
Anti-Semaphorin 4D Rescues Motor, Cognitive, and Respiratory Phenotypes in a Rett Syndrome Mouse Model

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Anti-SEMA4D Therapy Improves Clinical Scores in a Rett Syndrome Mouse Model
2.2. Anti-SEMA4D Therapy Prevents the Onset and Reverses Deficits of Coordination and Cognition
2.3. Anti-SEMA4D Therapy Prevents the Onset and Reverses Deficits of Key Locomotor Function
2.4. Anti-SEMA4D Therapy Prevents the Onset and Reverses Apnoea and Key Respiratory Deficits
2.5. SEMA4D Is Upregulated in Rett Syndrome Neurons and Anti-SEMA4D Therapy Reduces Activation of Receptor-Positive Glial Cells
2.6. Anti-SEMA4D Mitigates the Abnormal Cytoskeletal Structure in Astrocytes Isolated from Mecp2T158A/y Mutant Mice
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Antibody Treatment
4.3. Behavioural Assessments
4.4. Immunohistochemistry
4.5. Pharmacokinetics Analysis
4.6. Cell Culture
4.7. Immunofluorescence Assays
4.8. Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Ellaway, C.; Peat, J.; Leonard, H.; Christodoulou, J. Sleep dysfunction in Rett syndrome: Lack of age related decrease in sleep duration. Brain Dev. 2001, 23, S101–S103. [Google Scholar] [CrossRef]
- Laurvick, C.L.; de Klerk, N.; Bower, C.; Christodoulou, J.; Ravine, D.; Ellaway, C.; Williamson, S.; Leonard, H. Rett syndrome in Australia: A review of the epidemiology. J. Pediatr. 2006, 148, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Samaco, R.C.; Neul, J.L. Complexities of Rett Syndrome and MeCP2. J. Neurosci. 2011, 31, 7951–7959. [Google Scholar] [CrossRef]
- Cronk, J.C.; Derecki, N.C.; Litvak, V.; Kipnis, J. Unexpected cellular players in Rett syndrome pathology. Neurobiol. Dis. 2016, 92, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.-R.; Chen, X.-S.; Xiao, L. MeCP2 Deficiency in Neuroglia: New Progress in the Pathogenesis of Rett Syndrome. Front. Mol. Neurosci. 2017, 10, 316. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Swanberg, S.; Harvey, D.; LaSalle, J.M.; Jin, L.-W. Rett Syndrome Astrocytes Are Abnormal and Spread MeCP2 Deficiency through Gap Junctions. J. Neurosci. 2009, 29, 5051–5061. [Google Scholar] [CrossRef] [PubMed]
- Lioy, D.T.; Garg, S.K.; Monaghan, C.; Raber, J.; Foust, K.D.; Kaspar, B.K.; Hirrlinger, P.G.; Kirchhoff, F.; Bissonnette, J.M.; Ballas, N.; et al. A role for glia in the progression of Rett’s syndrome. Nat. Cell Biol. 2011, 475, 497–500. [Google Scholar] [CrossRef]
- Delepine, C.; Meziane, H.; Nectoux, J.; Opitz, M.; Smith, A.B.; Ballatore, C.; Saillour, Y.; Griscelli, A.B.; Chang, Q.; Williams, E.C.; et al. Altered microtubule dynamics and vesicular transport in mouse and human MeCP2-deficient astrocytes. Hum. Mol. Genet. 2016, 25, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, W.; Christodoulou, J. The Utility of Next-Generation Sequencing in Gene Discovery for Mutation-Negative Patients with Rett Syndrome. Front. Cell. Neurosci. 2015, 9, 266. [Google Scholar] [CrossRef] [Green Version]
- Okabe, Y.; Takahashi, T.; Mitsumasu, C.; Kosai, K.-I.; Tanaka, E.; Matsuishi, T. Alterations of Gene Expression and Glutamate Clearance in Astrocytes Derived from an MeCP2-Null Mouse Model of Rett Syndrome. PLoS ONE 2012, 7, e35354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noutel, J.; Hong, Y.K.; Leu, B.; Kang, E.; Chen, C. Experience-Dependent Retinogeniculate Synapse Remodeling Is Abnormal in MeCP2-Deficient Mice. Neuron 2011, 70, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Driscoll, C.M.; Lima, M.P.; Kaufmann, W.E.; Bressler, J.P. Methyl CpG binding protein 2 deficiency enhances expression of inflammatory cytokines by sustaining NF-κB signaling in myeloid derived cells. J. Neuroimmunol. 2015, 283, 23–29. [Google Scholar] [CrossRef]
