG, was shown to repress PTEN by directly binding to the PTEN promoter in prostate cancer cells [88].
3.2. PTEN and Tumor Microenviroment
The EMT is triggered not only by intrinsic but also by extrinsic factors. Tumor cells interact with the tumor microenvironment (TME), including stromal cells, the extracellular matrix, and secretome elements (cytokines, growth factors, hormones, and so on). There is much evidence accumulated to date suggesting that hypoxia and reactive oxygen species (ROS) affect the onset of EMT. A common feature of various solid tumors is intra-tumoral hypoxia or a low level of oxygen. Hypoxia correlates with altered gene expression, inhibition of apoptosis, activation of autophagy, and EMT. Hypoxia also favors the production of ROS via several mechanisms [89,90,91]. Increased ROS levels have been detected in many types of cancers, correlating with tumor development and progression. Moreover, the reverse scenario is also true, i.e., that hypoxia can be mediated by increased levels of ROS [92], and the mitochondrial ROS is considered to be instrumental for hypoxia-induced EMT [93]. PTEN itself contains nucleophilic cysteine residues in the active site and thus is considered to be sensitive to oxidation. Cysteine residues (Cys-124 in the catalytic domain) can form an intramolecular disulfide bridge with Cys 71, inhibiting the phosphatase activity [94].
EMT is promoted by direct interaction between tumor and stromal cells. The tumor stroma consists of an extracellular matrix, immune and endothelial cells, as well as cancer-associated fibroblasts (CAFs). CAFs represent the major cellular component among all the stromal cells. Normal fibroblasts can become CAFs under the influence of cancer-derived cytokines. Inactivation of PTEN in the stroma correlates with poor outcomes of cancer patients [95]. For example, mutations in the PTEN gene of stromal fibroblasts were detected in mammary carcinoma [96]. Although these PTEN mutations identified in stromal cells might be due to contaminations by carcinoma cells or to EMT, downregulation of PTEN in CAFs might contribute to neoplastic progression. Trimboli et al. [97] showed that the inactivation of PTEN in stromal fibroblasts leads to the progression of mammary epithelial tumors. Moreover, PTEN-specific signatures in the tumor stroma of patients with breast cancer were identified by gene expression profiling. Thus, it is prudent to say that PTEN can be considered a critical tumor-suppressive component of stroma-specific signaling pathways in solid tumors.
3.3. TGF-β and PTEN
Transforming growth factor β (TGF-β) is a multifunctional cytokine family that regulates cell proliferation, apoptosis, differentiation and migration. This signaling protein family is considered to be one of the key regulators of the EMT process. The TGF-β family in mammals includes three members, TGF-β1, TGF-β2, and TGF-β3. TGF-βs are expressed as pro-proteins, and furin proteases are necessary for proteolytic processing and the activation of TGF-βs. Activated TGF-β complexes bind to serine/threonine kinases-TGF-β receptors [98]. There are two signaling pathways triggered by TGF-β: the canonical pathway induces the phosphorylation of the SMAD family of transcription factors, while the non-canonical pathway includes MAPK, PI3K/AKT, and Rho GTPase. Upon phosphorylation, SMADs form a trimeric structure (SMAD2, SMAD3, and SMAD4) and translocate to the nucleus to regulate the transcription of target genes by directly binding to DNA [99] (Figure 4).
The regulation of PTEN expression by TGF-β signaling is multifaceted and depends on the cellular context. While PTEN was initially identified as a TGF-β-regulated and epithelial cell-enriched phosphatase (TEP1) [100], this concept was challenged by Kimbrough-Allah et al. [101], who reported that, in prostate cancer cells, TGF-β increased the protein level of PTEN, but had no effect on the PTEN mRNA expression level. However, it should be noted that the effect of TGF-β on the protein level of PTEN was very modest and is unlikely to be physiologically relevant. In turn, PTEN overexpression decreased AKT phosphorylation induced by TGF-β [101].
In contrast to the report of Kimbrough-Allah et al., in SMAD4-null pancreatic cancer cells, it was shown that TGF-β suppressed PTEN expression. It is important to note that the PTEN-coding gene is rarely mutated in pancreatic cancers and the authors hypothesized that PTEN regulation by TGF-β can confer changes in cell growth [102].
Furthermore, in SMAD4-null colon cancer cells, TGFβ also downregulated PTEN mRNA and concomitantly induced growth proliferation [103]. Collectively, the literature data strongly suggest that, in most cancer types, TGF-β attenuates PTEN expression.
Interplay between PI3K/AKT and TGF-β/SMAD signaling pathways was identified in wild-type mouse endometrial epithelial cells. Eritja et al. [104] demonstrated that TGF-β induced apoptosis via the activation of PTEN transcription and the inhibition of the PI3K/AKT pathway. Additionally, it was shown that both the expression of PTEN and AKT phosphorylation depend on SMAD3 activity. Thus, in this study, the link between AKT/PTEN/SMAD3 in response to TGF-β was demonstrated [104].
PTEN activity depends on various post-translational modifications such as oxidation, acetylation, and ubiquitination [105]. For example, phosphorylation at sites Ser380, Thr382, and Thr383 leads to the suppression of PTEN phosphatase activity [106]. Aoyama et al. [107] demonstrated that the activation of TGF-β increased the phosphorylation of PTEN at these residues. Moreover, the expression of PTEN with mutations in these inhibitory phosphorylation sites was able to repress EMT induced by TGF-β by blocking β-catenin translocation from E-cadherin complexes at the cell membrane into the cytoplasm [107].
Indeed, β-catenin plays the central role in the canonical Wnt/β-catenin signaling pathway, which regulates embryonic development and, in the event of abnormal activation, leads to metastases. There is a crosstalk between TGF-β and Wnt/β-catenin signaling pathways [108,109]. β-catenin is involved in various processes such as the regulation of cell adhesion and the transcription of genes. E-cadherin binds to β-catenin and promotes cell–cell adhesion. The activation of TGF-β induces the dissociation and stabilization of β-catenin in the cytoplasm, allowing β-catenin to be translocated to the nucleus. β-catenin is a critical element of Wnt signaling. Nuclear β-catenin reflects the aberrant activation of Wnt/β-catenin signaling in cancer cells, leading to dysregulated transcription and hence the activation of EMT [110,111].
3.4. NOTCH and PTEN
The Notch signaling pathway is transmitted through evolutionarily conserved Notch receptors, which are localized on the plasma membrane and affect the transcription of different genes. There are four isoforms of Notch proteins: Notch1, 2, 3, and 4, which work as heterodimeric receptors by binding two classes of ligands: Delta-like proteins and Jagged. The binding of ligands allows proteolytic cleavage to release active Notch intracellular domains (NICD). Active NICD translocate to the nucleus and bind transcription factors to regulate gene expression. The activity of Notch receptors is regulated not only by binding to ligands but also by post-translational modifications, epigenetic factors, and ligand cleavage [112]. Notch pathways take part in numerous embryonic developmental processes and are implicated in cancer progression and EMT (Figure 4). Saad et al. [113] demonstrated that the overexpression of Notch1 increases Snail1 expression and decreases the level of E-cadherin. Indeed, the Notch pathway regulates EMT in different types of cancer. For example, the activation of Notch1 enhanced EMT in gefitinib-resistant lung cancer cell lines. On the contrary, the inhibition of Notch1 reversed the mesenchymal phenotype of cancer cells to the epithelial one and increased the sensitivity of cancer cells to gefitinib [114]. At the same time, Notch3 has the opposite effect on EMT through the transactivation of Hippo/Yap signaling in breast cancer [115]. Zhang et al. [116] showed that the PTEN gene was transactivated by Notch3 in breast cancer cells, resulting in the inhibition of their migration and proliferation, thereby contributing to a favorable prognosis for breast cancer patients.
Some of the earlier proofs of the link between PTEN and EMT came from the study demonstrating that PTEN inhibits cell migration through the phosphatase-mediated dephosphorylation of the FAK (focal adhesion kinase) protein [117]. FAK is a cytoplasmic tyrosine kinase that plays an important role in EMT and mediates the signaling of growth factor receptors and integrins. Moreover, FAK induces a PI3K signaling pathway and regulates the transcriptions of vimentin and Snail. Thus, Li et al. [118] demonstrated that Notch1 signaling regulates the expression of PTEN, EMT-associated genes, and the activation of FAK through the transcriptional regulator RBP-Jκ in tongue cancer. The study by Baker et al. [119] confirmed that Notch1 downregulates PTEN expression leading to the inhibition of ERK1/2 signaling in HER2-positive breast cancer.
3.5. NF-κB and PTEN
NF-κB (Nuclear factor-κB) represents several inducible transcription factors, which regulate the transcription of genes involved in the immune response, survival, angiogenesis, and EMT. The NFκB family consists of five subunits: Rel (cRel), p65 (RelA, NFκB3), RelB, p50 (NFκB1), and p52 (NFκB2). These proteins contain the Rel-homology domain (RHD), which is responsible for homo- or hetero-dimerization, nuclear localization, and transcription [120]. Multiple links between the EMT and NFκB pathway have been demonstrated (Figure 4).
By using a luciferase reporter assay and chromatin immunoprecipitation, Pires et al. [121] showed that NF-κB/p65 directly binds promoters of the SLUG, TWIST1, and SIP1 genes in breast cancer cells. Several lines of evidence point to the link between NF-κB and EMT. The correlation between NF-κB expression and EMT-specific transcription factors was shown in breast cancer, renal carcinoma, prostate cancer, and other types of cancer [122,123,124]. Julien et al. [125] demonstrated that the AKT-dependent activation of NF-κB induces the expression of Snail, which in turn inhibits the expression of PTEN, thereby forming the PTEN/Akt/NF-κB/Snail feedback loop. Moreover, it was shown that the A-kinase-interacting protein 1 (AKIP1) induced NF-κB-mediated EMT by downregulating PTEN in cervical cancer [126].
