
Orchestrated Action of AMPK Activation and Combined VEGF/PD-1 Blockade with Lipid Metabolic Tunning as Multi-Target Therapeutics against Ovarian Cancers

 ,
,  and
and
Abstract
:1. Introduction—A Rapid Glance at Ovarian Cancer
2. Targeting AMPK as an Alternative Approach to Combat Ovarian Cancer
3. Limitations of Using AMPK Activators in Treating Malignancies
4. Complementary Targeted Therapeutics—A Double Hit Effect on Ovarian Cancer
5. Targeting Fatty Acid Metabolism for Cancer Immunotherapy
6. Ferroptosis Initiating Therapies (FITs) as the Achilles Heel in Cancer Treatment
7. Expanded Perspectives
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nguyen, L.; Cardenas-Goicoechea, S.J.; Gordon, P.; Curtin, C.; Momeni, M.; Chuang, L.; Fishman, D. Biomarkers for early detection of ovarian cancer. Women’s Health 2013, 9, 171–187. [Google Scholar] [CrossRef]
- Wang, X.; Yung, M.M.H.; Sharma, R.; Chen, F.; Poon, Y.T.; Lam, W.Y.; Li, B.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. Epigenetic Silencing of miR-33b Promotes Peritoneal Metastases of Ovarian Cancer by Modulating the TAK1/FASN/CPT1A/NF-kappaB Axis. Cancers 2021, 13, 4795. [Google Scholar] [CrossRef]
- Stewart, S.L.; Rim, S.H.; Richards, T.B. Gynecologic oncologists and ovarian cancer treatment: Avenues for improved survival. J. Women’s Health 2011, 20, 1257–1260. [Google Scholar] [CrossRef]
- Yu, L.; Deng, L.; Li, J.; Zhang, Y.; Hu, L. The prognostic value of vascular endothelial growth factor in ovarian cancer: A systematic review and meta-analysis. Gynecol. Oncol. 2013, 128, 391–396. [Google Scholar] [CrossRef]
- Chi, D.S.; Sabbatini, P. Advanced ovarian cancer. Curr. Treat. Options Oncol. 2000, 1, 139–146. [Google Scholar] [CrossRef]
- Han, C.Y.; Patten, D.A.; Kim, S.I.; Lim, J.J.; Chan, D.W.; Siu, M.K.Y.; Han, Y.; Carmona, E.; Parks, R.J.; Lee, C.; et al. Nuclear HKII-P-p53 (Ser15) Interaction is a Prognostic Biomarker for Chemoresponsiveness and Glycolytic Regulation in Epithelial Ovarian Cancer. Cancers 2021, 13, 3399. [Google Scholar] [CrossRef]
- Ngu, S.F.; Ngan, H.Y.; Chan, K.K. Role of adjuvant and post-surgical treatment in gynaecological cancer. Best Pr. Res. Clin. Obs. Gynaecol. 2022, 78, 2–13. [Google Scholar] [CrossRef]
- Bristow, R.E.; Tomacruz, R.S.; Armstrong, D.K.; Trimble, E.L.; Montz, F.J. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: A meta-analysis. J. Clin. Oncol. 2002, 20, 1248–1259. [Google Scholar] [CrossRef]
- Elit, L.; Hirte, H. Palliative systemic therapy for women with recurrent epithelial ovarian cancer: Current options. OncoTargets Ther. 2013, 6, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Hennessy, B.T.; Coleman, R.L.; Markman, M. Ovarian cancer. Lancet 2009, 374, 1371–1382. [Google Scholar] [CrossRef]
- Breedlove, G.; Busenhart, C. Screening and detection of ovarian cancer. J. Midwifery Women’s Health 2005, 50, 51–54. [Google Scholar] [CrossRef]
- Yung, M.M.; Ross, F.A.; Hardie, D.G.; Leung, T.H.; Zhan, J.; Ngan, H.Y.; Chan, D.W. Bitter Melon (Momordica charantia) Extract Inhibits Tumorigenicity and Overcomes Cisplatin-Resistance in Ovarian Cancer Cells Through Targeting AMPK Signaling Cascade. Integr. Cancer Ther. 2015, 15, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Macaluso, M.; Paggi, M.G.; Giordano, A. Genetic and epigenetic alterations as hallmarks of the intricate road to cancer. Oncogene 2003, 22, 6472–6478. [Google Scholar] [CrossRef] [Green Version]