- Smith, E.S.; Jonason, A.; Reilly, C.; Veeraraghavan, J.; Fisher, T.; Doherty, M.; Klimatcheva, E.; Mallow, C.; Cornelius, C.; Leonard, J.E.; et al. SEMA4D compromises blood–brain barrier, activates microglia, and inhibits remyelination in neurodegenerative disease. Neurobiol. Dis. 2015, 73, 254–268. [Google Scholar] [CrossRef] [Green Version]
- Toguchi, M.; Gonzalez, D.; Furukawa, S.; Inagaki, S. Involvement of Sema4D in the control of microglia activation. Neurochem. Int. 2009, 55, 573–580. [Google Scholar] [CrossRef]
- Chapoval, S.P.; Vadasz, Z.; Chapoval, A.I.; Toubi, E. Semaphorins 4A and 4D in chronic inflammatory diseases. Inflamm. Res. 2016, 66, 111–117. [Google Scholar] [CrossRef]
- Denis, H.L.; Lauruol, F.; Cicchetti, F. Are immunotherapies for Huntington’s disease a realistic option? Mol. Psychiatry 2018, 24, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Franciosi, S.; Villanueva, E.B.; Xie, Y.; Winter, L.A.; Veeraraghavan, J.; Jonason, A.; Felczak, B.; Zhang, W.; Kovalik, V.; et al. Anti-semaphorin 4D immunotherapy ameliorates neuropathology and some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 2015, 76, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, J.; Gao, Q.; Ye, F. The role of Sema4D/CD100 as a therapeutic target for tumor microenvironments and for autoimmune, neuroimmune and bone diseases. Expert Opin. Ther. Targets 2016, 20, 885–901. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, Y.; Jin, H.; Wu, J.; He, Q.; Wang, X.; Lei, H.; Hu, B. Sema4D/PlexinB1 inhibition ameliorates blood-brain barrier damage and improves outcome after stroke in rats. FASEB J. 2018, 32, 2181–2196. [Google Scholar] [CrossRef] [Green Version]
- Evans, E.E.; Jonason, A.S., Jr.; Bussler, H.; Torno, S.; Veeraraghavan, J.; Reilly, C.; Doherty, M.A.; Seils, J.; Winter, L.A.; Mallow, C.; et al. Antibody blockade of semaphorin 4D promotes immune infiltration into tumor and enhances response to other immunomodulatory therapies. Cancer Immunol. Res. 2015, 3, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Fisher, T.L.; Seils, J.; Reilly, C.; Litwin, V.; Green, L.; Salkowitz-Bokal, J.; Walsh, R.; Harville, S.; Leonard, J.E.; Smith, E.; et al. Saturation monitoring of VX15/2503, a novel semaphorin 4D-specific antibody, in clinical trials. Cytom. Part B Clin. Cytom. 2016, 90, 199–208. [Google Scholar] [CrossRef]
- Shah, D.K.; Betts, A.M. Antibody biodistribution coefficients: Inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. mAbs 2012, 5, 297–306. [Google Scholar] [CrossRef]
- Wang, Q.; Delva, L.; Weinreb, P.H.; Pepinsky, R.B.; Graham, D.; Veizaj, E.; Cheung, A.E.; Chen, W.; Nestorov, I.; Rohde, E.; et al. Monoclonal antibody exposure in rat and cynomolgus monkey cerebrospinal fluid following systemic administration. Fluids Barriers CNS 2018, 15, 10. [Google Scholar] [CrossRef]