Elegant experiments performed by Zhao [127] and colleagues established the link between PTEN and the chromatin-mediated regulation of the NF-κB pathway in prostate and breast cancers. Decreased expression of PTEN resulted in the stabilization of the chromatin helicase DNA-binding factor CHD1, which engages the trimethyl lysine-4 histone H3 modification to activate the transcription of cancer-promoting genes involved in the NF-κB pathway [127].
3.6. Integrins and PTEN
Integrins represent a family of transmembrane receptors that mediate cell adhesion to extracellular matrices. These receptors are presented by heterodimers composed of 18α and 8β subunits, which form at least 24 different integrin heterodimers. The general structure of α and β subunits appears to be similar. These subunits possess a globular N-terminally located extracellular domain, the transmembrane domain, and the small C-terminal cytoplasmic tail [128]. The α-subunits mediate ligand specificity, and the β subunits form a bridge between the extracellular matrix and integrins. These receptors control a variety of signaling pathways including actin polymerization, nucleation, PI3K, AKT, and MAPK [129]. Being key regulators of cellular adhesion, integrins also participate in the EMT process. Integrins support the colonization of metastatic cells and mediate the survival of circulating tumor cells [128]. Integrins act as receptors and transduce signals through additional proteins that connect to the cytoskeleton, transmembrane growth factor receptors, and cytoplasmic kinases [130]. Integrins themselves lack any enzymatic activity and all of the functions they exert are mediated via protein–protein interactions, resulting in the assembly of actin filaments [131]. The clustering of integrins and the reorganization of actin through adaptor proteins enhances the formation of stable aggregates named focal adhesion sites (FAS) [130]. These FASs mediate signal transduction to ensure different aspects of the EMT process such as proliferation, wound healing, and migration [132] (Figure 4).
Using several lung cancer cell lines with an ectopic expression of PTEN, Yu et al. identified differentially expressed genes by microarray analysis. One of the genes on top of the microarray list was αVβ6 integrin. The expression of PTEN decreased the protein level of αVβ6 integrin, thereby affecting the migration and invasion of lung cancer cell lines. As mentioned above, one of the earliest studies elucidating the impact of PTEN on the extracellular matrix (ECM) rearrangement during EMT was performed by Tamura et al. [133]. Importantly, PTEN-mediated dephosphorylation of FAK at Tyr397 is a key event ensuring the formation of intercellular contacts mediated by integrins. PTEN therefore negatively regulates cell interactions with the extracellular matrix [134]. Interestingly, on the other hand, PTEN is a substrate of FAK-mediated Tyr336 phosphorylation. This event, in turn, leads to increased phosphatase activity, an augmented protein–lipid interaction, and the stability of PTEN [135]. Thus, there is a crosstalk between the activity of FAK and the stability of PTEN.
Moreover, dephosphorylation by PTEN affects FAK adhesion to collagens. Collagens are the major fibrous proteins of ECM [136], while paxillin is a multidomain protein that transduces signals from integrins in order to reorganize the actin cytoskeleton. It also modulates actin filament dynamics and mediates the engagement of integrins with extracellular matrices [137]. It was demonstrated that the interactions of PTEN with FAK and paxillin weaken their interactions with ECM components, e.g., collagens I/IV [138]. Thus, PTEN may contribute to the process of destabilization of cell adhesion to ECM.
3.7. PTEN and Some Scaffolding Proteins
As mentioned above, one of the main hallmarks of EMT is the loss of epithelial markers, i.e., E-cadherin. The lipid phosphatase activity of PTEN is instrumental in stabilizing E-cadherin on the protein level. The expression of wild-type PTEN suppressed the invasive phenotype in an E-cadherin-dependent manner. Thus, the inactivation of PTEN can activate cell dissemination and metastasis [139]. While the extracellular region of E-cadherin extends from the cell surface and is involved in cell adhesion, the intracellular region interacts with catenins to form a complex E-cadherin/catenin. E-cadherin/catenin protein complexes are considered to be a part of bigger complexes that involve growth factor receptors and scaffolding molecules. For example, MAGI-1b, which belongs to the group of membrane-associated guanylate kinases (MAGUK), interacts with β-catenin. Kotelevets et al. showed that PTEN interacted with β-catenin by binding MAGI-1b, a scaffold protein with PDZ domains that mediate multiple protein–protein interactions [140]. Moreover, the expression of MAGI-1b is important for the interaction of junction complexes and PTEN, which promotes E-cadherin-dependent cell aggregation, thus suppressing invasion [140]. Interestingly, MAGI1 interacts with Thyroid Hormone Receptor Interactor 6 (TRIP6). In turn, enhanced levels of cytoplasmic TRIP6 compete away β-catenin from MAGI1. Thus, TRIP6 allows β-catenin translocation to the nucleus and subsequently activates Wnt/β-catenin signaling [141]. In addition, TRIP6 favors the dissociation of PTEN from the E-cadherin complexes, thereby activating invasiveness [142]. Thus, scaffold proteins play an important part in the participation of PTEN in EMT, affecting both protein–protein interactions and signaling pathways.
Furthermore, PTEN interacts with a scaffolding protein β-arrestin1, which regulates the signal transduction mediated by G protein-coupled receptor (GPCR). β-Arrestin1 regulates proliferation, apoptosis, angiogenesis, invasion, and migration in a variety of tumors. PTEN controls the multicellular assembly by controlling the localization and interaction of β-Arrestin1 [143]. On the other hand, β-arrestin is an important regulator of the lipid-phosphatase activity of PTEN and can regulate PTEN-dependent cell proliferation and migration [144].
Qi et al. identified a new substrate of PTEN, Abi1, which is a core adaptor protein of the WAVE regulatory complex (WRC). Abi1 is a scaffolding protein within the WRC and mediates the membrane recruitment and stabilization of WRC subunits [145]. WAVE proteins play an important role in immunity, neurogenesis, embryogenesis, cancer invasion, and metastasis [146]. It was shown that Abi1 is significantly upregulated in PTEN-deficient breast cancer cells [147]. The protein phosphatase activity of PTEN is required for the dephosphorylation and degradation of Abi1, which otherwise promotes EMT in breast cancer.
4. miRNA and PTEN
miRNAs are short non-coding RNAs that play an important role in the regulation of their target genes and are involved in different cellular processes such as differentiation, embryonic development, and EMT. In most cases, miRNAs interact with the 3′UTR (untranslated regions) of their target mRNAs and subsequently downregulate their expressions [148]. Notably, several microRNAs (miRNAs) have been recognized as regulators of PTEN [149].
Unsurprisingly, several miRNAs have been shown to regulate EMT by targeting PTEN/PI3K pathways. For example, miRNA-410 was shown to induce EMT and radioresistance in lung cancer cells by targeting the PI3K/mTOR pathway [150]. Another miRNA, miR-21 post-transcriptionally down-regulates the expression of PTEN and stimulates the invasiveness of non-small cell lung cancer cells [151]. In recent years, different miRNA regulators of PI3K/AKT/PTEN pathways have been discovered in various cancers. miRNA-221/222, miRNA-20a, and miRNA-200c were reported to target PTEN and modulate metastases in ovarian and breast cancer stem cells [152,153]. In addition, a number of microRNAs including miRNA-106b, miRNA-93, miRNA-144, and miRNA-202-5p regulate cell proliferation by targeting PTEN in breast cancer [154,155,156]. A list of various miRNAs is shown in Table 1. More information about the role of miRNAs in the regulation of the expression and activity of PTEN is available in the review by Ghafouri-Fard et al. [157].
5. lncRNA and PTEN
A number of recent publications have demonstrated the role of long-non-coding RNA (lncRNA) as important regulators of various cellular processes. Those RNAs are transcribed longer than 200 nucleotides but not translated into functional proteins. lncRNAs can regulate gene expression at different levels: chromatin organization, transcription, mRNA stability, miRNA sponging, and post-translation modifications of proteins. Moreover, they are involved in the formation of nuclear condensates and non-membrane organelles [174].
In this respect, the expression of a lncRNA named GAEA (Glucose Aroused for EMT Activation), which is induced by a variety of stimuli including high glucose, TGF-β, CTGF, SHH, and IL-6, was shown to enhance the enzymatic activity of the MEX3C RNA-binding E3 ligase that mediates the K27-linked polyubiquitination of PTEN. This covalent modification can change the specificity of PTEN activity from phosphatidylinositol/tyrosine phosphatase to serine/threonine phosphatase. Polyubiquitinated PTEN can dephosphorylate TWIST1, SNAIL, and YAP1, thereby mediating an accumulation of EMT transcription regulators [175].
Another lncRNA, Linc00702, is usually under-expressed in colorectal cancer. Importantly, the overexpression of Linc00702 reduced the invasion and migration of colorectal cancer cells. Mechanistically, Linc00702 induces PTEN expression and thus represses the PI3K/AKT pathway, thereby playing a tumor suppressive role in cancer [176]. Linc00702 is also downregulated in non-small cell lung carcinoma. The overexpression of this lncRNA in lung cancer cell lines inhibits cell growth and invasiveness by sponging miR-510, which targets PTEN transcripts [177].
On the contrary, Yan et al. [178] showed that Linc00470 mediated the degradation of PTEN mRNA in gastric cancer. Moreover, the expression of Linc00470 correlated with a poor prognosis and distant metastases in patients with gastric cancer. Thus, it can be concluded that lncRNAs differentially affect PTEN depending on their molecular targets and the cellular context and hence have different effects on the EMT and metastasis formation.
