Selected Aspects of Chemoresistance Mechanisms in Colorectal Carcinoma—A Focus on Epithelial-to-Mesenchymal Transition, Autophagy, and Apoptosis


{kind=link}
{kind=link}
{kind=link}
Abstract
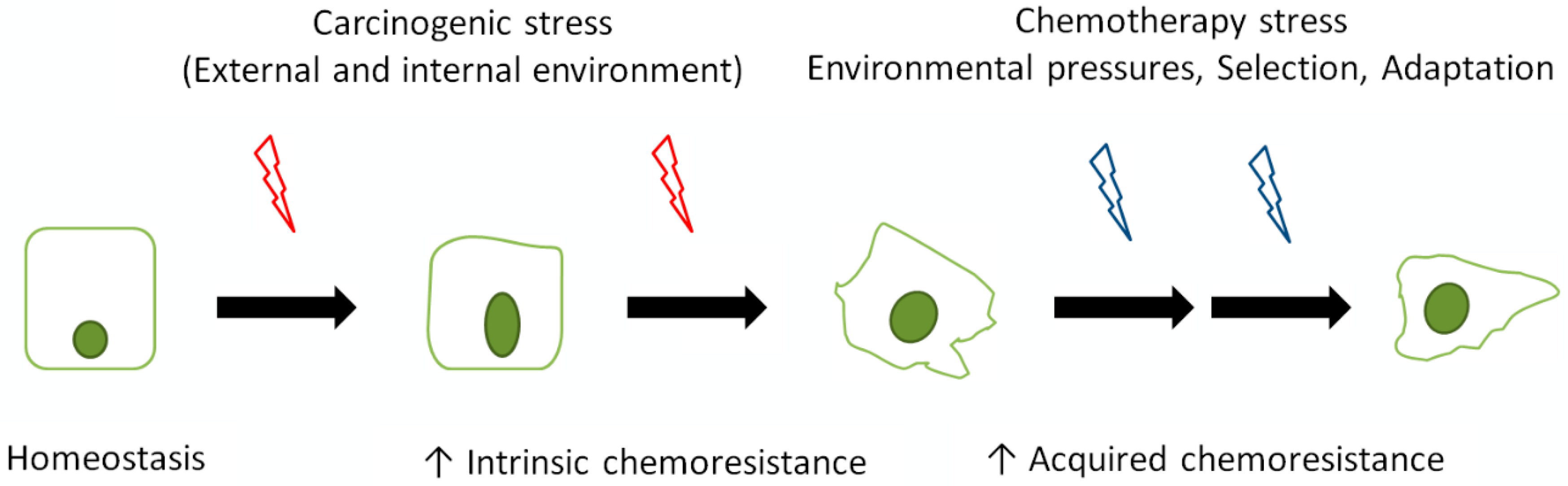
:1. Introduction
2. Chemoresistance of CRC Cells
3. Epithelial-to-Mesenchymal Transition
4. Autophagy
5. Cell Death
6. EMT, Autophagy, and Cell Death in Shaping Chemoresistance
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Marmol, I.; Sanchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Xu, X.; Liu, T. Hereditary nonpolyposis colorectal cancer and cancer syndromes: Recent basic and clinical discoveries. J. Oncol. 2018, 2018, 3979135. [Google Scholar] [CrossRef] [PubMed]
- Rustgi, A.K. The genetics of hereditary colon cancer. Genes Dev. 2007, 21, 2525–2538. [Google Scholar] [CrossRef] [Green Version]
- Radtke, F.; Clevers, H.; Riccio, O. From gut homeostasis to cancer. Curr. Mol. Med. 2006, 6, 275–289. [Google Scholar] [CrossRef]
- Takayama, T.; Katsuki, S.; Takahashi, Y.; Ohi, M.; Nojiri, S.; Sakamaki, S.; Kato, J.; Kogawa, K.; Miyake, H.; Niitsu, Y. Aberrant crypt foci of the colon as precursors of adenoma and cancer. N. Engl. J. Med. 1998, 339, 1277–1284. [Google Scholar] [CrossRef]
- Cummings, O.W. Pathology of the adenoma-carcinoma sequence: From aberrant crypt focus to invasive carcinoma. Semin. Gastrointest. Dis. 2000, 11, 229–237. [Google Scholar]
- Suehiro, Y.; Hinoda, Y. Genetic and epigenetic changes in aberrant crypt foci and serrated polyps. Cancer Sci. 2008, 99, 1071–1076. [Google Scholar] [CrossRef] [Green Version]
- Mundade, R.; Imperiale, T.F.; Prabhu, L.; Loehrer, P.J.; Lu, T. Genetic pathways, prevention, and treatment of sporadic colorectal cancer. Oncoscience 2014, 1, 400–406. [Google Scholar] [CrossRef]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. 2012, 5, 19–27. [Google Scholar]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Pitmon, E.; Wang, K. Microbiome, inflammation and colorectal cancer. Semin. Immunol. 2017, 32, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Bellam, N.; Pasche, B. Tgf-β signaling alterations and colon cancer. Cancer Treat. Res. 2010, 155, 85–103. [Google Scholar] [PubMed]
- Dziaman, T.; Gackowski, D.; Guz, J.; Linowiecka, K.; Bodnar, M.; Starczak, M.; Zarakowska, E.; Modrzejewska, M.; Szpila, A.; Szpotan, J.; et al. Characteristic profiles of DNA epigenetic modifications in colon cancer and its predisposing conditions-benign adenomas and inflammatory bowel disease. Clin. Epigenet. 2018, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Bahnassy, A.A.; Zekri, A.R.; Salem, S.E.; Abou-Bakr, A.A.; Sakr, M.A.; Abdel-Samiaa, A.G.; Al-Bradei, M. Differential expression of p53 family proteins in colorectal adenomas and carcinomas: Prognostic and predictive values. Histol. Histopathol. 2014, 29, 207–216. [Google Scholar] [PubMed]
- Amaro, A.; Chiara, S.; Pfeffer, U. Molecular evolution of colorectal cancer: From multistep carcinogenesis to the big bang. Cancer Metastasis Rev. 2016, 35, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G. Colorectal cancer: Genetic abnormalities, tumor progression, tumor heterogeneity, clonal evolution and tumor-initiating cells. Med. Sci. 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Borras, E.; San Lucas, F.A.; Chang, K.; Zhou, R.; Masand, G.; Fowler, J.; Mork, M.E.; You, Y.N.; Taggart, M.W.; McAllister, F.; et al. Genomic landscape of colorectal mucosa and adenomas. Cancer Prev. Res. 2016, 9, 417–427. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Vaque, J.P.; Martinez, N.; Varela, I.; Fernandez, F.; Mayorga, M.; Derdak, S.; Beltran, S.; Moreno, T.; Almaraz, C.; De Las Heras, G.; et al. Colorectal adenomas contain multiple somatic mutations that do not coincide with synchronous adenocarcinoma specimens. PLoS ONE 2015, 10, e0119946. [Google Scholar]