- Sawan, C.; Vaissiere, T.; Murr, R.; Herceg, Z. Epigenetic drivers and genetic passengers on the road to cancer. Mutat. Res. 2008, 642, 1–13. [Google Scholar] [CrossRef]
- Palaia, I.; Tomao, F.; Sassu, C.M.; Musacchio, L.; Benedetti Panici, P. Immunotherapy For Ovarian Cancer: Recent Advances And Combination Therapeutic Approaches. OncoTargets Ther. 2020, 13, 6109–6129. [Google Scholar] [CrossRef]
- Champiat, S.; Ileana, E.; Giaccone, G.; Besse, B.; Mountzios, G.; Eggermont, A.; Soria, J.C. Incorporating immune-checkpoint inhibitors into systemic therapy of NSCLC. J. Thorac. Oncol. 2014, 9, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Alme, A.K.; Karir, B.S.; Faltas, B.M.; Drake, C.G. Blocking immune checkpoints in prostate, kidney, and urothelial cancer: An overview. Urol. Oncol. 2016, 34, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients With Recurrent or Refractory Ovarian Cancer: Phase 1b Results from the JAVELIN Solid Tumor Trial. JAMA Oncol. 2019, 5, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.F.; Gordon, M.; Veneris, J.; Braiteh, F.; Balmanoukian, A.; Eder, J.P.; Oaknin, A.; Hamilton, E.; Wang, Y.; Sarkar, I.; et al. Safety, clinical activity and biomarker assessments of atezolizumab from a Phase I study in advanced/recurrent ovarian and uterine cancers. Gynecol. Oncol. 2019, 154, 314–322. [Google Scholar] [CrossRef]
- Ghisoni, E.; Imbimbo, M.; Zimmermann, S.; Valabrega, G. Ovarian Cancer Immunotherapy: Turning up the Heat. Int. J. Mol. Sci. 2019, 20, 2927. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Odunsi, K.; Edwards, R.P.; Kalinski, P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011, 71, 7463–7470. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.R.; Yung, M.M.H.; Xuan, Y.; Zhan, S.; Leung, L.L.; Liang, R.R.; Leung, T.H.Y.; Yang, H.; Xu, D.; Sharma, R.; et al. Targeting of lipid metabolism with a metabolic inhibitor cocktail eradicates peritoneal metastases in ovarian cancer cells. Commun. Biol. 2019, 2, 281. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Yang, J.; Wang, Y.; Xue, L.; Wang, J. Metabolic factors contribute to T-cell inhibition in the ovarian cancer ascites. Int. J. Cancer 2020, 147, 1768–1777. [Google Scholar] [CrossRef]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020, 9, e55185. [Google Scholar] [CrossRef]
- Pan, Q.; Law, C.O.K.; Yung, M.M.H.; Han, K.C.; Pon, Y.L.; Lau, T.C.K. Novel RNA aptamers targeting gastrointestinal cancer biomarkers CEA, CA50 and CA72-4 with superior affinity and specificity. PLoS ONE 2018, 13, e0198980. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Yung, M.M.; Ngan, H.Y.; Chan, D.W. Targeting AMPK signaling in combating ovarian cancers: Opportunities and challenges. Acta Biochim. Biophys. Sin. 2016, 48, 301–317. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, J.V.; Beckers, A.; Brusselmans, K.; Organe, S.; Segers, J.; Timmermans, L.; Vanderhoydonc, F.; Deboel, L.; Derua, R.; Waelkens, E.; et al. Mimicry of a cellular low energy status blocks tumor cell anabolism and suppresses the malignant phenotype. Cancer Res. 2005, 65, 2441–2448. [Google Scholar] [CrossRef] [Green Version]
- Meisse, D.; Van de Casteele, M.; Beauloye, C.; Hainault, I.; Kefas, B.A.; Rider, M.H.; Foufelle, F.; Hue, L. Sustained activation of AMP-activated protein kinase induces c-Jun N-terminal kinase activation and apoptosis in liver cells. FEBS Lett. 2002, 526, 38–42. [Google Scholar] [CrossRef]