6. Conclusions
Here, we attempted to summarize the current knowledge regarding the role of PTEN in the EMT process. PTEN, being the major inhibitor of the PI3K/AKT pathway, plays an important part in the regulation of cell proliferation, differentiation, metabolism, and migration. The coordination of different signaling pathways, including PI3K/AKT, TGF-β/Smad, Notch, and NF-κB, is required to control such cellular processes as EMT. The molecular particularities of crosstalk between these signaling pathways were not fully explored. In this review, we provided evidence suggesting that PTEN is involved in the regulation of EMT. Specifically, we found that the expression of PTEN is regulated by EMT transcription factors. In turn, PTEN regulates the activity of different signaling pathways that orchestrate the EMT and metastasis formation in cancer. Moreover, we referred to recent works demonstrating the influence of PTEN-targeting miRNAs and lncRNAs on the EMT.
To conclude, we would like to set the following questions that need to be answered in the future to decipher the molecular mechanism of PTEN participation in the EMT process:
- -
- Does complete ablation of PTEN or loss of its enzymatic function activate EMT on its own, or does this process require additional signaling?
- -
- Is PTEN required for the reverse process of EMT, mesenchymal-to-epithelial transition?
- -
- Can PTEN be a therapeutic target for the treatment of metastatic cancer?
Mutations in the tumor suppressor PTEN occur in different tumors. Understanding the role of PTEN in cancer invasion and in the development of metastasis provides new insights for drug development and in the search for promising new potential biomarkers. PTEN plays a crucial role in the activity of cancer stem cells, which are responsible for resistance to therapy and tumor metastatic spread [179].
Importantly, recent studies show that PTEN directly regulates resistance to radiation, chemo-, and immunotherapy [180]. The link between EMT and resistance to anticancer therapy has been reported in several reviews [10,181,182]. The general mechanism underlying EMT-driven drug resistance is related to an increased drug efflux by multiple cell membrane transporter proteins, especially the ABC transporter family of proteins, avoiding drug-induced apoptosis, slow cell proliferation, and the altered expression of molecules involved in immunosuppression [10,11]. Though cancer metastasis is the major cause of death from cancer, it is hard to overstate the problems caused by resistance to anticancer drugs. Thus, novel therapeutic strategies to activate PTEN in cancer cells may yield a more efficient way to treat drug-resistant cancers.
Author Contributions
Conceptualization, O.F. and N.A.B., writing—original draft preparation, O.F., A.D. and O.S. writing—review and editing, O.F., A.D., S.P. and N.A.B.; visualization, O.F. and S.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation (RSF), #18-75-10076.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulchinsky, E.; Demidov, O.; Kriajevska, M.; Barlev, N.A.; Imyanitov, E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Biophys. Acta 2018, 1871, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Garg, M. Epithelial-mesenchymal transition-activating transcription factors-multifunctional regulators in cancer. World J. Stem Cells 2013, 5, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Inoue, A.; Seidel, M.G.; Wu, W.; Kamizono, S.; Ferrando, A.A.; Bronson, R.T.; Iwasaki, H.; Akashi, K.; Morimoto, A.; Hitzler, J.K.; et al. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2002, 2, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Olmeda, D.; Moreno-Bueno, G.; Flores, J.M.; Fabra, A.; Portillo, F.; Cano, A. SNAI1 Is Required for Tumor Growth and Lymph Node Metastasis of Human Breast Carcinoma MDA-MB-231 Cells. Cancer Res. 2007, 67, 11721–11731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2018, 24, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer Stem Cells: The Promise and the Potential. Semin. Oncol. 2015, 42, S3–S17. [Google Scholar] [CrossRef]
- Chen, K.; Huang, Y.-H.; Chen, J.-L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, A.; Pandolfi, P.P. The PTEN–PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-Surface Receptors Transactivation Mediated by G Protein-Coupled Receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Meyers, R.E.; Cantley, L.C. PHOSPHOINOSITIDE KINASES. Annu. Rev. Biochem. 1998, 67, 481–507. [Google Scholar] [CrossRef]
- Dbouk, H.A.; Backer, J.M. A beta version of life: p110β takes center stage. Oncotarget 2010, 1, 729–733. [Google Scholar] [CrossRef] [Green Version]
- Fox, M.; Mott, H.R.; Owen, D. Class IA PI3K regulatory subunits: p110-independent roles and structures. Biochem. Soc. Trans. 2020, 48, 1397–1417. [Google Scholar] [CrossRef]
- Koch, P.A.; Dornan, G.L.; Hessenberger, M.; Haucke, V. The molecular mechanisms mediating class II PI 3-kinase function in cell physiology. FEBS J. 2021, 288, 7025–7042. [Google Scholar] [CrossRef] [PubMed]
- Vara, J.Á.F.; Casado, E.; De Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B.; et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J. Genet. Genom. 2015, 42, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.F.; Jakubowicz, T.; Hemmings, B.A. Molecular cloning of a second form of rac protein kinase. Cell Regul. 1991, 2, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.F.; Jakubowicz, T.; Pitossi, F.J.; Maurer, F.A.; Hemmings, B. Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc. Natl. Acad. Sci. USA 1991, 88, 4171–4175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masure, S.; Haefner, B.; Wesselink, J.-J.; Hoefnagel, E.; Mortier, E.; Verhasselt, P.; Tuytelaars, A.; Gordon, R.; Richardson, A. Molecular cloning, expression and characterization of the human serine/threonine kinase Akt-3. JBIC J. Biol. Inorg. Chem. 1999, 265, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Hers, I.; Vincent, E.E.; Tavaré, J.M. Akt signalling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef]
- Coffer, P.J.; Jin, J.; Woodgett, J. Protein kinase B (c-Akt): A multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998, 335, 1–13. [Google Scholar] [CrossRef]
- Scheid, M.P.; Woodgett, J.R. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003, 546, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Scheid, M.P.; Woodgett, J.R. PKB/AKT: Functional insights from genetic models. Nat. Rev. Mol. Cell Biol. 2001, 2, 760–768. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Delcommenne, M.; Tan, C.; Gray, V.; Rue, L.; Woodgett, J.; Dedhar, S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 11211–11216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebner, M.; Lučić, I.; Leonard, T.A.; Yudushkin, I. PI(3,4,5)P 3 Engagement Restricts Akt Activity to Cellular Membranes. Mol. Cell 2017, 65, 416–431. [Google Scholar] [CrossRef] [Green Version]
- Girardi, C.; James, P.; Zanin, S.; Pinna, L.A.; Ruzzene, M. Differential phosphorylation of Akt1 and Akt2 by protein kinase CK2 may account for isoform specific functions. Biochim. Biophys. Acta 2014, 1843, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.A.; Pinna, L.; Ruzzene, M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef]
- Liu, P.; Begley, M.J.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [Green Version]
- Burgering, B.M.T.; Medema, R.H. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J. Leukoc. Biol. 2003, 73, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; de Mattos, S.F.; van der Horst, A.; Klompmaker, R.; Kops, G.J.P.L.; Lam, E.W.-F.; Burgering, B.M.T.; Medema, R.H. Cell Cycle Inhibition by FoxO Forkhead Transcription Factors Involves Downregulation of Cyclin D. Mol. Cell. Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [Green Version]
- Ciechomska, I.; Pyrzynska, B.; Kazmierczak, P.; Kaminska, B. Inhibition of Akt kinase signalling and activation of Forkhead are indispensable for upregulation of FasL expression in apoptosis of glioma cells. Oncogene 2003, 22, 7617–7627. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 4629495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell. Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-Kinase Pathway in Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Paluch, B.E.; Wang, X.; Jiang, X. PTEN at a glance. J. Cell Sci. 2012, 125, 4687–4692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Kotelevets, L.; Trifault, B.; Chastre, E.; Scott, M.G.H. Posttranslational Regulation and Conformational Plasticity of PTEN. Cold Spring Harb. Perspect. Med. 2020, 10, a036095. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.L. Phosphatase and tensin homologue deleted on chromosome 10: Extending its PTENtacles. Int. J. Biochem. Cell Biol. 2009, 41, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Molinari, F.; Frattini, M. Functions and Regulation of the PTEN Gene in Colorectal Cancer. Front. Oncol. 2014, 3, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, T.; Thangada, S.; Wu, M.-T.; Kontos, C.D.; Wu, D.; Wu, H.; Hla, T. PTEN as an effector in the signaling of antimigratory G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 4312–4317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima-Fernandes, E.; Misticone, S.; Boularan, C.; Paradis, J.S.; Enslen, H.; Roux, P.P.; Bouvier, M.; Baillie, G.S.; Marullo, S.; Scott, M. A biosensor to monitor dynamic regulation and function of tumour suppressor PTEN in living cells. Nat. Commun. 2014, 5, 4431. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Y.; Wang, Y.; Chan, A.M. Multifaceted Regulation of PTEN Subcellular Distributions and Biological Functions. Cancers 2019, 11, 1247. [Google Scholar] [CrossRef] [Green Version]