- Blank, A.; Roberts, D.E., 2nd; Dawson, H.; Zlobec, I.; Lugli, A. Tumor heterogeneity in primary colorectal cancer and corresponding metastases. Does the apple fall far from the tree? Front. Med. 2018, 5, 234. [Google Scholar] [CrossRef] [PubMed]
- Twelves, C.; Scheithauer, W.; McKendrick, J.; Seitz, J.F.; Van Hazel, G.; Wong, A.; Diaz-Rubio, E.; Gilberg, F.; Cassidy, J. Capecitabine versus 5-fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: Final results from the x-act trial with analysis by age and preliminary evidence of a pharmacodynamic marker of efficacy. Ann. Oncol. 2012, 23, 1190–1197. [Google Scholar] [CrossRef]
- De Mattia, E.; Cecchin, E.; Toffoli, G. Pharmacogenomics of intrinsic and acquired pharmacoresistance in colorectal cancer: Toward targeted personalized therapy. Drug Resist. Updat. 2015, 20, 39–70. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Loprinzi, C.L.; Sargent, D.J.; Thome, S.D.; Alberts, S.R.; Haller, D.G.; Benedetti, J.; Francini, G.; Shepherd, L.E.; Francois Seitz, J.; et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: Who benefits and by how much? J. Clin. Oncol. 2004, 22, 1797–1806. [Google Scholar] [CrossRef]
- Haller, D.G.; Tabernero, J.; Maroun, J.; de Braud, F.; Price, T.; Van Cutsem, E.; Hill, M.; Gilberg, F.; Rittweger, K.; Schmoll, H.J. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage iii colon cancer. J. Clin. Oncol. 2011, 29, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Alberts, S.R.; Sargent, D.J.; Nair, S.; Mahoney, M.R.; Mooney, M.; Thibodeau, S.N.; Smyrk, T.C.; Sinicrope, F.A.; Chan, E.; Gill, S.; et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage iii colon cancer: A randomized trial. JAMA 2012, 307, 1383–1393. [Google Scholar]
- Vogel, A.; Hofheinz, R.D.; Kubicka, S.; Arnold, D. Treatment decisions in metastatic colorectal cancer-beyond first and second line combination therapies. Cancer Treat. Rev. 2017, 59, 54–60. [Google Scholar] [CrossRef]
- Schmoll, H.J.; Stein, A. Colorectal cancer in 2013: Towards improved drugs, combinations and patient selection. Nat. Rev. Clin. Oncol. 2014, 11, 79–80. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, B.H.; Wallmark, J.M.; Lorente, D.; Elez, E.; Raimbourg, J.; Gomez-Roca, C.; Ejadi, S.; Piha-Paul, S.A.; Stein, M.N.; Abdul Razak, A.R.; et al. Safety and antitumor activity of the anti-pd-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS ONE 2017, 12, e0189848. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B., 3rd; Venook, A.P.; Cederquist, L.; Chan, E.; Chen, Y.J.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; Enzinger, P.C.; Fichera, A.; et al. Colon cancer, version 1.2017, nccn clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 370–398. [Google Scholar] [CrossRef]
