
T-Cell Receptor Repertoire Sequencing and Its Applications: Focus on Infectious Diseases and Cancer

 ,
,  , , ,
, , ,  , , , and
, , , and
Abstract
:1. Introduction
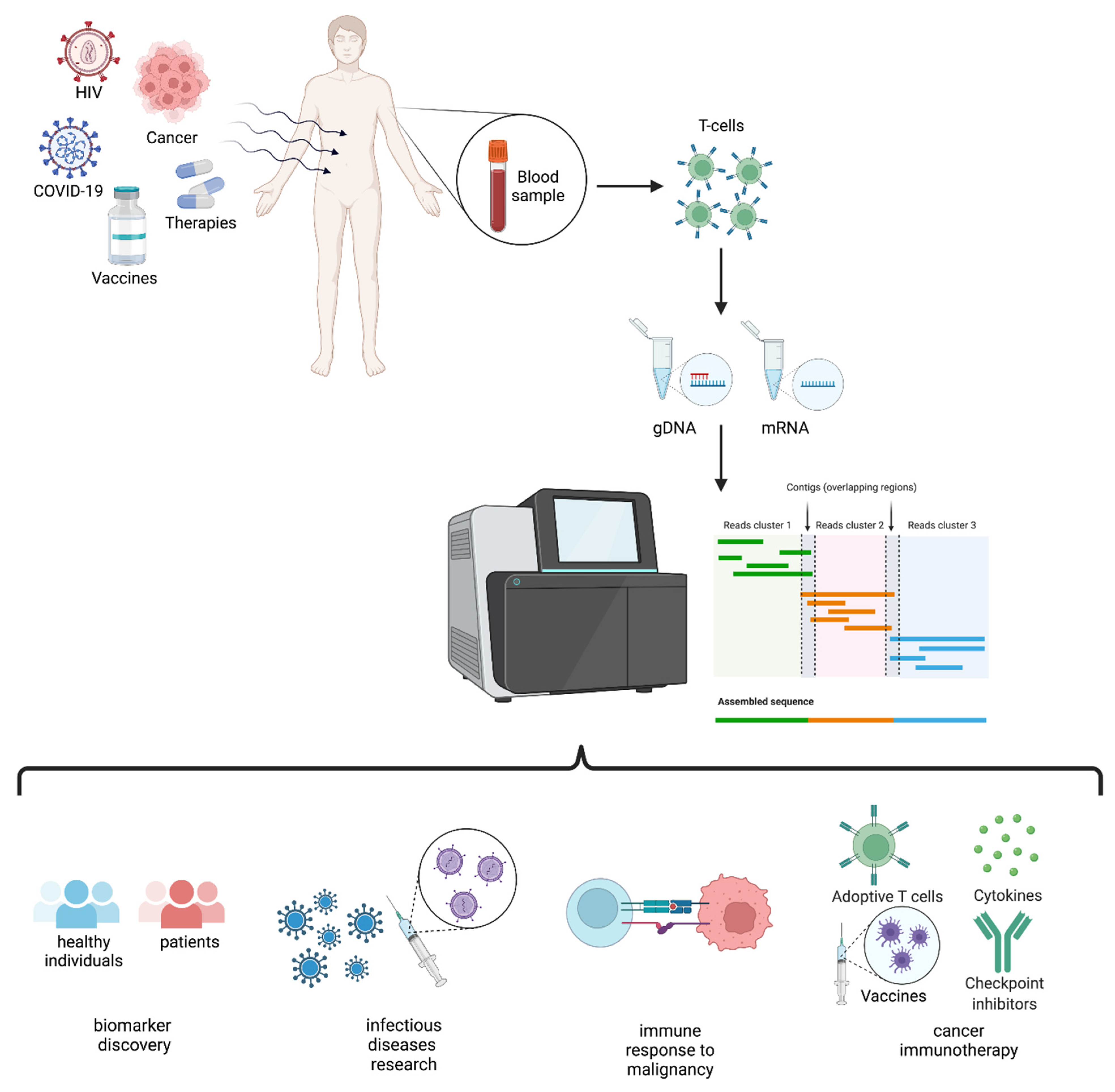
2. TCR Repertoire Analysis
3. TCR and HLA
4. TCR Repertoire via HTS: When Details Matter
5. TCR and Infectious Diseases
6. TCR and Cancer
7. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, H.; Pan, W.; Tang, C.; Tang, Y.; Wu, H.; Yoshimura, A.; Deng, Y.; He, N.; Li, S. The Methods and Advances of Adaptive Immune Receptors Repertoire Sequencing. Theranostics 2021, 11, 8945–8963. [Google Scholar] [CrossRef] [PubMed]
- Dahal-Koirala, S.; Balaban, G.; Neumann, R.S.; Scheffer, L.; Lundin, K.E.A.; Greiff, V.; Sollid, L.M.; Qiao, S.-W.; Sandve, G.K. TCRpower: Quantifying the Detection Power of T-Cell Receptor Sequencing with a Novel Computational Pipeline Calibrated by Spike-in Sequences. Brief. Bioinform. 2022, 23, bbab566. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Yuan, J.; Tian, W.; Meng, L.; Liu, Y. T-cell Receptor Repertoire Analysis for the Diagnosis and Treatment of Solid Tumor: A Methodology and Clinical Applications. Cancer Commun. 2020, 40, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Aversa, I.; Malanga, D.; Fiume, G.; Palmieri, C. Molecular T-Cell Repertoire Analysis as Source of Prognostic and Predictive Biomarkers for Checkpoint Blockade Immunotherapy. Int. J. Mol. Sci. 2020, 21, 2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, J.A.; Satpathy, A.T. High-Throughput and Single-Cell T Cell Receptor Sequencing Technologies. Nat. Methods 2021, 18, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Watkins, T.S.; Miles, J.J. The Human T-cell Receptor Repertoire in Health and Disease and Potential for Omics Integration. Immunol. Cell Biol. 2021, 99, 135–145. [Google Scholar] [CrossRef]
- Chiffelle, J.; Genolet, R.; Perez, M.A.; Coukos, G.; Zoete, V.; Harari, A. T-Cell Repertoire Analysis and Metrics of Diversity and Clonality. Curr. Opin. Biotechnol. 2020, 65, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Rosati, E.; Dowds, C.M.; Liaskou, E.; Henriksen, E.K.K.; Karlsen, T.H.; Franke, A. Overview of Methodologies for T-Cell Receptor Repertoire Analysis. BMC Biotechnol. 2017, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M. Mechanisms Underlying the Suppression of Chromosome Rearrangements by Ataxia-Telangiectasia Mutated. Genes 2021, 12, 1232. [Google Scholar] [CrossRef]
- Nishana, M.; Raghavan, S.C. Role of Recombination Activating Genes in the Generation of Antigen Receptor Diversity and Beyond. Immunology 2012, 137, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Ou, M.; Chen, H.; Guo, C.; Chen, J.; Lin, H.; Tang, D.; Xue, W.; Li, W.; Sui, W.; et al. Characteristic Analysis of TCR β-Chain CDR3 Repertoire for Pre- and Post-Liver Transplantation. Oncotarget 2018, 9, 34506–34519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassing, C.H.; Swat, W.; Alt, F.W. The Mechanism and Regulation of Chromosomal V(D)J Recombination. Cell 2002, 109, S45–S55. [Google Scholar] [CrossRef] [Green Version]