- Maddika, S.; Kavela, S.; Rani, N.; Palicharla, V.R.; Pokorny, J.L.; Sarkaria, J.N.; Chen, J. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 2011, 13, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.F.; Deb, S.; Paul, I.; Chatterjee, A.; Mandal, T.; Chatterjee, U.; Ghosh, M.K. The Chaperone-assisted E3 Ligase C Terminus of Hsc70-interacting Protein (CHIP) Targets PTEN for Proteasomal Degradation. J. Biol. Chem. 2012, 287, 15996–16006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-S.; Jeong, M.-H.; Lee, H.-W.; Han, H.-J.; Ko, A.; Hewitt, S.M.; Kim, J.-H.; Chun, K.-H.; Chung, J.-Y.; Lee, C.; et al. PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis. Nat. Commun. 2015, 6, 7769. [Google Scholar] [CrossRef]
- Van Themsche, C.; Leblanc, V.; Parent, S.; Asselin, E. X-linked Inhibitor of Apoptosis Protein (XIAP) Regulates PTEN Ubiquitination, Content, and Compartmentalization. J. Biol. Chem. 2009, 284, 20462–20466. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Trotman, L.C.; Koppie, T.; Alimonti, A.; Chen, Z.; Gao, Z.; Wang, J.; Erdjument-Bromage, H.; Tempst, P.; Cordon-Cardo, C.; et al. NEDD4-1 Is a Proto-Oncogenic Ubiquitin Ligase for PTEN. Cell 2007, 128, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotman, L.C.; Wang, X.; Alimonti, A.; Chen, Z.; Teruya-Feldstein, J.; Yang, H.; Pavletich, N.P.; Carver, B.S.; Cordon-Cardo, C.; Erdjument-Bromage, H.; et al. Ubiquitination Regulates PTEN Nuclear Import and Tumor Suppression. Cell 2007, 128, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Bassi, C.; Ho, J.; Srikumar, T.; Dowling, R.J.O.; Gorrini, C.; Miller, S.J.; Mak, T.W.; Neel, B.G.; Raught, B.; Stambolic, V. Nuclear PTEN Controls DNA Repair and Sensitivity to Genotoxic Stress. Science 2013, 341, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Minaguchi, T.; Waite, K.A.; Eng, C. Nuclear Localization of PTEN Is Regulated by Ca2+ through a Tyrosil Phosphorylation–Independent Conformational Modification in Major Vault Protein. Cancer Res. 2006, 66, 11677–11682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavela, S.; Shinde, S.R.; Ratheesh, R.; Viswakalyan, K.; Bashyam, M.D.; Gowrishankar, S.; Vamsy, M.; Pattnaik, S.; Rao, S.; Sastry, R.A.; et al. PNUTS Functions as a Proto-Oncogene by Sequestering PTEN. Cancer Res. 2013, 73, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.; Cruise, E.S.; Dowling, R.J.O.; Stambolic, V. PTEN Nuclear Functions. Cold Spring Harb. Perspect. Med. 2019, 10, a036079. [Google Scholar] [CrossRef]
- Déléris, P.; Gayral, S.; Breton-Douillon, M. Nuclear Ptdlns(3,4,5)P3 signaling: An ongoing story. J. Cell. Biochem. 2006, 98, 469–485. [Google Scholar] [CrossRef]
- Neri, L.M.; Borgatti, P.; Capitani, S.; Martelli, A.M. The nuclear phosphoinositide 3-kinase/AKT pathway: A new second messenger system. Biochim. Biophys. Acta. 2002, 1584, 73–80. [Google Scholar] [CrossRef]
- Tang, Y.; Eng, C. PTEN Autoregulates Its Expression by Stabilization of p53 in a Phosphatase-Independent Manner. Cancer Res. 2006, 66, 736–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN Transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.; Daks, A.; Petrova, V.; Petukhov, A.; Lezina, L.; Shuvalov, O.; Davidovich, P.; Kriger, D.; Lomert, E.; Tentler, D.; et al. Novel isatin-derived molecules activate p53 via interference with Mdm2 to promote apoptosis. Cell Cycle 2018, 17, 1917–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezina, L.; Aksenova, V.; Fedorova, O.; Malikova, D.; Shuvalov, O.; Antonov, A.V.; Tentler, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. KMT Set7/9 affects genotoxic stress response via the Mdm2 axis. Oncotarget 2015, 6, 25843–25855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chibaya, L.; Karim, B.; Zhang, H.; Jones, S.N. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2003193118. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. The PTEN, Mdm2, p53 tumor suppressor–oncoprotein network. Trends Biochem. Sci. 2002, 27, 462–467. [Google Scholar] [CrossRef]
- Chung, J.-H.; Eng, C. Nuclear-Cytoplasmic Partitioning of Phosphatase and Tensin Homologue Deleted on Chromosome 10 (PTEN) Differentially Regulates the Cell Cycle and Apoptosis. Cancer Res. 2005, 65, 8096–8100. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, D.W.; Mao, Y.; Sullivan, K.F. Centromeres and Kinetochores: From Epigenetics to Mitotic Checkpoint Signaling. Cell 2003, 112, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential Role for Nuclear PTEN in Maintaining Chromosomal Integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017, 45, 4590–4605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Guessous, F.; Kwon, S.; Kumar, M.; Ibidapo, O.; Fuller, L.; Johnson, E.; Lal, B.; Hussaini, I.; Bao, Y.; et al. PTEN Has Tumor-Promoting Properties in the Setting of Gain-of-Function p53 Mutations. Cancer Res. 2008, 68, 1723–1731. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Siles, L.; Lu, X.; Dean, K.C.; Cuatrecasas, M.; Postigo, A.; Dean, D.C. Mitotic polarization of transcription factors during asymmetric division establishes fate of forming cancer cells. Nat. Commun. 2018, 9, 2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Wan, G.; Yang, Y.; Liu, Y.; Yang, X.; Zheng, Y.; Jiang, L.; Zhang, P.; Liu, D.; Zhao, W.; et al. SLFN5 influences proliferation and apoptosis by upregulating PTEN transcription via ZEB1 and inhibits the purine metabolic pathway in breast cancer. Am. J. Cancer Res. 2020, 10, 2832–2850. [Google Scholar] [PubMed]
- Karreth, F.A.; Tay, Y.; Perna, D.; Ala, U.; Tan, S.M.; Rust, A.G.; DeNicola, G.; Webster, K.A.; Weiss, D.; Perez-Mancera, P.A.; et al. In Vivo Identification of Tumor-Suppressive PTEN ceRNAs in an Oncogenic BRAF-Induced Mouse Model of Melanoma. Cell 2011, 147, 382–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escrivà, M.; Peiró, S.; Herranz, N.; Villagrasa, P.; Dave, N.; Montserrat-Sentís, B.; Murray, S.A.; Francí, C.; Gridley, T.; Virtanen, I.; et al. Repression of PTEN Phosphatase by Snail1 Transcriptional Factor during Gamma Radiation-Induced Apoptosis. Mol. Cell. Biol. 2008, 28, 1528–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uygur, B.; Abramo, K.; Leikina, E.; Vary, C.; Liaw, L.; Wu, W.-S. SLUG is a direct transcriptional repressor of PTEN tumor suppressor. Prostate 2015, 75, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxidative Med. Cell. Longev. 2015, 2016, 3907147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Idelchik, M.d.P.S.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Proc. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Dada, L.A.; Wu, M.; Kelly, A.; Trejo, H.; Zhou, Q.; Varga, J.; Sznajder, J.I. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L1120–L1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Park, J.; Han, S.-J.; Yang, S.Y.; Yoon, H.J.; Park, I.; Woo, H.A.; Lee, S.-R. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biol. 2020, 34, 101553. [Google Scholar] [CrossRef]
- Lefler, J.E.; Seward, C.; Ostrowski, M.C. PTEN in cancer associated fibroblasts. Adv. Cancer Res. 2022, 154, 203–226. [Google Scholar] [CrossRef]
- Wallace, J.A.; Li, F.; Leone, G.; Ostrowski, M.C. Pten in the Breast Tumor Microenvironment: Modeling Tumor–Stroma Coevolution. Cancer Res. 2011, 71, 1203–1207. [Google Scholar] [CrossRef] [Green Version]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.-L.; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poniatowski, Ł.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming Growth Factor Beta Family: Insight into the Role of Growth Factors in Regulation of Fracture Healing Biology and Potential Clinical Applications. Mediat. Inflamm. 2015, 2015, 137823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Li, D.-M.; Sun, H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor β. Cancer Res. 1997, 57, 2124–2129. [Google Scholar] [PubMed]
- Kimbrough-Allah, M.N.; Millena, A.C.; Khan, S.A. Differential role of PTEN in transforming growth factor β (TGF-β) effects on proliferation and migration in prostate cancer cells. Prostate 2018, 78, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.Y.; Quach, K.T.; Cabrera, B.L.; Cabral, J.A.; Beck, S.E.; Carethers, J.M. RAS/ERK modulates TGFbeta-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis 2007, 28, 2321–2327. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.Y.; Cabral, J.A.; Chang, J.; Carethers, J.M. TGFβ modulates PTEN expression independently of SMAD signaling for growth proliferation in colon cancer cells. Cancer Biol. Ther. 2008, 7, 1694–1699. [Google Scholar] [CrossRef] [Green Version]