- Fakih, M.G. Metastatic colorectal cancer: Current state and future directions. J. Clin. Oncol. 2015, 33, 1809–1824. [Google Scholar] [CrossRef]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.R.; Longley, D.B.; Johnston, P.G. Chemoresistance in solid tumours. Ann. Oncol. 2006, 17 (Suppl. 10), x315–x324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathonnet, M.; Perraud, A.; Christou, N.; Akil, H.; Melin, C.; Battu, S.; Jauberteau, M.O.; Denizot, Y. Hallmarks in colorectal cancer: Angiogenesis and cancer stem-like cells. World J. Gastroenterol. 2014, 20, 4189–4196. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Mumenthaler, S.M.; Foo, J.; Choi, N.C.; Heise, N.; Leder, K.; Agus, D.B.; Pao, W.; Michor, F.; Mallick, P. The impact of microenvironmental heterogeneity on the evolution of drug resistance in cancer cells. Cancer Inform. 2015, 14, 19–31. [Google Scholar] [CrossRef]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA h19. Theranostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14 (Suppl. 1), 35–48. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Lippert, T.H.; Ruoff, H.J.; Volm, M. Current status of methods to assess cancer drug resistance. Int. J. Med. Sci. 2011, 8, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [PubMed]
- Kosuri, K.V.; Wu, X.; Wang, L.; Villalona-Calero, M.A.; Otterson, G.A. An epigenetic mechanism for capecitabine resistance in mesothelioma. Biochem. Biophys. Res. Commun. 2010, 391, 1465–1470. [Google Scholar] [CrossRef]
- Lubner, S.J.; Uboha, N.V.; Deming, D.A. Primary and acquired resistance to biologic therapies in gastrointestinal cancers. J. Gastrointest. Oncol. 2017, 8, 499–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Grossi, V.; Peserico, A.; Tezil, T.; Simone, C. P38alpha mapk pathway: A key factor in colorectal cancer therapy and chemoresistance. World J. Gastroenterol. 2014, 20, 9744–9758. [Google Scholar] [CrossRef]
- Jensen, N.F.; Stenvang, J.; Beck, M.K.; Hanakova, B.; Belling, K.C.; Do, K.N.; Viuff, B.; Nygard, S.B.; Gupta, R.; Rasmussen, M.H.; et al. Establishment and characterization of models of chemotherapy resistance in colorectal cancer: Towards a predictive signature of chemoresistance. Mol. Oncol. 2015, 9, 1169–1185. [Google Scholar] [CrossRef] [Green Version]
- Fanale, D.; Castiglia, M.; Bazan, V.; Russo, A. Involvement of non-coding rnas in chemo- and radioresistance of colorectal cancer. Adv. Exp. Med. Biol. 2016, 937, 207–228. [Google Scholar]
- Brachtendorf, S.; Wanger, R.A.; Birod, K.; Thomas, D.; Trautmann, S.; Wegner, M.S.; Fuhrmann, D.C.; Brune, B.; Geisslinger, G.; Grosch, S. Chemosensitivity of human colon cancer cells is influenced by a p53-dependent enhancement of ceramide synthase 5 and induction of autophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1214–1227. [Google Scholar] [CrossRef]
- Crea, F.; Nobili, S.; Paolicchi, E.; Perrone, G.; Napoli, C.; Landini, I.; Danesi, R.; Mini, E. Epigenetics and chemoresistance in colorectal cancer: An opportunity for treatment tailoring and novel therapeutic strategies. Drug Resist. Updat. 2011, 14, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Loe, D.W.; Deeley, R.G.; Cole, S.P. Biology of the multidrug resistance-associated protein, mrp. Eur. J. Cancer 1996, 32A, 945–957. [Google Scholar] [CrossRef]
- Brule, S.Y.; Jonker, D.J.; Karapetis, C.S.; O’Callaghan, C.J.; Moore, M.J.; Wong, R.; Tebbutt, N.C.; Underhill, C.; Yip, D.; Zalcberg, J.R.; et al. Location of colon cancer (right-sided versus left-sided) as a prognostic factor and a predictor of benefit from cetuximab in ncic co.17. Eur. J. Cancer 2015, 51, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Sipos, F.; Galamb, O. Epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions in the colon. World J. Gastroenterol. 2012, 18, 601–608. [Google Scholar] [CrossRef]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. Emt as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 2012, 22, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Joyce, T.; Cantarella, D.; Isella, C.; Medico, E.; Pintzas, A. A molecular signature for epithelial to mesenchymal transition in a human colon cancer cell system is revealed by large-scale microarray analysis. Clin. Exp. Metastasis 2009, 26, 569–587. [Google Scholar] [CrossRef]