- Calis, J.J.A.; Rosenberg, B.R. Characterizing Immune Repertoires by High Throughput Sequencing: Strategies and Applications. Trends Immunol. 2014, 35, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Valkiers, S.; de Vrij, N.; Gielis, S.; Verbandt, S.; Ogunjimi, B.; Laukens, K.; Meysman, P. Recent Advances in T-Cell Receptor Repertoire Analysis: Bridging the Gap with Multimodal Single-Cell RNA Sequencing. ImmunoInformatics 2022, 5, 100009. [Google Scholar] [CrossRef]
- Pauken, K.E.; Lagattuta, K.A.; Lu, B.Y.; Lucca, L.E.; Daud, A.I.; Hafler, D.A.; Kluger, H.M.; Raychaudhuri, S.; Sharpe, A.H. TCR-Sequencing in Cancer and Autoimmunity: Barcodes and Beyond. Trends Immunol. 2022, 43, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Robins, H.S.; Campregher, P.V.; Srivastava, S.K.; Wacher, A.; Turtle, C.J.; Kahsai, O.; Riddel, S.R.; Warren, E.H.; Carlson, C.S. Comprehensive Assessment of T-Cell Receptor β-Chain Diversity in Aβ T Cells. Blood 2009, 114, 4099–4107. [Google Scholar] [CrossRef]
- Warren, R.L.; Freeman, J.D.; Zeng, T.; Choe, G.; Munro, S.; Moore, R.; Webb, J.R.; Holt, R.A. Exhaustive T-Cell Repertoire Sequencing of Human Peripheral Blood Samples Reveals Signatures of Antigen Selection and a Directly Measured Repertoire Size of at Least 1 Million Clonotypes. Genome Res. 2011, 21, 790–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolich-Žugich, J.; Slifka, M.K.; Messaoudi, I. The Many Important Facets of T-Cell Repertoire Diversity. Nat. Rev. Immunol. 2004, 4, 123–132. [Google Scholar] [CrossRef]
- Dupic, T.; Marcou, Q.; Walczak, A.M.; Mora, T. Genesis of the Aβ T-Cell Receptor. PLoS Comput. Biol. 2019, 15, e1006874. [Google Scholar] [CrossRef] [Green Version]
- Sewell, A.K. Why Must T Cells Be Cross-Reactive? Nat. Rev. Immunol. 2012, 12, 669–677. [Google Scholar] [CrossRef]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, L.; Beckford, J.; Alachkar, H. Deciphering the TCR Repertoire to Solve the COVID-19 Mystery. Trends Pharmacol. Sci. 2020, 41, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Wherry, E.J.; Goldrath, A.W. Molecular Regulation of Effector and Memory T Cell Differentiation. Nat. Immunol. 2014, 15, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.A.; Teixeiro, E. TCR Signaling in T Cell Memory. Front. Immunol. 2015, 6, 617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zvyagin, I.V.; Pogorelyy, M.V.; Ivanova, M.E.; Komech, E.A.; Shugay, M.; Bolotin, D.A.; Shelenkov, A.A.; Kurnosov, A.A.; Staroverov, D.B.; Chudakov, D.M.; et al. Distinctive Properties of Identical Twins’ TCR Repertoires Revealed by High-Throughput Sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 5980–5985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Yang, X.; Ko, A.; Sun, X.; Gao, M.; Zhang, Y.; Shi, A.; Mariuzza, R.A.; Weng, N. Sequence and Structural Analyses Reveal Distinct and Highly Diverse Human CD8 + TCR Repertoires to Immunodominant Viral Antigens. Cell Rep. 2017, 19, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, G.; Demarest, J.F.; Soudeyns, H.; Graziosi, C.; Denis, F.; Adelsberger, J.W.; Borrow, P.; Saag, M.S.; Shaw, G.M.; Sekaly, R.P.; et al. Major Expansion of CD8+ T Cells with a Predominant vp Usage during the Primary Immune Response to HIV. Nature 1994, 370, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, T.; Pignon, J.-C.; Wang, B.; Wang, J.; Shukla, S.A.; Dou, R.; Chen, Q.; Hodi, F.S.; Choueiri, T.K.; et al. Landscape of Tumor-Infiltrating T Cell Repertoire of Human Cancers. Nat. Genet. 2016, 48, 725–732. [Google Scholar] [CrossRef]