- Eritja, N.; Felip, I.; Dosil, M.A.; Vigezzi, L.; Mirantes, C.; Yeramian, A.; Navaridas, R.; Santacana, M.; Llobet-Navas, D.; Yoshimura, A.; et al. A Smad3-PTEN regulatory loop controls proliferation and apoptotic responses to TGF-β in mouse endometrium. Cell Death Differ. 2017, 24, 1443–1458. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhen, Y.; Zhou, S.-F.; Lu, N. Posttranslational regulation of phosphatase and tensin homolog (PTEN) and its functional impact on cancer behaviors. Drug Des. Dev. Ther. 2014, 8, 1745–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, F.; Ramaswamy, S.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN Tail Regulates Protein Stability and Function. Mol. Cell. Biol. 2000, 20, 5010–5018. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, D.; Hashimoto, N.; Sakamoto, K.; Kohnoh, T.; Kusunose, M.; Kimura, M.; Ogata, R.; Imaizumi, K.; Kawabe, T.; Hasegawa, Y. Involvement of TGFβ-Induced Phosphorylation of the PTEN C-Terminus on TGFβ-Induced Acquisition of Malignant Phenotypes in Lung Cancer Cells. PLoS ONE 2013, 8, e81133. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions Between β-Catenin and Transforming Growth Factor-β Signaling Pathways Mediate Epithelial-Mesenchymal Transition and Are Dependent on the Transcriptional Co-activator cAMP-response Element-binding Protein (CREB)-binding Protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.P.; Herazo-Maya, J.D.; Sennello, J.A.; Flozak, A.S.; Russell, S.; Mutlu, G.M.; Budinger, G.R.S.; DasGupta, R.; Varga, J.; Kaminski, N.; et al. Wnt Coreceptor Lrp5 Is a Driver of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-Cadherin/β-Catenin Complex and the Epithelial Barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [Green Version]
- Masszi, A.; Fan, L.; Rosivall, L.; McCulloch, C.A.; Rotstein, O.D.; Mucsi, I.; Kapus, A. Integrity of cell-cell contacts is a critical regulator of TGF-β1-induced epithelial-to-myofibroblast transition: Role for β-catenin. Am. J. Pathol. 2004, 165, 1955–1967. [Google Scholar] [CrossRef]
- Wang, M.M. Notch signaling and Notch signaling modifiers. Int. J. Biochem. Cell Biol. 2011, 43, 1550–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, S.; Stanners, S.R.; Yong, R.; Tang, O.; Pollock, C.A. Notch mediated epithelial to mesenchymal transformation is associated with increased expression of the Snail transcription factor. Int. J. Biochem. Cell Biol. 2010, 42, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhang, L.; He, C.-S.; Xu, F.; Liu, J.-L.; Hu, Z.-H.; Zhao, L.-P.; Tian, Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell. Biochem. 2011, 113, 1501–1513. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Luo, J.; Xiao, W.; Ye, X.; Chen, M.; Li, Y.; Zhang, G.-J. Notch3 inhibits epithelial–mesenchymal transition by activating Kibra-mediated Hippo/YAP signaling in breast cancer epithelial cells. Oncogenesis 2016, 5, e269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Q.; Liang, Y.-K.; Wu, Y.; Chen, M.; Chen, W.-L.; Li, R.-H.; Zeng, Y.-Z.; Huang, W.-H.; Wu, J.-D.; Zeng, D.; et al. Notch3 inhibits cell proliferation and tumorigenesis and predicts better prognosis in breast cancer through transactivating PTEN. Cell Death Dis. 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.-I.; Parsons, R.; Yamada, K.M. Inhibition of Cell Migration, Spreading, and Focal Adhesions by Tumor Suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Huang, W.-X.; Zhou, X.; Chen, J.; Li, Z. Numb inhibits epithelial-mesenchymal transition via RBP-Jκ-dependent Notch1/PTEN/FAK signaling pathway in tongue cancer. BMC Cancer 2019, 19, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, A.; Wyatt, D.; Bocchetta, M.; Li, J.; Filipovic, A.; Green, A.; Peiffer, D.S.; Fuqua, S.; Miele, L.; Albain, K.S.; et al. Notch-1-PTEN-ERK1/2 signaling axis promotes HER2+ breast cancer cell proliferation and stem cell survival. Oncogene 2018, 37, 4489–4504. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; De Souza, W.F.; Morgado-Díaz, J.A.; Maia, A.M.; Corrêa, S.; Abdelhay, E.S.F.W. NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef] [Green Version]
- Neil, J.R.; Schiemann, W.P. Altered TAB1:I κB kinase interaction promotes transforming growth factor beta-mediated nuclear factor-kappaB activation during breast cancer progression. Cancer Res. 2008, 68, 1462–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantuck, A.J.; An, J.; Liu, H.; Rettig, M.B. NF-κB–Dependent Plasticity of the Epithelial to Mesenchymal Transition Induced by Von Hippel-Lindau Inactivation in Renal Cell Carcinomas. Cancer Res. 2010, 70, 752–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Helfand, B.T.; Jang, T.L.; Zhu, L.J.; Chen, L.; Yang, X.J.; Kozlowski, J.; Smith, N.; Kundu, S.D.; Yang, G.; et al. Nuclear Factor-κB-Mediated Transforming Growth Factor-β-Induced Expression of Vimentin Is an Independent Predictor of Biochemical Recurrence after Radical Prostatectomy. Clin. Cancer Res. 2009, 15, 3557–3567. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Baritaki, S.; Militello, L.; Malaponte, G.; Bevelacqua, Y.; Bonavida, B. The Role of B-RAF Mutations in Melanoma and the Induction of EMT via Dysregulation of the NF-κB/Snail/RKIP/PTEN Circuit. Genes Cancer 2010, 1, 409–420. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, S.; Zhu, Y. A-kinase-interacting protein 1 promotes EMT and metastasis via PI3K/Akt/IKKβ pathway in cervical cancer. Cell Biochem. Funct. 2020, 38, 782–791. [Google Scholar] [CrossRef]
- Zhao, D.; Lu, X.; Wang, G.; Lan, Z.; Liao, W.; Li, J.; Liang, X.; Chen, J.R.; Shah, S.; Shang, X.; et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature 2017, 542, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, K.K.; Pal, S.; Moulik, S.; Chatterjee, A. Integrins and metastasis. Cell Adhes. Migr. 2013, 7, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2009, 339, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Giancotti, F.G.; Ruoslahti, E. Integrin Signaling. Science 1999, 285, 1028–1033. [Google Scholar] [CrossRef]
- Taherian, A.; Li, X.; Liu, Y.; Haas, T.A. Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer 2011, 11, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniak, M.A.; Modzelewska, K.; Kwong, L.; Keely, P.J. Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta 2004, 1692, 103–119. [Google Scholar] [CrossRef]
- Tamura, M.; Gu, J.; Tran, H.; Yamada, K.M. PTEN Gene and Integrin Signaling in Cancer. JNCI J. Natl. Cancer Inst. 1999, 91, 1820–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perumal, E.; Youn, K.S.; Sun, S.; Seung-Hyun, J.; Suji, M.; Jieying, L.; Yeun-Jun, C. PTEN inactivation induces epithelial-mesenchymal transition and metastasis by intranuclear translocation of β-catenin and snail/slug in non-small cell lung carcinoma cells. Lung Cancer 2019, 130, 25–34. [Google Scholar] [CrossRef]
- Tzenaki, N.; Aivaliotis, M.; Papakonstanti, E.A. Focal adhesion kinase phosphorylates the phosphatase and tensin homolog deleted on chromosome 10 under the control of p110δ phosphoinositide-3 kinase. FASEB J. 2015, 29, 4840–4852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.; Mojares, E.; del Río Hernández, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haier, J.; Nicolson, G.L. PTEN regulates tumor cell adhesion of colon carcinoma cells under dynamic conditions of fluid flow. Oncogene 2002, 21, 1450–1460. [Google Scholar] [CrossRef] [Green Version]
- Kotelevets, L.; Van Hengel, J.; Bruyneel, E.; Mareel, M.; Van Roy, F.; Chastre, E. The lipid phosphatase activity of PTEN is critical for stabilizing intercellular junctions and reverting invasiveness. J. Cell Biol. 2001, 155, 1129–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotelevets, L.; Van Hengel, J.; Bruyneel, E.; Mareel, M.; Van Roy, F.; Chastre, E. Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. FASEB J. 2004, 19, 115–117. [Google Scholar] [CrossRef]
- Gou, H.; Liang, J.Q.; Zhang, L.; Chen, H.; Zhang, Y.; Li, R.; Wang, X.; Ji, J.; Tong, J.H.; To, K.-F.; et al. TTPAL Promotes Colorectal Tumorigenesis by Stabilizing TRIP6 to Activate Wnt/β-Catenin Signaling. Cancer Res. 2019, 79, 3332–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chastre, E.; Abdessamad, M.; Kruglov, A.; Bruyneel, E.; Bracke, M.; Di Gioia, Y.; Beckerle, M.C.; Van Roy, F.; Kotelevets, L. TRIP6, a novel molecular partner of the MAGI-1 scaffolding molecule, promotes invasiveness. FASEB J. 2008, 23, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Javadi, A.; Deevi, R.K.; Evergren, E.; Blondel-Tepaz, E.; Baillie, G.S.; Scott, M.G.; Campbell, F.C. PTEN controls glandular morphogenesis through a juxtamembrane β-Arrestin1/ARHGAP21 scaffolding complex. eLife 2017, 6, e24578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima-Fernandes, E.; Enslen, H.; Camand, E.; Kotelevets, L.; Boularan, C.; Achour, L.; Benmerah, A.; Gibson, L.C.D.; Baillie, G.S.; Pitcher, J.A.; et al. Distinct functional outputs of PTEN signalling are controlled by dynamic association with β-arrestins. EMBO J. 2011, 30, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Liu, J.; Chao, J.; Greer, P.A.; Li, S. PTEN dephosphorylates Abi1 to promote epithelial morphogenesis. J. Cell Biol. 2020, 219, e201910041. [Google Scholar] [CrossRef]