- Li, J.; Jiang, P.; Robinson, M.; Lawrence, T.S.; Sun, Y. AMPK-beta1 subunit is a p53-independent stress responsive protein that inhibits tumor cell growth upon forced expression. Carcinogenesis 2003, 24, 827–834. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.Y.; Chan, D.W.; Liu, V.W.; Ngan, H.Y. Inhibition of cervical cancer cell growth through activation of upstream kinases of AMP-activated protein kinase. Tumour Biol. 2009, 30, 80–85. [Google Scholar] [CrossRef]
- Yung, M.M.; Chan, D.W.; Liu, V.W.; Yao, K.M.; Ngan, H.Y. Activation of AMPK inhibits cervical cancer cell growth through AKT/FOXO3a/FOXM1 signaling cascade. BMC Cancer 2013, 13, 327. [Google Scholar] [CrossRef] [Green Version]
- Kwan, H.T.; Chan, D.W.; Cai, P.C.; Mak, C.S.; Yung, M.M.; Leung, T.H.; Wong, O.G.; Cheung, A.N.; Ngan, H.Y. AMPK activators suppress cervical cancer cell growth through inhibition of DVL3 mediated Wnt/beta-catenin signaling activity. PLoS ONE 2013, 8, e53597. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.W.; Yung, M.M.; Chan, Y.S.; Xuan, Y.; Yang, H.; Xu, D.; Zhan, J.B.; Chan, K.K.; Ng, T.B.; Ngan, H.Y. MAP30 protein from Momordica charantia is therapeutic and has synergic activity with cisplatin against ovarian cancer in vivo by altering metabolism and inducing ferroptosis. Pharm. Res. 2020, 161, 105157. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; He, S.; Wang, X.L.; Peng, W.; Chen, Q.Y.; Chi, D.M.; Chen, J.R.; Han, B.W.; Lin, G.W.; Li, Y.Q.; et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat. Commun. 2021, 12, 741. [Google Scholar] [CrossRef]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Alessi, D.R. LKB1 and AMPK and the cancer-metabolism link—Ten years after. BMC Biol. 2013, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M.; Hay, N. The dark face of AMPK as an essential tumor promoter. Cell Logist. 2012, 2, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sun, R.; Zou, J.; Ying, Y.; Luo, Z. Dual Roles of the AMP-Activated Protein Kinase Pathway in Angiogenesis. Cells 2019, 8, 752. [Google Scholar] [CrossRef] [Green Version]
- Sadria, M.; Seo, D.; Layton, A.T. The mixed blessing of AMPK signaling in Cancer treatments. BMC Cancer 2022, 22, 105. [Google Scholar] [CrossRef]
- Vara-Ciruelos, D.; Russell, F.M.; Hardie, D.G. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019, 9, 190099. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Eichner, L.J.; Brun, S.N.; Herzig, S.; Young, N.P.; Curtis, S.D.; Shackelford, D.B.; Shokhirev, M.N.; Leblanc, M.; Vera, L.I.; Hutchins, A.; et al. Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab. 2019, 29, 285–302.e7. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.; Singh, H. Bevacizumab and ovarian cancer. Adv. Med. Oncol. 2013, 5, 133–141. [Google Scholar] [CrossRef] [Green Version]
- McClung, E.C.; Wenham, R.M. Profile of bevacizumab in the treatment of platinum-resistant ovarian cancer: Current perspectives. Int. J. Women’s Health 2016, 8, 59–75. [Google Scholar] [CrossRef] [Green Version]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- McGonigle, K.F.; Muntz, H.G.; Vuky, J.; Paley, P.J.; Veljovich, D.S.; Greer, B.E.; Goff, B.A.; Gray, H.J.; Malpass, T.W. Combined weekly topotecan and biweekly bevacizumab in women with platinum-resistant ovarian, peritoneal, or fallopian tube cancer: Results of a phase 2 study. Cancer 2011, 117, 3731–3740. [Google Scholar] [CrossRef]