- McGill, B.E.; Bundle, S.F.; Yaylaoglu, M.B.; Carson, J.; Thaller, C.; Zoghbi, H.Y. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 18267–18272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temudo, T.; Ramos, E.; Dias, K.; Barbot, C.; Vieira, J.P.; Moreira, A.; Calado, E.; Carrilho, I.; Oliveira, G.G.; Levy, A.; et al. Movement disorders in Rett syndrome: An analysis of 60 patients with detected MECP2 mutation and correlation with mutation type. Mov. Disord. 2008, 23, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Vignoli, A.; la Briola, F.; Canevini, M.P. Evolution of stereotypies in adolescents and women with Rett syndrome. Mov. Disord. 2009, 24, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Goffin, D.; Allen, M.; Zhang, L.; de Amorim, M.G.; Wang, I.-T.J.; Reyes, A.-R.S.; Mercado-Berton, A.; Ong, C.; Cohen, S.; Hu, L.; et al. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat. Neurosci. 2011, 15, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.L.; Ma, Y.L.; Liu, Y.C.; Tai, D.J.; Lee, E.H. Restoring Wnt6 signaling ameliorates behavioral deficits in MeCP2 T158A mouse model of Rett syndrome. Sci. Rep. 2020, 10, 1074. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Goffin, D. Modeling Rett Syndrome with MeCP2 T158A Knockin Mice. Compr. Guide Autism 2014, 2723–2739. [Google Scholar] [CrossRef]
- Anderson, A.; Wong, K.; Jacoby, P.; Downs, J.; Leonard, H. Twenty years of surveillance in Rett syndrome: What does this tell us? Orphanet J. Rare Dis. 2014, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, M.; Gray, L.J.; Pelka, G.J.; Christodoulou, J.; Tam, P.P.L.; Hannan, A.J. Environmental enrichment ameliorates a motor coordination deficit in a mouse model of Rett syndromeMecp2gene dosage effects and BDNF expression. Eur. J. Neurosci. 2008, 27, 3342–3350. [Google Scholar] [CrossRef]
- Bellot-Saez, A.; Kékesi, O.; Morley, J.; Buskila, Y. Astrocytic modulation of neuronal excitability through K+ spatial buffering. Neurosci. Biobehav. Rev. 2017, 77, 87–97. [Google Scholar] [CrossRef]
- Kahanovitch, U.; Patterson, K.C.; Hernandez, R.; Olsen, M.L. Glial Dysfunction in MeCP2 Deficiency Models: Implications for Rett Syndrome. Int. J. Mol. Sci. 2019, 20, 3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, N.L.; Heaven, M.R.; Holt, L.M.; Crossman, D.K.; Boggio, K.J.; Shaffer, S.A.; Flint, D.L.; Olsen, M.L. RNA sequencing and proteomics approaches reveal novel deficits in the cortex of Mecp2-deficient mice, a model for Rett syndrome. Mol. Autism 2017, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Anderson, C.M. Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front. Cell. Neurosci. 2013, 7, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Mokhtari, R.; Pedrosa, E.; Birnbaum, R.; Zheng, D.; Lachman, H.M. Transcriptome analysis of microglia in a mouse model of Rett syndrome: Differential expression of genes associated with microglia/macrophage activation and cellular stress. Mol. Autism 2017, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Katz, D.M.; Dutschmann, M.; Ramirez, J.-M.; Hilaire, G. Breathing disorders in Rett syndrome: Progressive neurochemical dysfunction in the respiratory network after birth. Respir. Physiol. Neurobiol. 2009, 168, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turovsky, E.; Karagiannis, A.; Abdala, A.P.; Gourine, A.V. Impaired CO2 sensitivity of astrocytes in a mouse model of Rett syndrome. J. Physiol. 2015, 593, 3159–3168. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Su, J.; Cui, N.; Gai, H.; Wu, Z.; Jiang, C. The disruption of central CO2 chemosensitivity in a mouse model of Rett syndrome. Am. J. Physiol. Physiol. 2011, 301, C729–C738. [Google Scholar] [CrossRef] [Green Version]