- Chen, B.; Brinkmann, K.; Chen, Z.; Pak, C.W.; Liao, Y.; Shi, S.; Henry, L.; Grishin, N.V.; Bogdan, S.; Rosen, M.K. The WAVE Regulatory Complex Links Diverse Receptors to the Actin Cytoskeleton. Cell 2014, 156, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Liu, J.; Chao, J.; Scheuerman, M.P.; Rahimi, S.A.; Lee, L.Y.; Li, S. PTEN suppresses epithelial–mesenchymal transition and cancer stem cell activity by downregulating Abi1. Sci. Rep. 2020, 10, 12685. [Google Scholar] [CrossRef]
- Annese, T.; Tamma, R.; De Giorgis, M.; Ribatti, D. microRNAs Biogenesis, Functions and Role in Tumor Angiogenesis. Front. Oncol. 2020, 10, 581007. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Abak, A.; Shoorei, H.; Mohaqiq, M.; Majidpoor, J.; Sayad, A.; Taheri, M. Regulatory role of microRNAs on PTEN signaling. Biomed. Pharmacother. 2020, 133, 110986. [Google Scholar] [CrossRef]
- Yuan, Y.; Liao, H.; Pu, Q.; Ke, X.; Hu, X.; Ma, Y.; Luo, X.; Jiang, Q.; Gong, Y.; Wu, M.; et al. miR-410 induces both epithelial–mesenchymal transition and radioresistance through activation of the PI3K/mTOR pathway in non-small cell lung cancer. Signal Transduct. Target. Ther. 2020, 5, 85. [Google Scholar] [CrossRef]
- Zhang, J.-G.; Wang, J.-J.; Zhao, F.; Liu, Q.; Jiang, K.; Yang, G.-H. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin. Chim. Acta 2010, 411, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Dong, Z.; Chen, Y.; Yang, L.; Lai, D. Enrichment of ovarian cancer stem-like cells is associated with epithelial to mesenchymal transition through an miRNA-activated AKT pathway. Cell Prolif. 2013, 46, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lu, Y.; Wang, H.; Han, X.; Mao, J.; Li, J.; Yu, L.; Wang, B.; Fan, S.; Yu, X.; et al. miR-221/222 enhance the tumorigenicity of human breast cancer stem cells via modulation of PTEN/Akt pathway. Biomed. Pharmacother. 2016, 79, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kia, V.; Beigli, M.S.; Hosseini, V.; Koochaki, A.; Paryan, M.; Mohammadi-Yeganeh, S. Is miR-144 an effective inhibitor of PTEN mRNA: A controversy in breast cancer. In Vitro Cell Dev. Biol. Anim. 2018, 54, 621–628. [Google Scholar] [CrossRef]
- Li, N.; Miao, Y.; Shan, Y.; Liu, B.; Li, Y.; Zhao, L.; Jia, L. MiR-106b and miR-93 regulate cell progression by suppression of PTEN via PI3K/Akt pathway in breast cancer. Cell Death Dis. 2017, 8, e2796. [Google Scholar] [CrossRef]
- Liu, T.; Guo, J.; Zhang, X. MiR-202-5p/PTEN mediates doxorubicin-resistance of breast cancer cells via PI3K/Akt signaling pathway. Cancer Biol. Ther. 2019, 20, 989–998. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Sasi, A.K.; Abak, A.; Shoorei, H.; Khoshkar, A.; Taheri, M. Contribution of miRNAs in the Pathogenesis of Breast Cancer. Front. Oncol. 2021, 11, 768949. [Google Scholar] [CrossRef]
- Liu, Z.; Liang, X.; Li, X.; Liu, X.; Zhu, M.; Gu, Y.; Zhou, P. MiRNA-21 functions in ionizing radiation-induced epithelium-to-mesenchymal transition (EMT) by downregulating PTEN. Toxicol. Res. 2019, 8, 328–340. [Google Scholar] [CrossRef]
- Guo, Y.; Yuan, X.; Hong, L.; Wang, Q.; Liu, S.; Li, Z.; Huang, L.; Jiang, S.; Shi, J. Promotor Hypomethylation Mediated Upregulation of miR-23b-3p Targets PTEN to Promote Bronchial Epithelial-Mesenchymal Transition in Chronic Asthma. Front. Immunol. 2022, 12, 771216. [Google Scholar] [CrossRef]
- Ouyang, L.; Liu, R.-D.; Lei, D.-Q.; Shang, Q.-C.; Li, H.-F.; Hu, X.-G.; Zheng, H.; Jin, G. MiR-499a-5p promotes 5-FU resistance and the cell proliferation and migration through activating PI3K/Akt signaling by targeting PTEN in pancreatic cancer. Ann. Transl. Med. 2021, 9, 1798. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, Q.; Liu, Y.; Wang, X.; Ma, C.; Zhu, W. LncRNA HOTAIR Promotes Chemoresistance by Facilitating Epithelial to Mesenchymal Transition through miR-29b/PTEN/PI3K Signaling in Cervical Cancer. Cells Tissues Organs 2021, 211, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Gurbuz, V.; Sozen, S.; Bilen, C.Y.; Konac, E. miR-148a, miR-152 and miR-200b promote prostate cancer metastasis by targeting DNMT1 and PTEN expression. Oncol. Lett. 2021, 22, 805. [Google Scholar] [CrossRef]
- Qian, X.-L.; Zhou, F.; Xu, S.; Jiang, J.; Chen, Z.-P.; Wang, S.-K.; Zuo, Y.; Ni, C. MiR-454-3p Promotes Oxaliplatin Resistance by Targeting PTEN in Colorectal Cancer. Front. Oncol. 2021, 11, 638537. [Google Scholar] [CrossRef]
- Wu, Z.; Luo, J.; Huang, T.; Yi, R.; Ding, S.; Xie, C.; Xu, A.; Zeng, Y.; Wang, X.; Song, Y.; et al. MiR-4310 induced by SP1 targets PTEN to promote glioma progression. Cancer Cell Int. 2020, 20, 567. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, L.; Ding, X.; Sui, X. LINC00861 inhibits the progression of cervical cancer cells by functioning as a ceRNA for miR-513b-5p and regulating the PTEN/AKT/mTOR signaling pathway. Mol. Med. Rep. 2020, 23, 11662. [Google Scholar] [CrossRef]
- Cao, H.-L.; Gu, M.-Q.; Sun, Z.; Chen, Z.-J. miR-144-3p Contributes to the Development of Thyroid Tumors Through the PTEN/PI3K/AKT Pathway. Cancer Manag. Res. 2020, 12, 9845–9855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, G.-X.; Che, L.-S.; Shi, S.-H.; Lin, W.-Y. miR-19 promotes development of renal fibrosis by targeting PTEN-mediated epithelial-mesenchymal transition. Int. J. Clin. Exp. Pathol. 2020, 13, 642–654. [Google Scholar] [PubMed]
- Zhang, J.; Chen, D.; Liang, S.; Wang, J.; Liu, C.; Nie, C.; Shan, Z.; Wang, L.; Fan, Q.; Wang, F. miR-106b promotes cell invasion and metastasis via PTEN mediated EMT in ESCC. Oncol. Lett. 2018, 15, 4619–4626. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.-Y.; Yan, M.-D.; Wu, A.T.; Yuan, K.S.-P.; Liu, S.H. Brown Seaweed Fucoidan Inhibits Cancer Progression by Dual Regulation of mir-29c/ADAM12 and miR-17-5p/PTEN Axes in Human Breast Cancer Cells. J. Cancer 2016, 7, 2408–2419. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Zhang, S.; Chen, S.; Wu, H.; Jiang, M.; Liu, A. Exosomal miR-552-5p promotes tumorigenesis and disease progression via the PTEN/TOB1 axis in gastric cancer. J. Cancer 2022, 13, 890–905. [Google Scholar] [CrossRef]
- Yang, Y.; Ban, D.; Zhang, C.; Shen, L. Downregulation of circ_0000673 Promotes Cell Proliferation and Migration in Endometriosis via the Mir-616-3p/PTEN Axis. Int. J. Med Sci. 2021, 18, 3506–3515. [Google Scholar] [CrossRef]
- Chang, S.; Chen, B.; Wang, X.; Wu, K.; Sun, Y. Long non-coding RNA XIST regulates PTEN expression by sponging miR-181a and promotes hepatocellular carcinoma progression. BMC Cancer 2017, 17, 248. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yao, L.; Li, G.; Ma, D.; Sun, C.; Gao, S.; Zhang, P.; Gao, F. miR-221 Promotes Epithelial-Mesenchymal Transition through Targeting PTEN and Forms a Positive Feedback Loop with β-catenin/c-Jun Signaling Pathway in Extra-Hepatic Cholangiocarcinoma. PLoS ONE 2015, 10, e0141168. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.-W.; Wang, Y.; Chen, L.-L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Hu, Q.; Li, C.; Wang, S.; Li, Y.; Wen, B.; Zhang, Y.; Liang, K.; Yao, J.; Ye, Y.; Hsiao, H.; et al. LncRNAs-directed PTEN enzymatic switch governs epithelial–mesenchymal transition. Cell Res. 2019, 29, 286–304. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Wang, X.-Y.; Jin, Z.-L. Linc00702 inhibits cell growth and metastasis through regulating PTEN in colorectal cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3624–3632. [Google Scholar]
- Yu, W.; Li, D.; Ding, X.; Sun, Y.; Liu, Y.; Cong, J.; Yang, J.; Sun, J.; Ning, X.; Wang, H. LINC00702 suppresses proliferation and invasion in non-small cell lung cancer through regulating miR-510/PTEN axis. Aging 2019, 11, 1471. [Google Scholar] [CrossRef]
- Yan, J.; Huang, X.; Zhang, X.; Chen, Z.; Ye, C.; Xiang, W.; Huang, Z. LncRNA LINC00470 promotes the degradation of PTEN mRNA to facilitate malignant behavior in gastric cancer cells. Biochem. Biophys. Res. Commun. 2019, 521, 887–893. [Google Scholar] [CrossRef]
- Luongo, F.; Colonna, F.; Calapà, F.; Vitale, S.; Fiori, M.E.; De Maria, R. PTEN Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers 2019, 11, 1076. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.; Hartmann, O.; Reissland, M.; Prieto-Garcia, C.; Klann, K.; Pahor, N.; Schülein-Völk, C.; Baluapuri, A.; Polat, B.; Abazari, A.; et al. PTEN mutant non-small cell lung cancer require ATM to suppress pro-apoptotic signalling and evade radiotherapy. Cell Biosci. 2022, 12, 50. [Google Scholar] [CrossRef]
- Rivas, J.D.L.; Brozovic, A.; Izraely, S.; Casas-Pais, A.; Witz, I.P.; Figueroa, A. Cancer drug resistance induced by EMT: Novel therapeutic strategies. Arch. Toxicol. 2021, 95, 2279–2297. [Google Scholar] [CrossRef] [PubMed]