- Olmeda, D.; Jorda, M.; Peinado, H.; Fabra, A.; Cano, A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene 2007, 26, 1862–1874. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, zeb and bhlh factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Suman, S.; Kurisetty, V.; Das, T.P.; Vadodkar, A.; Ramos, G.; Lakshmanaswamy, R.; Damodaran, C. Activation of akt signaling promotes epithelial-mesenchymal transition and tumor growth in colorectal cancer cells. Mol. Carcinog. 2014, 53 (Suppl. 1), E151–E160. [Google Scholar] [CrossRef]
- Yang, A.D.; Fan, F.; Camp, E.R.; van Buren, G.; Liu, W.; Somcio, R.; Gray, M.J.; Cheng, H.; Hoff, P.M.; Ellis, L.M. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin. Cancer Res. 2006, 12, 4147–4153. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.; Yu, J.; Yu, H. Chemoresistance to doxorubicin induces epithelial-mesenchymal transition via upregulation of transforming growth factor β signaling in hct116 colon cancer cells. Mol. Med. Rep. 2015, 12, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.Y.; Kwak, J.H.; Je, N.K.; Lee, Y.H.; Jung, Y.S. Epithelial-mesenchymal transition is associated with acquired resistance to 5-fluorocuracil in ht-29 colon cancer cells. Toxicol. Res. 2015, 31, 151–156. [Google Scholar] [CrossRef]
- Bao, Y.; Lu, Y.; Wang, X.; Feng, W.; Sun, X.; Guo, H.; Tang, C.; Zhang, X.; Shi, Q.; Yu, H. Eukaryotic translation initiation factor 5a2 (eif5a2) regulates chemoresistance in colorectal cancer through epithelial mesenchymal transition. Cancer Cell Int. 2015, 15, 109. [Google Scholar] [CrossRef] [PubMed]
- Tato-Costa, J.; Casimiro, S.; Pacheco, T.; Pires, R.; Fernandes, A.; Alho, I.; Pereira, P.; Costa, P.; Castelo, H.B.; Ferreira, J.; et al. Therapy-induced cellular senescence induces epithelial-to-mesenchymal transition and increases invasiveness in rectal cancer. Clin. Colorectal Cancer 2016, 15, 170–178.e3. [Google Scholar] [CrossRef] [PubMed]
- Tsoumas, D.; Nikou, S.; Giannopoulou, E.; Champeris Tsaniras, S.; Sirinian, C.; Maroulis, I.; Taraviras, S.; Zolota, V.; Kalofonos, H.P.; Bravou, V. Ilk expression in colorectal cancer is associated with emt, cancer stem cell markers and chemoresistance. Cancer Genom. Proteom. 2018, 15, 127–141. [Google Scholar]
- Yang, Y.; Wang, G.; Zhu, D.; Huang, Y.; Luo, Y.; Su, P.; Chen, X.; Wang, Q. Epithelial-mesenchymal transition and cancer stem cell-like phenotype induced by twist1 contribute to acquired resistance to irinotecan in colon cancer. Int. J. Oncol. 2017, 51, 515–524. [Google Scholar] [CrossRef]
- Hu, T.; Li, Z.; Gao, C.Y.; Cho, C.H. Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J. Gastroenterol. 2016, 22, 6876–6889. [Google Scholar] [CrossRef]
- Findlay, V.J.; Wang, C.; Nogueira, L.M.; Hurst, K.; Quirk, D.; Ethier, S.P.; Staveley O’Carroll, K.F.; Watson, D.K.; Camp, E.R. Snai2 modulates colorectal cancer 5-fluorouracil sensitivity through mir145 repression. Mol. Cancer Ther. 2014, 13, 2713–2726. [Google Scholar] [CrossRef]