- Okuno, T.; Nakatsuji, Y.; Moriya, M.; Takamatsu, H.; Nojima, S.; Takegahara, N.; Toyofuku, T.; Nakagawa, Y.; Kang, S.; Friedel, R.H.; et al. Roles of Sema4D–plexin-B1 interactions in the central nervous system for pathogenesis of experimental auto-immune encephalomyelitis. J. Immunol. 2010, 184, 1499–1506. [Google Scholar] [CrossRef] [Green Version]
- Garré, J.M.; Silva, H.M.; Lafaille, J.J.; Yang, G. P2X7 receptor inhibition ameliorates dendritic spine pathology and social behavioral deficits in Rett syndrome mice. Nat. Commun. 2020, 11, 1784. [Google Scholar] [CrossRef] [Green Version]
- Blank, T.; Prinz, M. Microglia as modulators of cognition and neuropsychiatric disorders. Glia 2013, 61, 62–70. [Google Scholar] [CrossRef]
- Fisher, T.L.; Reilly, C.A.; Winter, L.A.; Pandina, T.; Jonason, A.; Scrivens, M.; Balch, L.; Bussler, H.; Torno, S.; Seils, J.; et al. Generation and preclinical characterization of an antibody specific for SEMA4D. mAbs 2016, 8, 150–162. [Google Scholar] [CrossRef]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases Improves the Behavioral Phenotype and Reverses Astrocytic Deficits in a Mouse Model of Rett Syndrome. Neuropsychopharmacology 2011, 37, 1152–1163. [Google Scholar] [CrossRef]
- Nag, N.; Moriuchi, J.M.; Peitzman, C.G.; Ward, B.C.; Kolodny, N.H.; Berger-Sweeney, J.E. Environmental enrichment alters locomotor behaviour and ventricular volume in Mecp21lox mice. Behav. Brain Res. 2009, 196, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Stearns, N.; Schaevitz, L.; Bowling, H.; Nag, N.; Berger, U.; Berger-Sweeney, J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: A model for Rett syndrome. Neuroscience 2007, 146, 907–921. [Google Scholar] [CrossRef]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schanen, C.; Houwink, E.J.; Dorrani, N.; Lane, J.; Everett, R.; Feng, A.; Cantor, R.M.; Percy, A. Phenotypic manifestations ofMECP2 mutations in classical and atypical rett syndrome. Am. J. Med. Genet. 2004, 126A, 129–140. [Google Scholar] [CrossRef]
- Vacca, M.; Filippini, F.; Budillon, A.; Rossi, V.; della Ragione, F.; de Bonis, M.L.; Mercadante, G.; Manzati, E.; Gualandi, F.; Bigoni, S.; et al. MECP2 gene mutation analysis in the British and Italian Rett Syndrome patients: Hot spot map of the most re-current mutations and bioinformatic analysis of a new MECP2 conserved region. Brain Dev. 2001, 23 (Suppl. 1), S246–S250. [Google Scholar] [CrossRef]
- Komada, M.; Takao, K.; Miyakawa, T. Elevated Plus Maze for Mice. J. Vis. Exp. 2008, 2008, e1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
 C57BL/6 mice treated with control antibody (WT),
C57BL/6 mice treated with control antibody (WT),  Mecp2T158A/y mice treated with control antibody (P),
Mecp2T158A/y mice treated with control antibody (P),  Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001.
C57BL/6 mice treated with control antibody (WT), Mecp2T158A/y mice treated with control antibody (P), Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001.
Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001.
C57BL/6 mice treated with control antibody (WT), Mecp2T158A/y mice treated with control antibody (P), Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001. C57BL/6 mice treated with control antibody (WT), Mecp2T158A/y mice treated with control antibody (P), Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001.
C57BL/6 mice treated with control antibody (WT), Mecp2T158A/y mice treated with control antibody (P), Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001.