- Tillmanns, T.D.; Lowe, M.P.; Walker, M.S.; Stepanski, E.J.; Schwartzberg, L.S. Phase II clinical trial of bevacizumab with albumin-bound paclitaxel in patients with recurrent, platinum-resistant primary epithelial ovarian or primary peritoneal carcinoma. Gynecol. Oncol. 2013, 128, 221–228. [Google Scholar] [CrossRef]
- Verschraegen, C.F.; Czok, S.; Muller, C.Y.; Boyd, L.; Lee, S.J.; Rutledge, T.; Blank, S.; Pothuri, B.; Eberhardt, S.; Muggia, F. Phase II study of bevacizumab with liposomal doxorubicin for patients with platinum- and taxane-resistant ovarian cancer. Ann. Oncol. 2012, 23, 3104–3110. [Google Scholar] [CrossRef]
- Kudoh, K.; Takano, M.; Kouta, H.; Kikuchi, R.; Kita, T.; Miyamoto, M.; Watanabe, A.; Kato, M.; Goto, T.; Kikuchi, Y. Effects of bevacizumab and pegylated liposomal doxorubicin for the patients with recurrent or refractory ovarian cancers. Gynecol. Oncol. 2011, 122, 233–237. [Google Scholar] [CrossRef]
- Hagemann, A.R.; Novetsky, A.P.; Zighelboim, I.; Gao, F.; Massad, L.S.; Thaker, P.H.; Powell, M.A.; Mutch, D.G.; Wright, J.D. Phase II study of bevacizumab and pemetrexed for recurrent or persistent epithelial ovarian, fallopian tube or primary peritoneal cancer. Gynecol. Oncol. 2013, 131, 535–540. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ren, Z.; Xu, S.; Bai, H.; Ma, N.; Wang, F. Low-dose-intensity bevacizumab with weekly irinotecan for platinum- and taxanes-resistant epithelial ovarian cancer. Cancer Chemother. Pharm. 2015, 75, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Takano, M.; Oda, K.; Kouta, H.; Goto, T.; Kudoh, K.; Sasaki, N.; Kita, T.; Kikuchi, Y. Weekly administration of bevacizumab, gemcitabine, and oxaliplatin in patients with recurrent and refractory ovarian cancer: A preliminary result of 19 cases. Int. J. Gynecol. Cancer 2013, 23, 355–360. [Google Scholar] [CrossRef]
- Tewari, K.S.; Burger, R.A.; Enserro, D.; Norquist, B.M.; Swisher, E.M.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Huang, H.; Homesley, H.D.; et al. Final Overall Survival of a Randomized Trial of Bevacizumab for Primary Treatment of Ovarian Cancer. J. Clin. Oncol. 2019, 37, 2317–2328. [Google Scholar] [CrossRef]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Theodoropoulou, S.; Brodowska, K.; Kayama, M.; Morizane, Y.; Miller, J.W.; Gragoudas, E.S.; Vavvas, D.G. Aminoimidazole carboxamide ribonucleotide (AICAR) inhibits the growth of retinoblastoma in vivo by decreasing angiogenesis and inducing apoptosis. PLoS ONE 2013, 8, e52852. [Google Scholar] [CrossRef] [Green Version]
- Cauchy, F.; Mebarki, M.; Leporq, B.; Laouirem, S.; Albuquerque, M.; Lambert, S.; Bourgoin, P.; Soubrane, O.; Van Beers, B.E.; Faivre, S.; et al. Strong antineoplastic effects of metformin in preclinical models of liver carcinogenesis. Clin. Sci. 2017, 131, 27–36. [Google Scholar] [CrossRef]
- Daugan, M.; Dufay Wojcicki, A.; d’Hayer, B.; Boudy, V. Metformin: An anti-diabetic drug to fight cancer. Pharm. Res. 2016, 113, 675–685. [Google Scholar] [CrossRef]
- Wang, J.C.; Li, X.X.; Sun, X.; Li, G.Y.; Sun, J.L.; Ye, Y.P.; Cong, L.L.; Li, W.M.; Lu, S.Y.; Feng, J.; et al. Activation of AMPK by simvastatin inhibited breast tumor angiogenesis via impeding HIF-1alpha-induced pro-angiogenic factor. Cancer Sci. 2018, 109, 1627–1637. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.L.; Wong, J.H.; Fang, E.F.; Chan, Y.S.; Ng, T.B.; Cheung, R.C. Preferential cytotoxicity of the type I ribosome inactivating protein alpha-momorcharin on human nasopharyngeal carcinoma cells under normoxia and hypoxia. Biochem. Pharm. 2014, 89, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Sur, S.; Ray, R.B. Bitter Melon (Momordica Charantia), a Nutraceutical Approach for Cancer Prevention and Therapy. Cancers 2020, 12, 64. [Google Scholar] [CrossRef]