- Holtmeier, W.; Kabelitz, D.; Gammadelta, T. Cells Link Innate and Adaptive Immune Responses. Chem. Immunol. Allergy 2005, 86, 151–183. [Google Scholar]
- Woodsworth, D.J.; Castellarin, M.; Holt, R.A. Sequence Analysis of T-Cell Repertoires in Health and Disease. Genome Med. 2013, 5, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosaad, Y.M. Clinical Role of Human Leukocyte Antigen in Health and Disease. Scand. J. Immunol. 2015, 82, 283–306. [Google Scholar] [CrossRef]
- DeWitt, W.S.; Smith, A.; Schoch, G.; Hansen, J.A.; Matsen, F.A.; Bradley, P. Human T Cell Receptor Occurrence Patterns Encode Immune History, Genetic Background, and Receptor Specificity. eLife 2018, 7, e38358. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA Variation and Disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Amiel, J. Study of the Leukocyte Phenotypes in Hodgkin’s Disease in Histocompatibility Testing; Teraski, P.I., Ed.; Munksgaard: Copenhagen, Denmark, 1967; pp. 79–81. [Google Scholar]
- Trowsdale, J.; Knight, J.C. Major Histocompatibility Complex Genomics and Human Disease. Annu. Rev. Genom. Hum. Genet. 2013, 14, 301–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaslow, R.A.; Carrington, M.; Apple, R.; Park, L.; Muñoz, A.; Saah, A.J.; Goedert, J.J.; Winkler, C.; O’Brien, S.J.; Rinaldo, C.; et al. Influence of Combinations of Human Major Histocompatibility Complex Genes on the Course of HIV-1 Infection. Nat. Med. 1996, 2, 405–411. [Google Scholar] [CrossRef]
- Simmonds, M.; Gough, S. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr. Genom. 2007, 8, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Robles, B.-J.; Labrador-Sánchez, E.; Andrés-Trasahedo, E.; Pinillos-Aransay, V.; Joven-Zapata, M.-Y.; Torrecilla Lerena, L.; Salazar-Asencio, O.-A.; López-Martín, J.-A. Concurrence of Rheumatoid Arthritis and Ankylosing Spondylitis: Analysis of Seven Cases and Literature Review. Case Rep. Rheumatol. 2022, 2022, 8500567. [Google Scholar] [CrossRef] [PubMed]
- Padyukov, L. Genetics of Rheumatoid Arthritis. Semin. Immunopathol. 2022, 44, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hussein, S.S.; Ali, E.N.; Zaki, N.H.; Ad’hiah, A.H. Genetic Polymorphism of HLA-G Gene (G*01:03, G*01:04, and G*01:05N) in Iraqi Patients with Inflammatory Bowel Disease (Ulcerative Colitis and Crohn’s Disease). Egypt J. Med. Hum. Genet. 2021, 22, 34. [Google Scholar] [CrossRef]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Simone, M.; Rossetti, G.; Pagani, M. Single Cell T Cell Receptor Sequencing: Techniques and Future Challenges. Front. Immunol. 2018, 9, 1638. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing Technologies—The next Generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, J.M.; Buck, M.J. Key Principles and Clinical Applications of “Next-Generation” DNA Sequencing. Cancer Prev. Res. 2012, 5, 887–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takara Bio Blog Team. 4 Factors to Consider for Immune Repertoire Profiling; Web Document Reprint; Takara Bio USA, Inc.: San Jose, CA, USA, 2019. [Google Scholar]