- Ikeguchi, M.; Taniguchi, T.; Makino, M.; Kaibara, N. Reduced e-cadherin expression and enlargement of cancer nuclei strongly correlate with hematogenic metastasis in colorectal adenocarcinoma. Scand. J. Gastroenterol. 2000, 35, 839–846. [Google Scholar]
- Lugli, A.; Zlobec, I.; Minoo, P.; Baker, K.; Tornillo, L.; Terracciano, L.; Jass, J.R. Prognostic significance of the wnt signalling pathway molecules apc, β-catenin and e-cadherin in colorectal cancer: A tissue microarray-based analysis. Histopathology 2007, 50, 453–464. [Google Scholar] [CrossRef]
- Roca, F.; Mauro, L.V.; Morandi, A.; Bonadeo, F.; Vaccaro, C.; Quintana, G.O.; Specterman, S.; de Kier Joffe, E.B.; Pallotta, M.G.; Puricelli, L.I.; et al. Prognostic value of e-cadherin, β-catenin, mmps (7 and 9), and timps (1 and 2) in patients with colorectal carcinoma. J. Surg. Oncol. 2006, 93, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhou, M.; Tian, B.; Wu, B.; Li, J. Expression of lncrna-ccat1, e-cadherin and n-cadherin in colorectal cancer and its clinical significance. Int. J. Clin. Exp. Med. 2015, 8, 3707–3715. [Google Scholar] [PubMed]
- Shan, Z.Z.; Yan, X.B.; Yan, L.L.; Tian, Y.; Meng, Q.C.; Qiu, W.W.; Zhang, Z.; Jin, Z.M. Overexpression of tbx3 is correlated with epithelial-mesenchymal transition phenotype and predicts poor prognosis of colorectal cancer. Am. J. Cancer Res. 2015, 5, 344–353. [Google Scholar] [PubMed]
- Fan, F.; Samuel, S.; Evans, K.W.; Lu, J.; Xia, L.; Zhou, Y.; Sceusi, E.; Tozzi, F.; Ye, X.C.; Mani, S.A.; et al. Overexpression of snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med. 2012, 1, 5–16. [Google Scholar] [CrossRef]
- Busch, E.L.; Keku, T.O.; Richardson, D.B.; Cohen, S.M.; Eberhard, D.A.; Avery, C.L.; Sandler, R.S. Evaluating markers of epithelial-mesenchymal transition to identify cancer patients at risk for metastatic disease. Clin. Exp. Metastasis 2016, 33, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef]
- Panda, P.K.; Mukhopadhyay, S.; Das, D.N.; Sinha, N.; Naik, P.P.; Bhutia, S.K. Mechanism of autophagic regulation in carcinogenesis and cancer therapeutics. Semin. Cell Dev. Biol. 2015, 39, 43–55. [Google Scholar] [CrossRef]
- Bhutia, S.K.; Mukhopadhyay, S.; Sinha, N.; Das, D.N.; Panda, P.K.; Patra, S.K.; Maiti, T.K.; Mandal, M.; Dent, P.; Wang, X.Y.; et al. Autophagy: Cancer’s friend or foe? Adv. Cancer Res. 2013, 118, 61–95. [Google Scholar]
- Roy, S.; Debnath, J. Autophagy and tumorigenesis. Semin. Immunopathol. 2010, 32, 383–396. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, hif1 induction and nfkappab activation in the tumor stromal microenvironment. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Delomenie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Burada, F.; Nicoli, E.R.; Ciurea, M.E.; Uscatu, D.C.; Ioana, M.; Gheonea, D.I. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, M.; Cho, H.J.; Paul, S.; Jakhar, R.; Khan, I.; Lee, S.J.; Kim, B.Y.; Krishnan, M.; Khaket, T.P.; Lee, H.G.; et al. Vitexin induces apoptosis by suppressing autophagy in multi-drug resistant colorectal cancer cells. Oncotarget 2018, 9, 3278–3291. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Takeuchi, T.; Kuwano, H. Inhibition of autophagy by 3-ma enhances the effect of 5-fu-induced apoptosis in colon cancer cells. Ann. Surg. Oncol. 2009, 16, 761–771. [Google Scholar] [CrossRef]
- Choi, J.H.; Yoon, J.S.; Won, Y.W.; Park, B.B.; Lee, Y.Y. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. APMIS 2012, 120, 597–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370. [Google Scholar] [CrossRef]