- Gallo, C.; Dallaglio, K.; Bassani, B.; Rossi, T.; Rossello, A.; Noonan, D.M.; D’Uva, G.; Bruno, A.; Albini, A. Hop derived flavonoid xanthohumol inhibits endothelial cell functions via AMPK activation. Oncotarget 2016, 7, 59917–59931. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.J.; Siu, M.K.; Jiang, Y.X.; Leung, T.H.; Chan, D.W.; Cheng, R.R.; Cheung, A.N.; Ngan, H.Y.; Chan, K.K. Aberrant upregulation of PDK1 in ovarian cancer cells impairs CD8(+) T cell function and survival through elevation of PD-L1. Oncoimmunology 2019, 8, e1659092. [Google Scholar] [CrossRef] [Green Version]
- Sweis, R.F.; Luke, J.J. Mechanistic and pharmacologic insights on immune checkpoint inhibitors. Pharm. Res. 2017, 120, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, E.; Romero, P. The therapeutic promise of disrupting the PD-1/PD-L1 immune checkpoint in cancer: Unleashing the CD8 T cell mediated anti-tumor activity results in significant, unprecedented clinical efficacy in various solid tumors. J. Immunother. Cancer 2015, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Yang, H.; Zhang, Y.; Wei, H.; Zhu, Z.; Zhu, B.; Yang, M.; Cao, W.; Wang, L.; Wu, Z. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 2017, 36, 5829–5839. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Kythreotou, A.; Siddique, A.; Mauri, F.A.; Bower, M.; Pinato, D.J. Pd-L1. J. Clin. Pathol. 2018, 71, 189–194. [Google Scholar] [CrossRef]
- Wang, J.J.; Siu, M.K.; Jiang, Y.X.; Leung, T.H.; Chan, D.W.; Wang, H.G.; Ngan, H.Y.; Chan, K.K. A Combination of Glutaminase Inhibitor 968 and PD-L1 Blockade Boosts the Immune Response against Ovarian Cancer. Biomolecules 2021, 11, 1749. [Google Scholar] [CrossRef]
- Pokhrel, R.H.; Acharya, S.; Ahn, J.H.; Gu, Y.; Pandit, M.; Kim, J.O.; Park, Y.Y.; Kang, B.; Ko, H.J.; Chang, J.H. AMPK promotes antitumor immunity by downregulating PD-1 in regulatory T cells via the HMGCR/p38 signaling pathway. Mol. Cancer 2021, 20, 133. [Google Scholar] [CrossRef]
- Shen, X.; Zhao, Y.; Liu, G.; Zhou, H.L.; Fan, J.; Zhang, L.; Li, Y.L.; Wang, Y.; Liang, J.; Xu, Z.X. Upregulation of programmed death ligand 1 by liver kinase B1 and its implication in programmed death 1 blockade therapy in non-small cell lung cancer. Life Sci. 2020, 256, 117923. [Google Scholar] [CrossRef]
- Sur, S.; Ray, R.B. Diverse roles of bitter melon (Momordica charantia) in prevention of oral cancer. J. Cancer Metastasis Treat. 2021, 7, 12. [Google Scholar] [CrossRef]
- Kubo, T.; Ninomiya, T.; Hotta, K.; Kozuki, T.; Toyooka, S.; Okada, H.; Fujiwara, T.; Udono, H.; Kiura, K. Study Protocol: Phase-Ib Trial of Nivolumab Combined With Metformin for Refractory/Recurrent Solid Tumors. Clin. Lung Cancer 2018, 19, e861–e864. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Dragnev, K.; Sarwar, T.; Shirai, K. Clinical outcomes in non-small-cell lung cancer patients receiving concurrent metformin and immune checkpoint inhibitors. Lung Cancer Manag. 2019, 8, LMT11. [Google Scholar] [CrossRef] [Green Version]