- Kockelbergh, H.; Evans, S.; Deng, T.; Clyne, E.; Kyriakidou, A.; Economou, A.; Luu Hoang, K.N.; Woodmansey, S.; Foers, A.; Fowler, A.; et al. Utility of Bulk T-Cell Receptor Repertoire Sequencing Analysis in Understanding Immune Responses to COVID-19. Diagnostics 2022, 12, 1222. [Google Scholar] [CrossRef] [PubMed]
- Barennes, P.; Quiniou, V.; Shugay, M.; Egorov, E.S.; Davydov, A.N.; Chudakov, D.M.; Uddin, I.; Ismail, M.; Oakes, T.; Chain, B.; et al. Benchmarking of T Cell Receptor Repertoire Profiling Methods Reveals Large Systematic Biases. Nat. Biotechnol. 2021, 39, 236–245. [Google Scholar] [CrossRef]
- Shugay, M.; Britanova, O.V.; Merzlyak, E.M.; Turchaninova, M.A.; Mamedov, I.Z.; Tuganbaev, T.R.; Bolotin, D.A.; Staroverov, D.B.; Putintseva, E.K.; Plevova, K.; et al. Towards Error-Free Profiling of Immune Repertoires. Nat. Methods 2014, 11, 653–655. [Google Scholar] [CrossRef]
- Logan, A.C.; Gao, H.; Wang, C.; Sahaf, B.; Jones, C.D.; Marshall, E.L.; Buno, I.; Armstrong, R.; Fire, A.Z.; Weinberg, K.I.; et al. High-Throughput VDJ Sequencing for Quantification of Minimal Residual Disease in Chronic Lymphocytic Leukemia and Immune Reconstitution Assessment. Proc. Natl. Acad. Sci. USA 2011, 108, 21194–21199. [Google Scholar] [CrossRef] [Green Version]
- Tiller, T.; Busse, C.E.; Wardemann, H. Cloning and Expression of Murine Ig Genes from Single B Cells. J. Immunol. Methods 2009, 350, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wilkinson, M.F. Nonsense Surveillance in Lymphocytes? Immunity 1998, 8, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, Y.; Xu, L.T.; Jackson, K.J.L.; Roskin, K.M.; Pham, T.D.; Laserson, J.; Marshall, E.L.; Seo, K.; Lee, J.; et al. Effects of Aging, Cytomegalovirus Infection, and EBV Infection on Human B Cell Repertoires. J. Immunol. 2014, 192, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Mamedov, I.Z.; Britanova, O.V.; Zvyagin, I.V.; Turchaninova, M.A.; Bolotin, D.A.; Putintseva, E.V.; Lebedev, Y.B.; Chudakov, D.M. Preparing Unbiased T-Cell Receptor and Antibody CDNA Libraries for the Deep Next Generation Sequencing Profiling. Front. Immunol. 2013, 4, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trück, J.; Eugster, A.; Barennes, P.; Tipton, C.M.; Luning Prak, E.T.; Bagnara, D.; Soto, C.; Sherkow, J.S.; Payne, A.S.; Lefran, M.; et al. Biological Controls for Standardization and Interpretation of Adaptive Immune Receptor Repertoire Profiling. eLife 2021, 10, e66274. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.H.; Sham, P.C.; Wang, J. Evaluation of Tools for Highly Variable Gene Discovery from Single-Cell RNA-Seq Data. Brief. Bioinform. 2019, 20, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.K.; Baryawno, N. Introduction to Single-Cell RNA Sequencing. Curr. Protoc. Mol. Biol. 2018, 122, e57. [Google Scholar] [CrossRef] [PubMed]