- Ma, Q.; Chang, Z.; Wang, W.; Wang, B. Rapamycin-mediated mtor inhibition reverses drug resistance to adriamycin in colon cancer cells. Hepatogastroenterology 2015, 62, 880–886. [Google Scholar] [PubMed]
- Levy, J.; Cacheux, W.; Bara, M.A.; L’Hermitte, A.; Lepage, P.; Fraudeau, M.; Trentesaux, C.; Lemarchand, J.; Durand, A.; Crain, A.M.; et al. Intestinal inhibition of atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol. 2015, 17, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Ahn, C.H.; Jeong, E.G.; Lee, J.W.; Kim, M.S.; Kim, S.H.; Kim, S.S.; Yoo, N.J.; Lee, S.H. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS 2007, 115, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Xue, X.F.; Shen, H.G.; Guo, X.B.; Wang, X.; Yuan, B.; Guo, X.P.; Kuang, Y.T.; Zhi, Q.M.; Zhao, H. Prognostic significance of beclin-1 expression in colorectal cancer: A meta-analysis. Asian Pac. J. Cancer Prev. 2014, 15, 4583–4587. [Google Scholar] [CrossRef] [PubMed]
- Scopa, C.D.; Tsamandas, A.C.; Zolota, V.; Kalofonos, H.P.; Batistatou, A.; Vagianos, C. Potential role of bcl-2 and ki-67 expression and apoptosis in colorectal carcinoma: A clinicopathologic study. Dig. Dis. Sci. 2003, 48, 1990–1997. [Google Scholar] [CrossRef]
- Park, J.M.; Huang, S.; Wu, T.T.; Foster, N.R.; Sinicrope, F.A. Prognostic impact of beclin 1, p62/sequestosome 1 and lc3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol. Ther. 2013, 14, 100–107. [Google Scholar] [CrossRef]
- Wu, S.; Sun, C.; Tian, D.; Li, Y.; Gao, X.; He, S.; Li, T. Expression and clinical significances of beclin1, lc3 and mtor in colorectal cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 3882–3891. [Google Scholar]
- Cai, Z.; Ke, J.; He, X.; Yuan, R.; Chen, Y.; Wu, X.; Wang, L.; Wang, J.; Lan, P.; Wu, X. Significance of mtor signaling and its inhibitor against cancer stem-like cells in colorectal cancer. Ann. Surg. Oncol. 2014, 21, 179–188. [Google Scholar] [CrossRef]
- Cho, D.H.; Jo, Y.K.; Kim, S.C.; Park, I.J.; Kim, J.C. Down-regulated expression of atg5 in colorectal cancer. Anticancer Res. 2012, 32, 4091–4096. [Google Scholar]
- Choi, J.H.; Cho, Y.S.; Ko, Y.H.; Hong, S.U.; Park, J.H.; Lee, M.A. Absence of autophagy-related proteins expression is associated with poor prognosis in patients with colorectal adenocarcinoma. Gastroenterol. Res. Pract. 2014, 2014, 179586. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.; Killingsworth, M.C.; Lee, C.S. The significance of autophagy in colorectal cancer pathogenesis and implications for therapy. J. Clin. Pathol. 2014, 67, 854–858. [Google Scholar] [CrossRef]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhao, H.; Guo, L.; Zhang, Q.; Zhao, L.; Bai, S.; Zhang, M.; Xu, S.; Wang, F.; Wang, X.; et al. Autophagy-based survival prognosis in human colorectal carcinoma. Oncotarget 2015, 6, 7084–7103. [Google Scholar] [CrossRef] [Green Version]
- Gil, J.; Pesz, K.A.; Sasiadek, M.M. May autophagy be a novel biomarker and antitumor target in colorectal cancer? Biomark. Med. 2016, 10, 1081–1094. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Orlowski, G.M.; Chan, F.K. Differential roles of ripk1 and ripk3 in tnf-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef] [PubMed]
- Vachon, P.H. Methods for assessing apoptosis and anoikis in normal intestine/colon and colorectal cancer. Methods Mol. Biol. 2018, 1765, 99–137. [Google Scholar] [PubMed]