- Pyaram, K.; Kumar, A.; Kim, Y.H.; Noel, S.; Reddy, S.P.; Rabb, H.; Chang, C.H. Keap1-Nrf2 System Plays an Important Role in Invariant Natural Killer T Cell Development and Homeostasis. Cell Rep. 2019, 27, 699–707.e4. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, B.; Vuerich, M.; Daneshmandi, S.; Seth, P. Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-cancer Therapy. Front. Oncol. 2018, 8, 284. [Google Scholar] [CrossRef] [Green Version]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Morales, D.R.; Morris, A.D. Metformin in cancer treatment and prevention. Annu. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.W.; Kim, T.; Chang, S.S.; Lee, H.H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Yang, W.H.; Jung, Y.S.; Cha, J.H. A new aspect of an old friend: The beneficial effect of metformin on anti-tumor immunity. BMB Rep. 2020, 53, 512–520. [Google Scholar] [CrossRef]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vazquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.H.; Oh, M.H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef] [Green Version]
- Rolf, J.; Zarrouk, M.; Finlay, D.K.; Foretz, M.; Viollet, B.; Cantrell, D.A. AMPKalpha1: A glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 2013, 43, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Bourne, P.E. Developing multi-target therapeutics to fine-tune the evolutionary dynamics of the cancer ecosystem. Front. Pharm. 2015, 6, 209. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Lee, D.K.; Chen, K.Y.; Chen, J.Y.; Zhang, K.; Silva, A.; Ho, C.M.; Ho, D. Mechanism-independent optimization of combinatorial nanodiamond and unmodified drug delivery using a phenotypically driven platform technology. ACS Nano 2015, 9, 3332–3344. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Li, P.; Cai, J.; Ma, J.; Zhang, X.; Song, Y.; Liu, Y. Lipid Metabolism: Immune Regulation and Therapeutic Prospectives in Systemic Lupus Erythematosus. Front. Immunol. 2022, 13, 860586. [Google Scholar] [CrossRef]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef]
- Elia, I.; Schmieder, R.; Christen, S.; Fendt, S.M. Organ-Specific Cancer Metabolism and Its Potential for Therapy. Handb. Exp. Pharm. 2016, 233, 321–353. [Google Scholar] [CrossRef] [Green Version]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Wu, H.; Han, Y.; Rodriguez Sillke, Y.; Deng, H.; Siddiqui, S.; Treese, C.; Schmidt, F.; Friedrich, M.; Keye, J.; Wan, J.; et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol. Med. 2019, 11, e10698. [Google Scholar] [CrossRef]
- Wang, H.; Yung, M.M.H.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression. Int. J. Mol. Sci. 2021, 22, 6560. [Google Scholar] [CrossRef]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R.; et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015, 36, 81–91. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8(+) T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol. Res. 2018, 6, 1375–1387. [Google Scholar] [CrossRef]
- Voss, K.; Luthers, C.R.; Pohida, K.; Snow, A.L. Fatty Acid Synthase Contributes to Restimulation-Induced Cell Death of Human CD4 T Cells. Front. Mol. Biosci. 2019, 6, 106. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharm. 2017, 8, 992. [Google Scholar] [CrossRef]
- Sun, L.L.; Linghu, D.L.; Hung, M.C. Ferroptosis: A promising target for cancer immunotherapy. Am. J. Cancer Res. 2021, 11, 5856–5863. [Google Scholar] [PubMed]