- Prakadan, S.M.; Shalek, A.K.; Weitz, D.A. Scaling by Shrinking: Empowering Single-Cell “omics” with Microfluidic Devices. Nat. Rev. Genet. 2017, 18, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.; Kaczorowski, D.; Valdes-Mora, F.; Nordon, R.E.; Neild, A.; Farbehi, N.; Bartonicekcg, N.; Gallego-Ortega, D. Droplet-Based Single Cell RNAseq Tools: A Practical Guide. Lab Chip 2019, 19, 1706–1727. [Google Scholar] [CrossRef]
- Zhang, J.; Song, C.; Tian, Y.; Yang, X. Single-Cell RNA Sequencing in Lung Cancer: Revealing Phenotype Shaping of Stromal Cells in the Microenvironment. Front. Immunol. 2022, 12, 802080. [Google Scholar] [CrossRef]
- Carlson, C.S.; Emerson, R.O.; Sherwood, A.M.; Desmarais, C.; Chung, M.-W.; Parsons, J.M.; Steen, M.S.; LaMadrid-Herrmannsfeldt, M.A.; Williamson, D.W.; Livingston, R.J.; et al. Using Synthetic Templates to Design an Unbiased Multiplex PCR Assay. Nat. Commun. 2013, 4, 2680. [Google Scholar] [CrossRef] [Green Version]
- Wulf, M.G.; Maguire, S.; Humbert, P.; Dai, N.; Bei, Y.; Nichols, N.M.; Correa, I.R., Jr.; Guan, S. Non-Templated Addition and Template Switching by Moloney Murine Leukemia Virus (MMLV)-Based Reverse Transcriptases Co-Occur and Compete with Each Other. J. Biol. Chem. 2019, 294, 18220–18231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamopoulos, P.G.; Tsiakanikas, P.; Stolidi, I.; Scorilas, A. A Versatile 5′ RACE-Seq Methodology for the Accurate Identification of the 5′ Termini of MRNAs. BMC Genom. 2022, 23, 163. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, W.; Zeng, X.; Zhang, R.; Du, Y.; Hong, X.; Cao, H.; Su, Z.; Wang, C.; Wu, J.; et al. Systematic Comparative Evaluation of Methods for Investigating the TCRβ Repertoire. PLoS ONE 2016, 11, e0152464. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Shichino, S.; Matsushima, K.; Ueha, S. Revealing Clonal Responses of Tumor-Reactive T-Cells Through T Cell Receptor Repertoire Analysis. Front. Immunol. 2022, 13, 807696. [Google Scholar] [CrossRef]
- Ye, J.; Ma, N.; Madden, T.L.; Ostell, J.M. IgBLAST: An Immunoglobulin Variable Domain Sequence Analysis Tool. Nucleic Acids Res. 2013, 41, W34–W40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamyar, E.; Duroux, P.; Lefranc, M.P.; Giudicelli, V. IMGT ® Tools for the Nucleotide Analysis of Immunoglobulin (IG) and T Cell Receptor (TR) V- (D)-J Repertoires, Polymorphisms, and IG Mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol. Biol. 2012, 882, 569–604. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, D.A.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Chudakov, D.M. MiXCR: Software for Comprehensive Adaptive Immunity Profiling. Nat. Methods 2015, 12, 380–381. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.R.; Akbar, R.; Yermanos, A.; Pavlović, M.; Snapkov, I.; Sandve, G.K.; Reddy, S.T.; Greiff, V. ImmuneSIM: Tunable Multi-Feature Simulation of B- and T-Cell Receptor Repertoires for Immunoinformatics Benchmarking. Bioinformatics 2020, 36, 3594–3596. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, B.; Pandit, A.; Andeweg, A.C.; de Boer, R.J. RTCR: A Pipeline for Complete and Accurate Recovery of T Cell Repertoires from High Throughput Sequencing Data. Bioinformatics 2016, 32, 3098–3106. [Google Scholar] [CrossRef] [Green Version]