- Song, K.-A.; Faber, A.C. Epithelial-to-mesenchymal transition and drug resistance: Transitioning away from death. J. Thorac. Dis. 2019, 11, E82–E85. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic of the canonical PI3K/AKT signaling pathway. Light blue arrows denote the activating signal transduction events between the members of a particular pathway (shown as circles of different color). Red crossbars denote the inhibitory events. Upon activation of RTK (receptor tyrosine kinase), PI3K (phosphatidylinositol 3-kinase) phosphorylates and produces PIP3 (phosphatidylinositol (3,4,5)) from PIP2 (phosphatidylinositol (4,5) bisphosphate). PTEN (phosphatase) promotes the reverse reaction thus negatively regulates the pathway. PIP3 facilitates the phosphorylation of AKT (protein kinase B). Activating AKT phosphorylates different target proteins and mediates multiple cellular events and processes such as cell cycle, metabolism, survival, apoptosis, etc. Schematic representation of an alternative non-AKT-dependent activation pathway via the Ras/MAPK signaling. Upon exposure of cancer cells to extracellular signals sensed by RTKs, the signal is transferred via the Grb2/SOS protein complex to specific receptor(s) rat sarcoma virus (Ras), which binds GTP and recruits a protein kinase, rapidly accelerated fibrosarcoma (Raf). The latter relays the signal to another kinase, mitogen-activated protein kinase kinase (MEK), followed by the transfer to yet another kinase, extracellular signal-regulated kinase (ERK). ERK mediates a variety of cellular events via phosphorylation of a number of transcription factors (cell cycle, survival, apoptosis, etc.). AKT phosphorylates TSC2, which forms a functional complex with TSC1. Phosphorylation of TSC2 impairs the ability of the TSC1-TSC2 complex to act as a GAP toward the small GTPase Rheb. Rheb-GTP potently activates mTORC1. Phosphorylation of PRAS40 by Akt and by mTORC1 itself results in dissociation of PRAS40 from mTORC1 and may relieve an inhibitory constraint on mTORC1 activity. Only the key upstream phosphorylation events are shown.
Figure 1.
Schematic of the canonical PI3K/AKT signaling pathway. Light blue arrows denote the activating signal transduction events between the members of a particular pathway (shown as circles of different color). Red crossbars denote the inhibitory events. Upon activation of RTK (receptor tyrosine kinase), PI3K (phosphatidylinositol 3-kinase) phosphorylates and produces PIP3 (phosphatidylinositol (3,4,5)) from PIP2 (phosphatidylinositol (4,5) bisphosphate). PTEN (phosphatase) promotes the reverse reaction thus negatively regulates the pathway. PIP3 facilitates the phosphorylation of AKT (protein kinase B). Activating AKT phosphorylates different target proteins and mediates multiple cellular events and processes such as cell cycle, metabolism, survival, apoptosis, etc. Schematic representation of an alternative non-AKT-dependent activation pathway via the Ras/MAPK signaling. Upon exposure of cancer cells to extracellular signals sensed by RTKs, the signal is transferred via the Grb2/SOS protein complex to specific receptor(s) rat sarcoma virus (Ras), which binds GTP and recruits a protein kinase, rapidly accelerated fibrosarcoma (Raf). The latter relays the signal to another kinase, mitogen-activated protein kinase kinase (MEK), followed by the transfer to yet another kinase, extracellular signal-regulated kinase (ERK). ERK mediates a variety of cellular events via phosphorylation of a number of transcription factors (cell cycle, survival, apoptosis, etc.). AKT phosphorylates TSC2, which forms a functional complex with TSC1. Phosphorylation of TSC2 impairs the ability of the TSC1-TSC2 complex to act as a GAP toward the small GTPase Rheb. Rheb-GTP potently activates mTORC1. Phosphorylation of PRAS40 by Akt and by mTORC1 itself results in dissociation of PRAS40 from mTORC1 and may relieve an inhibitory constraint on mTORC1 activity. Only the key upstream phosphorylation events are shown.

Figure 2.
Schematic relationships between PTEN, MDM2, and p53. In addition to being the main negative regulator of the PI3K/AKT pathway, PTEN also regulates stability and transcriptional activity of p53 (tumor suppressor). Moreover, PTEN itself is a transcriptional target of p53. Another transcriptional target of p53 is a E3-ubiquitin ligase, MDM2. Importantly, MDM2 mediates the proteasomal degradation of p53. In addition, MDM2 also mediates autoubiquitination thus rendering itself an unstable protein. AKT can phosphorylate MDM2 thereby stabilizing the latter and promoting the degradation of the tumor suppressor p53. Expression of the constitutively active form of AKT augments the nuclear entry of MDM2. Light blue arrows denote activating transduction events between the members of a particular pathway (shown as circles of different colors). Red crossbars denote the inhibitory events.
Figure 2.
Schematic relationships between PTEN, MDM2, and p53. In addition to being the main negative regulator of the PI3K/AKT pathway, PTEN also regulates stability and transcriptional activity of p53 (tumor suppressor). Moreover, PTEN itself is a transcriptional target of p53. Another transcriptional target of p53 is a E3-ubiquitin ligase, MDM2. Importantly, MDM2 mediates the proteasomal degradation of p53. In addition, MDM2 also mediates autoubiquitination thus rendering itself an unstable protein. AKT can phosphorylate MDM2 thereby stabilizing the latter and promoting the degradation of the tumor suppressor p53. Expression of the constitutively active form of AKT augments the nuclear entry of MDM2. Light blue arrows denote activating transduction events between the members of a particular pathway (shown as circles of different colors). Red crossbars denote the inhibitory events.

Figure 3.
Schematic diagram of the epithelial–mesenchymal transition (EMT) and EMT transcription factors. There are a limited number of critical transcription factors that activate EMT. This core set of EMT master regulators includes zinc-finger E-box-binding homeobox 1 (ZEB1) and ZEB2, Snail (SNAI1), Slug (SNAI2), and Twist-related protein 1 (TWIST1). The EMT is a process by which epithelial cells lose their apicobasal polarity and intercellular contacts and transform into mesenchymal cells, acquiring the ability to migrate. The process of EMT causes suppressed expression of epithelial markers (such as E-cadherin) and upregulation of mesenchymal markers (such as N-cadherin). As a result, cells acquire motility and invasive properties.
Figure 3.
Schematic diagram of the epithelial–mesenchymal transition (EMT) and EMT transcription factors. There are a limited number of critical transcription factors that activate EMT. This core set of EMT master regulators includes zinc-finger E-box-binding homeobox 1 (ZEB1) and ZEB2, Snail (SNAI1), Slug (SNAI2), and Twist-related protein 1 (TWIST1). The EMT is a process by which epithelial cells lose their apicobasal polarity and intercellular contacts and transform into mesenchymal cells, acquiring the ability to migrate. The process of EMT causes suppressed expression of epithelial markers (such as E-cadherin) and upregulation of mesenchymal markers (such as N-cadherin). As a result, cells acquire motility and invasive properties.

Figure 4.
Positive and negative effects of PTEN on TGF-β, Notch, TNF, and integrin signaling pathways. Upon binding of TGF-β, the TGF-β receptor (TGF-βR) induces the phosphorylation of and nuclear translocation of SMADs, while the latter regulate transcription of EMT factors. TGF-β may potentially increase the protein level of PTEN via its phosphorylation. Notch signaling also regulates the expression of PTEN. The cleaved part of Notch receptors affects the transcription of different genes, including PTEN, which is a direct transcriptional target of Notch3. Notch1 downregulates PTEN expression. The NF-κB (Nuclear factor-κB) pathway is activated via LTRs, TNF receptors, and receptors for cytokines (e.g., IL-1R). The NFκB family consists of five subunits: Rel (c-Rel), p65 (RelA, NFκB3), RelB, p50 (NFκB1), and p52 (NFκB2). AKT-dependent activation of NF-κB induces the expression of Snail, which in turn inhibits the expression of PTEN, thereby forming the PTEN/Akt/NF-κB/Snail feedback loop. Integrins are involved in the regulation of cell adhesion to an extracellular matrix. The expression of PTEN inhibits migration and invasion through the αVβ6 integrin signaling pathway. There is a crosstalk between PTEN and FAK. On the one hand, PTEN interacts with and de-phosphorylates FAK tyrosine kinase thereby inhibiting the integrin-mediated migration and invasion of cancer cells. On the other hand, FAK phosphorylates PTEN at Tyr336, which increases the phosphatase activity and protein stability of PTEN. Interactions of PTEN with FAK and paxillin, respectively, weaken their interactions with ECM components. Thus, PTEN may contribute to the process of destabilization of cell adhesion to ECM.