- Zuo, S.; Yu, J.; Pan, H.; Lu, L. Novel insights on targeting ferroptosis in cancer therapy. Biomark. Res. 2020, 8, 50. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Wang, H.; Yung, M.M.; Chen, F.; Chan, W.S.; Chan, Y.S.; Tsui, S.W.; Ngan, Y.S.; Chan, K.L.; Chan, D.W. SCD1/FADS2 fatty acid desaturases equipoise lipid metabolic activity and redox-driven ferroptosis in ascites-derived ovarian cancer cells. Theranostics 2022, 12, 3534–3552. [Google Scholar] [CrossRef]
- Luo, X.; Gong, H.B.; Gao, H.Y.; Wu, Y.P.; Sun, W.Y.; Li, Z.Q.; Wang, G.; Liu, B.; Liang, L.; Kurihara, H.; et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021, 28, 1971–1989. [Google Scholar] [CrossRef]
- Kloditz, K.; Fadeel, B. Three cell deaths and a funeral: Macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019, 5, 65. [Google Scholar] [CrossRef]
- Efimova, I.; Catanzaro, E.; Van der Meeren, L.; Turubanova, V.D.; Hammad, H.; Mishchenko, T.A.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J. Immunother. Cancer 2020, 8, e001369. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Kusnick, J.; Bruneau, A.; Tacke, F.; Hammerich, L. Ferroptosis in Cancer Immunotherapy—Implications for Hepatocellular Carcinoma. Immuno 2022, 2, 185–217. [Google Scholar] [CrossRef]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef] [Green Version]
- Sacco, A.; Battaglia, A.M.; Botta, C.; Aversa, I.; Mancuso, S.; Costanzo, F.; Biamonte, F. Iron Metabolism in the Tumor Microenvironment-Implications for Anti-Cancer Immune Response. Cells 2021, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, H.; Evstatiev, R.; Kornek, G.; Aapro, M.; Bauernhofer, T.; Buxhofer-Ausch, V.; Fridrik, M.; Geissler, D.; Geissler, K.; Gisslinger, H.; et al. Iron metabolism and iron supplementation in cancer patients. Wien. Klin. Wochenschr. 2015, 127, 907–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifhofer-Obermair, C.; Tymoszuk, P.; Petzer, V.; Weiss, G.; Nairz, M. Iron in the Tumor Microenvironment-Connecting the Dots. Front. Oncol. 2018, 8, 549. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012.e5. [Google Scholar] [CrossRef]
- Xu, S.; Chaudhary, O.; Rodriguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.M.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 2021, 54, 1561–1577.e7. [Google Scholar] [CrossRef]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc(-) Activity. Curr. Biol. 2018, 28, 2388–2399.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Z.; Tsai, H.I.; Liu, Y.; Gao, J.; Wang, M.; Song, L.; Cao, X.; Xu, Z.; Chen, H.; et al. Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells. Cell Death Differ. 2021, 28, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef] [Green Version]
- Alberts, D.S.; Jiang, C.; Liu, P.Y.; Wilczynski, S.; Markman, M.; Rothenberg, M.L. Long-term follow-up of a phase II trial of oral altretamine for consolidation of clinical complete remission in women with stage III epithelial ovarian cancer in the Southwest Oncology Group. Int. J. Gynecol. Cancer 2004, 14, 224–228. [Google Scholar] [CrossRef]
- Mendiola, C.; Valdiviezo, N.; Vega, E.; Sanchez Munoz, A.; Ciruelos, E.M.; Manso, L.; Ghanem, I.; Dorta, M.; Manneh, R.; Flores, C.J.; et al. Altretamine for recurrent ovarian cancer or as maintenance after response to second-line therapy. J. Clin. Oncol. 2011, 29, e15589. [Google Scholar] [CrossRef]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for cancer prevention and treatment. Oncotarget 2015, 6, 7365–7378. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, T.K.; Leclerc, G.M.; Hsieh-Kinser, T.T.; Leclerc, G.J.; Singh, I.; Barredo, J.C. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: Implication for targeted therapy. Mol. Cancer 2007, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Liang, R.; Yung, M.M.H.; He, F.; Jiao, P.; Chan, K.K.L.; Ngan, H.Y.S.; Chan, D.W. The Stress-Inducible BCL2A1 Is Required for Ovarian Cancer Metastatic Progression in the Peritoneal Microenvironment. Cancers 2021, 13, 4577. [Google Scholar] [CrossRef] [PubMed]