- Tobin, N.H.; Learn, G.H.; Holte, S.E.; Wang, Y.; Melvin, A.J.; McKernan, J.L.; Pawluk, D.M.; Mohan, K.M.; Lewis, P.F.; Mullins, J.I.; et al. Evidence That Low-Level Viremias during Effective Highly Active Antiretroviral Therapy Result from Two Processes: Expression of Archival Virus and Replication of Virus. J. Virol. 2005, 79, 9625–9634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jost, S.; Tomezsko, P.J.; Rands, K.; Toth, I.; Lichterfeld, M.; Gandhi, R.T.; Altfeld, M. CD4+ T-Cell Help Enhances NK Cell Function Following Therapeutic HIV-1 Vaccination. J. Virol. 2014, 88, 8349–8354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding Roles for CD4+ T Cells in Immunity to Viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Serroukh, Y.; Gu-Trantien, C.; Hooshiar Kashani, B.; Defrance, M.; Vu Manh, T.-P.; Azouz, A.; Detavernier, A.; Hoyois, A.; Das, J.; Bizet, M.; et al. The Transcription Factors Runx3 and ThPOK Cross-Regulate Acquisition of Cytotoxic Function by Human Th1 Lymphocytes. eLife 2018, 7, e30496. [Google Scholar] [CrossRef] [Green Version]
- Juno, J.A.; van Bockel, D.; Kent, S.J.; Kelleher, A.D.; Zaunders, J.J.; Munier, C.M.L. Cytotoxic CD4 T Cells—Friend or Foe during Viral Infection? Front. Immunol. 2017, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Pantaleo, G.; Stanley, S.; Weissman, D. Immunopathogenic Mechanisms of HIV Infection. Ann. Intern. Med. 1996, 124, 654–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klatt, N.R.; Chomont, N.; Douek, D.C.; Deeks, S.G. Immune Activation and HIV Persistence: Implications for Curative Approaches to HIV Infection. Immunol. Rev. 2013, 254, 326–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 Expression on HIV-Specific T Cells Is Associated with T-Cell Exhaustion and Disease Progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Bengsch, B.; Ohtani, T.; Khan, O.; Setty, M.; Manne, S.; O’Brien, S.; Gherardini, P.F.; Herati, R.S.; Huang, A.C.; Chang, K.; et al. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity 2018, 48, 1029–1045.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, R.; Uldrick, T.S. HIV-Associated Cancers and Related Diseases. N. Engl. J. Med. 2018, 378, 1029–1041. [Google Scholar] [CrossRef]
- Leung, V.; Gillis, J.; Raboud, J.; Cooper, C.; Hogg, R.S.; Loutfy, M.R.; Machouf, N.; Montaner, J.S.G.; Rourke, S.B.; Tsoukas, C.; et al. Predictors of CD4:CD8 Ratio Normalization and Its Effect on Health Outcomes in the Era of Combination Antiretroviral Therapy. PLoS ONE 2013, 8, e77665. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M.; Wells, D.W.; Zerbato, J.M.; Kuruc, J.D.; Guo, S.; Luke, B.T.; Eron, J.J.; Bale, M.; Spindler, J.; Simonetti, F.R.; et al. Clones of Infected Cells Arise Early in HIV-Infected Individuals. JCI Insight 2019, 4, e128432. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.M.; et al. HIV Reservoir Size and Persistence Are Driven by T Cell Survival and Homeostatic Proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Jaafoura, S.; de Goër de Herve, M.G.; Hernandez-Vargas, E.A.; Hendel-Chavez, H.; Abdoh, M.; Mateo, M.C.; Krzysiek, R.; Merad, M.; Seng, R.; Tardieu, M.; et al. Progressive Contraction of the Latent HIV Reservoir around a Core of Less-Differentiated CD4+ Memory T Cells. Nat. Commun. 