- Pandurangan, A.K.; Divya, T.; Kumar, K.; Dineshbabu, V.; Velavan, B.; Sudhandiran, G. Colorectal carcinogenesis: Insights into the cell death and signal transduction pathways: A review. World J. Gastrointest. Oncol. 2018, 10, 244–259. [Google Scholar] [PubMed]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, L. The recent progress of the mechanism and regulation of tumor necrosis in colorectal cancer. J. Cancer Res. Clin. Oncol. 2016, 142, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Yu, L.C. Pathophysiological mechanisms of death resistance in colorectal carcinoma. World J. Gastroenterol. 2015, 21, 11777–11792. [Google Scholar] [CrossRef]
- Shanmugathasan, M.; Jothy, S. Apoptosis, anoikis and their relevance to the pathobiology of colon cancer. Pathol. Int. 2000, 50, 273–279. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, J. Role of apoptosis in colon cancer biology, therapy, and prevention. Curr. Colorectal Cancer Rep. 2013, 9, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Miquel, C.; Borrini, F.; Grandjouan, S.; Auperin, A.; Viguier, J.; Velasco, V.; Duvillard, P.; Praz, F.; Sabourin, J.C. Role of bax mutations in apoptosis in colorectal cancers with microsatellite instability. Am. J. Clin. Pathol. 2005, 123, 562–570. [Google Scholar] [CrossRef]
- Yamamoto, H.; Imai, K. Microsatellite instability: An update. Arch. Toxicol. 2015, 89, 899–921. [Google Scholar] [CrossRef]
- Kim, M.R.; Jeong, E.G.; Chae, B.; Lee, J.W.; Soung, Y.H.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Pro-apoptotic puma and anti-apoptotic phospho-bad are highly expressed in colorectal carcinomas. Dig. Dis. Sci. 2007, 52, 2751–2756. [Google Scholar] [CrossRef]
- Wilson, T.R.; McLaughlin, K.M.; McEwan, M.; Sakai, H.; Rogers, K.M.; Redmond, K.M.; Johnston, P.G.; Longley, D.B. C-flip: A key regulator of colorectal cancer cell death. Cancer Res. 2007, 67, 5754–5762. [Google Scholar] [CrossRef]
- Xiang, G.; Wen, X.; Wang, H.; Chen, K.; Liu, H. Expression of x-linked inhibitor of apoptosis protein in human colorectal cancer and its correlation with prognosis. J. Surg. Oncol. 2009, 100, 708–712. [Google Scholar]
- Krajewska, M.; Kim, H.; Kim, C.; Kang, H.; Welsh, K.; Matsuzawa, S.; Tsukamoto, M.; Thomas, R.G.; Assa-Munt, N.; Piao, Z.; et al. Analysis of apoptosis protein expression in early-stage colorectal cancer suggests opportunities for new prognostic biomarkers. Clin. Cancer Res. 2005, 11, 5451–5461. [Google Scholar] [CrossRef]
- Endo, K.; Kohnoe, S.; Watanabe, A.; Tashiro, H.; Sakata, H.; Morita, M.; Kakeji, Y.; Maehara, Y. Clinical significance of smac/diablo expression in colorectal cancer. Oncol. Rep. 2009, 21, 351–355. [Google Scholar] [CrossRef]
- Shintani, M.; Sangawa, A.; Yamao, N.; Kamoshida, S. Smac/diablo expression in human gastrointestinal carcinoma: Association with clinicopathological parameters and survivin expression. Oncol. Lett. 2014, 8, 2581–2586. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Rumio, C. How tumor cells choose between epithelial-mesenchymal transition and autophagy to resist stress-therapeutic implications. Front. Pharmacol. 2018, 9, 714. [Google Scholar] [CrossRef]
- De Krijger, I.; Mekenkamp, L.J.; Punt, C.J.; Nagtegaal, I.D. Micrornas in colorectal cancer metastasis. J. Pathol. 2011, 224, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Koehler, B.C.; Jassowicz, A.; Scherr, A.L.; Lorenz, S.; Radhakrishnan, P.; Kautz, N.; Elssner, C.; Weiss, J.; Jaeger, D.; Schneider, M.; et al. Pan-bcl-2 inhibitor obatoclax is a potent late stage autophagy inhibitor in colorectal cancer cells independent of canonical autophagy signaling. BMC Cancer 2015, 15, 919. [Google Scholar] [CrossRef]