C57BL/6 mice treated with control antibody (WT), Mecp2T158A/y mice treated with control antibody (P), Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001.
C57BL/6 mice treated with control antibody (WT), Mecp2T158A/y mice treated with control antibody (P), Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001. C57BL/6 mice treated with control antibody (WT), Mecp2T158A/y mice treated with control antibody (P), Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001.
C57BL/6 mice treated with control antibody (WT), Mecp2T158A/y mice treated with control antibody (P), Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001.
C57BL/6 mice treated with control antibody (WT), Mecp2T158A/y mice treated with control antibody (P), Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001.
C57BL/6 mice treated with control antibody (WT), Mecp2T158A/y mice treated with control antibody (P), Mecp2T158A/y mice treated with anti-semaphorin 4D antibody (T). * WT vs. P, ~ WT vs. T, ^ P vs. T. *, ~, and ^ p < 0.05; **, ~~, and ^^ p < 0.01; ***, ~~~, and ^^^ p < 0.001.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
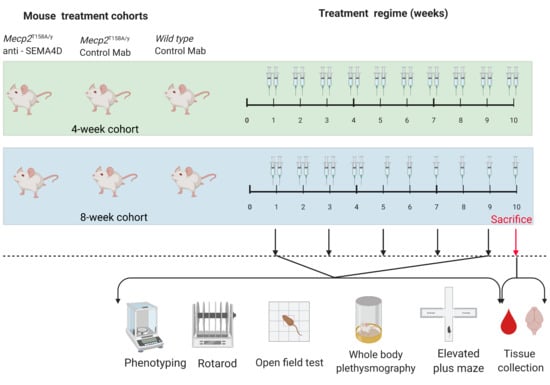
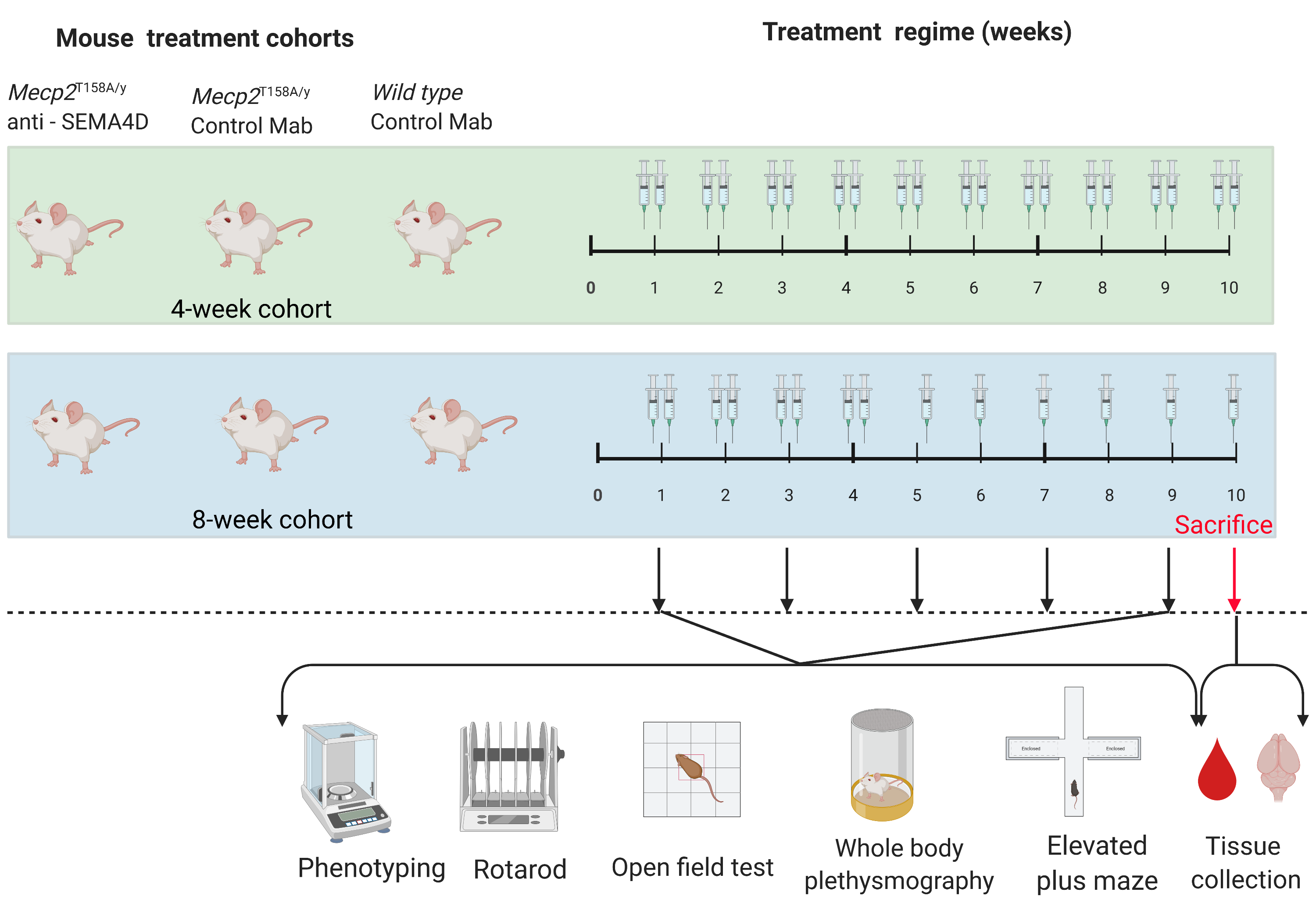
| Mouse Group | Age at Start of Treatment | Treatment Type | Treatment Duration | N = |
|---|---|---|---|---|
| Wild type | 4 weeks old | Isotype control | 10 weeks | 10 |
| Pre-symptomatic Mecp2T158A/y | 4 weeks old | Isotype control | 10 weeks | 11 |
| Pre-symptomatic Mecp2T158A/y | 4 weeks old | Anti-SEMA4D | 10 weeks | 11 |