Figure 4.
Positive and negative effects of PTEN on TGF-β, Notch, TNF, and integrin signaling pathways. Upon binding of TGF-β, the TGF-β receptor (TGF-βR) induces the phosphorylation of and nuclear translocation of SMADs, while the latter regulate transcription of EMT factors. TGF-β may potentially increase the protein level of PTEN via its phosphorylation. Notch signaling also regulates the expression of PTEN. The cleaved part of Notch receptors affects the transcription of different genes, including PTEN, which is a direct transcriptional target of Notch3. Notch1 downregulates PTEN expression. The NF-κB (Nuclear factor-κB) pathway is activated via LTRs, TNF receptors, and receptors for cytokines (e.g., IL-1R). The NFκB family consists of five subunits: Rel (c-Rel), p65 (RelA, NFκB3), RelB, p50 (NFκB1), and p52 (NFκB2). AKT-dependent activation of NF-κB induces the expression of Snail, which in turn inhibits the expression of PTEN, thereby forming the PTEN/Akt/NF-κB/Snail feedback loop. Integrins are involved in the regulation of cell adhesion to an extracellular matrix. The expression of PTEN inhibits migration and invasion through the αVβ6 integrin signaling pathway. There is a crosstalk between PTEN and FAK. On the one hand, PTEN interacts with and de-phosphorylates FAK tyrosine kinase thereby inhibiting the integrin-mediated migration and invasion of cancer cells. On the other hand, FAK phosphorylates PTEN at Tyr336, which increases the phosphatase activity and protein stability of PTEN. Interactions of PTEN with FAK and paxillin, respectively, weaken their interactions with ECM components. Thus, PTEN may contribute to the process of destabilization of cell adhesion to ECM.


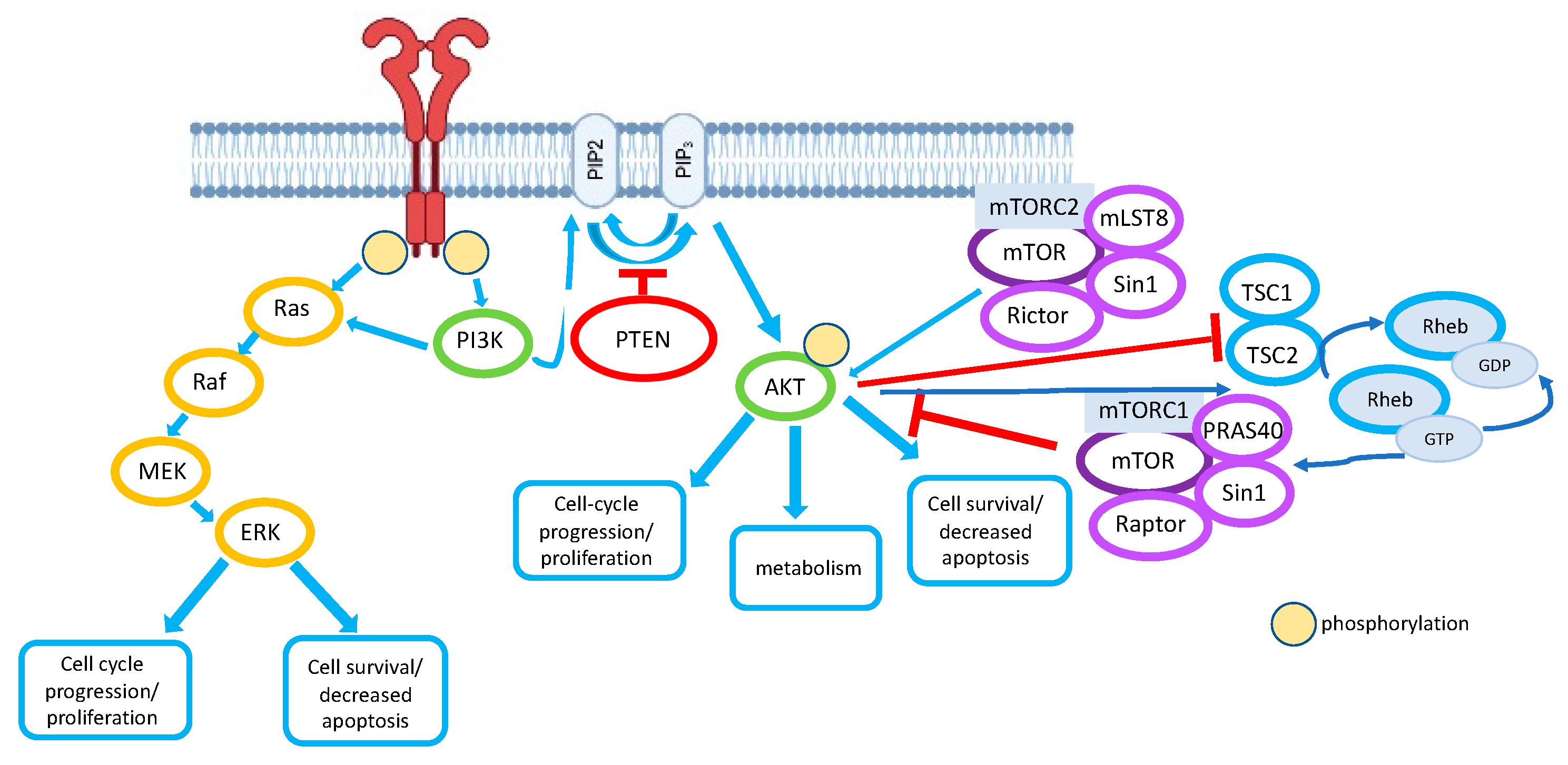{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
miRNAs targeting PTEN transcripts.
| miRs | Interaction with PTEN | Role in EMT Process |
|---|---|---|
| miR-21 | downregulation of PTEN | promotes EMT progression in lung epithelial cells [158] |
| miR-410 | downregulation of PTEN | activation of EMT in non-small cell lung cancer [150] |
| miR-106b and miR-93 | inhibits PTEN | promotes cell migration, invasion, and proliferation in vitro (in breast cancer cells) and tumor growth in vivo (breast cancer) [155] |
| miR-202-5p | downregulation of PTEN | increases DOX resistance and cell proliferation as well as inhibiting apoptosis (in breast cancer cells) [156] |
| miR-23b-3p | silence of PTEN | promotes EMT and migration in bronchial epithelium (lung) [159] |
| miR-499a-5p | decreases the expression levels of PTEN mRNA and protein | promotes 5-FU resistance and cell proliferation and migration in pancreatic cancer [160] |
| miR-29b | targeting the 3′-UTR of PTEN mRNA upregulation of PTEN and downregulation of PI3K in cervical cancer cells | decreases cell proliferation, migration, and invasion abilities of NSCLC cells [161] |
| miR-148a, miR-152 and miR-200b | downregulation of PTEN | presence of these miRs correlates with metastasis in patients with prostate cancer and metastatic prostate cancer [162] |
| miR-454-3p | suppresses PTEN | promotes oxaliplatin resistance and inhibits apoptosis in Colorectal cancer cell line [163] |
| miR-4310 | suppresses PTEN SP1 activates miR-4310 gene expression | promotes proliferation, migration, and invasion in glioma tissues [164] |
| miR-513b-5p | decreases the level of PTEN | migration and invasion in cervical cancer tissues and cell lines [165] |
| miR-144-3p | inhibition of miR-144-3p expression can up-regulate PTEN | induces cell proliferation and invasion, and reduces apoptosis, in thyroid cancer [166] |
| miR-19 | inhibits PTEN mRNA expression | decreases the expression of E-cadherin and increases the expression of α-SMA and fibronectin, while inhibition of miR-19 reverses TGF-β1-induced EMT in renal tubular epithelial cells, thereby promoting renal fibrosis [167] |
| miR-106b | suppresses the expression of PTEN | induces EMT in esophageal squamous cell carcinoma [168] |
| miR-17-5p | inhibits PTEN | enchases survival of breast cancer cells [169] |
| miR-552-5p | inhibits PTEN | promotes proliferation and migration and inhibits apoptosis in gastric cancer cells and stimulates metastasis in vivo; upregulation of miR-552-5p led to an increase in N-cadherin and vimentin and a reduction in E-cadherin [170] |
| miR-616-3p | reduced the mRNA and protein levels of PTEN | promotes proliferation and migration of endometrial stromal cells [171] |
| miR-181a | inhibits PTEN | promotes the proliferation and metastasis of Hepatocellular carcinoma cells in vitro and in vivo [172] |
| miR-221 | regulates PTEN | promotes invasion and metastasis in Extrahepatic cholangiocarcinoma [173] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fedorova, O.; Parfenyev, S.; Daks, A.; Shuvalov, O.; Barlev, N.A. The Role of PTEN in Epithelial–Mesenchymal Transition. Cancers 2022, 14, 3786. https://doi.org/10.3390/cancers14153786
AMA Style
Fedorova O, Parfenyev S, Daks A, Shuvalov O, Barlev NA. The Role of PTEN in Epithelial–Mesenchymal Transition. Cancers. 2022; 14(15):3786. https://doi.org/10.3390/cancers14153786
Chicago/Turabian StyleFedorova, Olga, Sergey Parfenyev, Alexandra Daks, Oleg Shuvalov, and Nickolai A. Barlev. 2022. "The Role of PTEN in Epithelial–Mesenchymal Transition" Cancers 14, no. 15: 3786. https://doi.org/10.3390/cancers14153786
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.