- Gulhati, P.; Bowen, K.A.; Liu, J.; Stevens, P.D.; Rychahou, P.G.; Chen, M.; Lee, E.Y.; Weiss, H.L.; O’Connor, K.L.; Gao, T.; et al. Mtorc1 and mtorc2 regulate emt, motility, and metastasis of colorectal cancer via rhoa and rac1 signaling pathways. Cancer Res. 2011, 71, 3246–3256. [Google Scholar] [CrossRef]
- Gugnoni, M.; Sancisi, V.; Manzotti, G.; Gandolfi, G.; Ciarrocchi, A. Autophagy and epithelial-mesenchymal transition: An intricate interplay in cancer. Cell Death Dis. 2016, 7, e2520. [Google Scholar] [CrossRef]
- Qian, H.R.; Shi, Z.Q.; Zhu, H.P.; Gu, L.H.; Wang, X.F.; Yang, Y. Interplay between apoptosis and autophagy in colorectal cancer. Oncotarget 2017, 8, 62759–62768. [Google Scholar] [CrossRef] [Green Version]
- Stanislav, J.; Mls, J.; Cervinka, M.; Rudolf, E. The role of autophagic cell death and apoptosis in irinotecan-treated p53 null colon cancer cells. Anticancer Agents Med. Chem. 2013, 13, 811–820. [Google Scholar]
- Fujikawa, H.; Tanaka, K.; Toiyama, Y.; Saigusa, S.; Inoue, Y.; Uchida, K.; Kusunoki, M. High trkb expression levels are associated with poor prognosis and emt induction in colorectal cancer cells. J. Gastroenterol. 2012, 47, 775–784. [Google Scholar] [CrossRef]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing emt in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, L.; Palmans, S.; Andel, D.; Govaere, O.; Boeckx, B.; Smeets, D.; Galle, E.; Wouters, J.; Barras, D.; Suffiotti, M.; et al. Expression profiling of budding cells in colorectal cancer reveals an emt-like phenotype and molecular subtype switching. Br. J. Cancer 2017, 116, 58–65. [Google Scholar] [CrossRef]
- Ren, B.J.; Zhou, Z.W.; Zhu, D.J.; Ju, Y.L.; Wu, J.H.; Ouyang, M.Z.; Chen, X.W.; Zhou, S.F. Alisertib induces cell cycle arrest, apoptosis, autophagy and suppresses emt in ht29 and caco-2 cells. Int. J. Mol. Sci. 2016, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, M.; Petit, V.; Jain, A.; Amsellem, R.; Johansen, T.; Larue, L.; Codogno, P.; Beau, I. Sqstm1/p62 regulates the expression of junctional proteins through epithelial-mesenchymal transition factors. Cell Cycle 2015, 14, 364–374. [Google Scholar] [CrossRef]
- Shen, H.; Yin, L.; Deng, G.; Guo, C.; Han, Y.; Li, Y.; Cai, C.; Fu, Y.; Liu, S.; Zeng, S. Knockdown of beclin-1 impairs epithelial-mesenchymal transition of colon cancer cells. J. Cell. Biochem. 2018, 119, 7022–7031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Wang, L.Y.; Zhao, S.; Guo, X.C.; Xu, Y.Q.; Zheng, Z.H.; Lu, H.; Zheng, H.C. Effects of beclin 1 overexpression on aggressive phenotypes of colon cancer cells. Oncol. Lett. 2019, 17, 2441–2450. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skarkova, V.; Kralova, V.; Vitovcova, B.; Rudolf, E. Selected Aspects of Chemoresistance Mechanisms in Colorectal Carcinoma—A Focus on Epithelial-to-Mesenchymal Transition, Autophagy, and Apoptosis. Cells 2019, 8, 234. https://doi.org/10.3390/cells8030234
Skarkova V, Kralova V, Vitovcova B, Rudolf E. Selected Aspects of Chemoresistance Mechanisms in Colorectal Carcinoma—A Focus on Epithelial-to-Mesenchymal Transition, Autophagy, and Apoptosis. Cells. 2019; 8(3):234. https://doi.org/10.3390/cells8030234
Chicago/Turabian StyleSkarkova, Veronika, Vera Kralova, Barbora Vitovcova, and Emil Rudolf. 2019. "Selected Aspects of Chemoresistance Mechanisms in Colorectal Carcinoma—A Focus on Epithelial-to-Mesenchymal Transition, Autophagy, and Apoptosis" Cells 8, no. 3: 234. https://doi.org/10.3390/cells8030234