| Wild type | 8 weeks old | Isotype control | 10 weeks | 10 |
| Symptomatic Mecp2T158A/y | 8 weeks old | Isotype control | 10 weeks | 12 |
| Symptomatic Mecp2T158A/y | 8 weeks old | Anti-SEMA4D | 10 weeks | 10 |
| Score | Tremor | Hindlimb Clasping |
|---|---|---|
| 0 | Not visible or sensible when held in hands | Hindlimbs consistently splayed outward, away from the abdomen |
| 1 | Not visible but sensible when held in hands | One hindlimb retracted or both hindlimbs partially retracted toward the abdomen |
| 2 | Visible and sensible when held in hands | Hindlimbs entirely retracted and touching the abdomen |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, Y.; Evans, E.E.; Mishra, V.; Balch, L.; Eberhardt, A.; Zauderer, M.; Gold, W.A. Anti-Semaphorin 4D Rescues Motor, Cognitive, and Respiratory Phenotypes in a Rett Syndrome Mouse Model. Int. J. Mol. Sci. 2021, 22, 9465. https://doi.org/10.3390/ijms22179465
Mao Y, Evans EE, Mishra V, Balch L, Eberhardt A, Zauderer M, Gold WA. Anti-Semaphorin 4D Rescues Motor, Cognitive, and Respiratory Phenotypes in a Rett Syndrome Mouse Model. International Journal of Molecular Sciences. 2021; 22(17):9465. https://doi.org/10.3390/ijms22179465
Chicago/Turabian StyleMao, Yilin, Elizabeth E. Evans, Vikas Mishra, Leslie Balch, Allison Eberhardt, Maurice Zauderer, and Wendy A. Gold. 2021. "Anti-Semaphorin 4D Rescues Motor, Cognitive, and Respiratory Phenotypes in a Rett Syndrome Mouse Model" International Journal of Molecular Sciences 22, no. 17: 9465. https://doi.org/10.3390/ijms22179465