- Korolev, K.S.; Xavier, J.B.; Gore, J. Turning ecology and evolution against cancer. Nat. Rev. Cancer 2014, 14, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Hilvo, M.; de Santiago, I.; Gopalacharyulu, P.; Schmitt, W.D.; Budczies, J.; Kuhberg, M.; Dietel, M.; Aittokallio, T.; Markowetz, F.; Denkert, C.; et al. Accumulated Metabolites of Hydroxybutyric Acid Serve as Diagnostic and Prognostic Biomarkers of Ovarian High-Grade Serous Carcinomas. Cancer Res. 2016, 76, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]





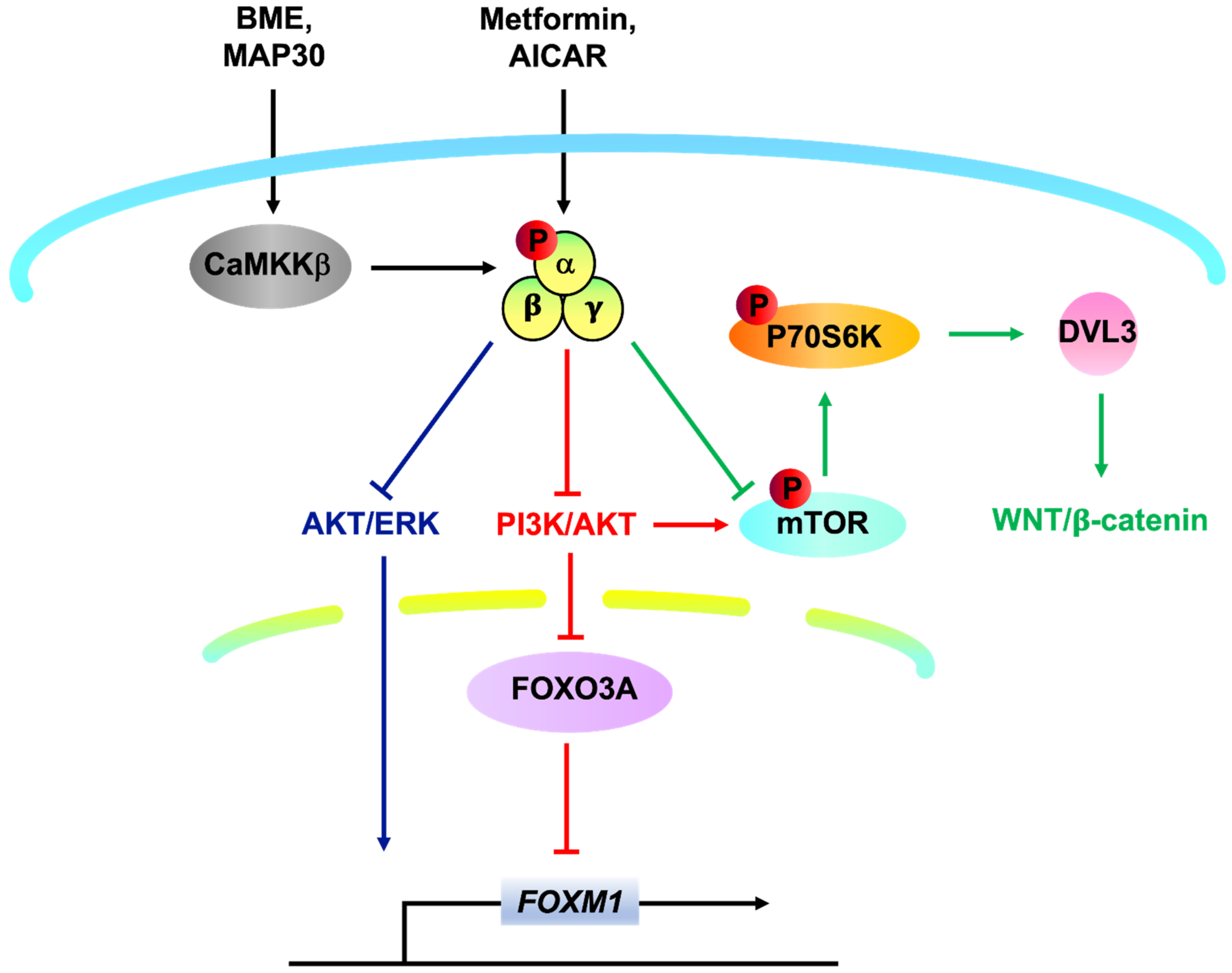{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Trials | No. of Patient | Disease | Intervention/ Treatment | Phase | Recruitment Status | Sponsor |
|---|---|---|---|---|---|---|
| Nivolumab and Metformin Hydrochloride in Treating Patients with Stage III-IV Non-small Cell Lung Cancer That Cannot Be Removed by Surgery (NCT03048500) | N = 17 | NSCLC (Stage III, IIIA, IIIB & IV) | Metformin & Nivolumab | Phase II | Active, not recruiting | Northwestern University |
| Nivolumab and Metformin in Patients with Treatment Refractory MSS Colorectal Cancer (NCT03800602) | N = 24 | Colorectal Cancer (Stage IVA, IVB & IVC) | Metformin & Nivolumab | Phase II | Active, not recruiting | Emory University |
| Anti-PD-1 mAb Plus Metabolic Modulator in Solid Tumor Malignancies (NCT04114136) | N = 108 | Melanoma, NSCLC, HCC, Urothelial Cancer, Gastric Adenocarcinoma, HNSCC, Esophageal Adenocarcinoma & MSI-High solid tumors | Metformin & Nivolumab or Pembrolizumab (dependent upon approved indication) | Phase II | Recruiting | Dan Zandberg, University of Pittsburgh |
| Combining Pembrolizumab and Metformin in Metastatic Head and Neck Cancer Patients (NCT04414540) | N = 20 | HNSCC | Metformin & Pembrolizumab | Phase II | Recruiting | Trisha Wise-Draper, University of Cincinnati |
| A Trial of Pembrolizumab and Metformin Versus Pembrolizumab Alone in Advanced Melanoma (NCT03311308) | N = 30 | Advanced Melanoma | Metformin & Pembrolizumab | Phase I | Recruiting | Yana Najjar, University of Pittsburgh |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yung, M.M.H.; Siu, M.K.Y.; Ngan, H.Y.S.; Chan, D.W.; Chan, K.K.L. Orchestrated Action of AMPK Activation and Combined VEGF/PD-1 Blockade with Lipid Metabolic Tunning as Multi-Target Therapeutics against Ovarian Cancers. Int. J. Mol. Sci. 2022, 23, 6857. https://doi.org/10.3390/ijms23126857
Yung MMH, Siu MKY, Ngan HYS, Chan DW, Chan KKL. Orchestrated Action of AMPK Activation and Combined VEGF/PD-1 Blockade with Lipid Metabolic Tunning as Multi-Target Therapeutics against Ovarian Cancers. International Journal of Molecular Sciences. 2022; 23(12):6857. https://doi.org/10.3390/ijms23126857
Chicago/Turabian StyleYung, Mingo M. H., Michelle K. Y. Siu, Hextan Y. S. Ngan, David W. Chan, and Karen K. L. Chan. 2022. "Orchestrated Action of AMPK Activation and Combined VEGF/PD-1 Blockade with Lipid Metabolic Tunning as Multi-Target Therapeutics against Ovarian Cancers" International Journal of Molecular Sciences 23, no. 12: 6857. https://doi.org/10.3390/ijms23126857