2014, 5, 5407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiener, B.; Horsburgh, B.A.; Eden, J.-S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4 + T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Chomont, N.; DaFonseca, S.; Vandergeeten, C.; Ancuta, P.; Sékaly, R.-P. Maintenance of CD4+ T-Cell Memory and HIV Persistence: Keeping Memory, Keeping HIV. Curr. Opin. HIV AIDS 2011, 6, 30–36. [Google Scholar] [CrossRef]
- von Stockenstrom, S.; Odevall, L.; Lee, E.; Sinclair, E.; Bacchetti, P.; Killian, M.; Epling, L.; Shao, W.; Hoh, R.; Ho, T.; et al. Longitudinal Genetic Characterization Reveals That Cell Proliferation Maintains a Persistent HIV Type 1 DNA Pool During Effective HIV Therapy. J. Infect. Dis. 2015, 212, 596–607. [Google Scholar] [CrossRef]
- Wang, Z.; Gurule, E.E.; Brennan, T.P.; Gerold, J.M.; Kwon, K.J.; Hosmane, N.N.; Kumar, M.R.; Beg, S.A.; Capoferri, A.A.; Ray, S.C.; et al. Expanded Cellular Clones Carrying Replication-Competent HIV-1 Persist, Wax, and Wane. Proc. Natl. Acad. Sci. USA 2018, 115, E2575–E2584. [Google Scholar] [CrossRef] [Green Version]
- Josefsson, L.; von Stockenstrom, S.; Faria, N.R.; Sinclair, E.; Bacchetti, P.; Killian, M.; Epling, L.; Tan, A.; Ho, T.; Lemey, P.; et al. The HIV-1 Reservoir in Eight Patients on Long-Term Suppressive Antiretroviral Therapy Is Stable with Few Genetic Changes over Time. Proc. Natl. Acad. Sci. USA 2013, 110, E4987–E4996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gantner, P.; Morand-Joubert, L.; Sueur, C.; Raffi, F.; Fagard, C.; Lascoux-Combe, C.; Salmon, D.; Amiel, C.; Lambert-Niclot, S.; Fofana, D.B.; et al. Drug Resistance and Tropism as Markers of the Dynamics of HIV-1 DNA Quasispecies in Blood Cells of Heavily Pretreated Patients Who Achieved Sustained Virological Suppression. J. Antimicrob. Chemother. 2016, 71, 751–761. [Google Scholar] [CrossRef]
- Gantner, P.; Lee, G.Q.; Rey, D.; Mesplede, T.; Partisani, M.; Cheneau, C.; Beck-Wirth, G.; Faller, J.-P.; Mohseni-Zadeh, M.; Martinot, M.; et al. Dolutegravir Reshapes the Genetic Diversity of HIV-1 Reservoirs. J. Antimicrob. Chemother. 2018, 73, 1045–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connors, M.; Kovacs, J.A.; Krevat, S.; Gea-Banacloche, J.C.; Sneller, M.C.; Flanigan, M.; Metcalf, J.A.; Walker, R.E.; Falloon, J.; Baseler, M.; et al. HIV Infection Induces Changes in CD4+ T-Cell Phenotype and Depletions within the CD4+ T-Cell Repertoire That Are Not Immediately Restored by Antiviral or Immune-Based Therapies. Nat. Med. 1997, 3, 533–540. [Google Scholar] [CrossRef]
- Gorochov, G.; Neumann, A.U.; Kereveur, A.; Parizot, C.; Li, T.; Katlama, C.; Karmochkine, M.; Raguin, G.; Autran, B.; Debré, P. Perturbation of CD4+ and CD8+ T-Cell Repertoires during Progression to AIDS and Regulation of the CD4+ Repertoire during Antiviral Therapy. Nat. Med. 1998, 4, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhou, T.; Zhu, J.; Zhang, B.; Georgiev, I.; Wang, C.; Chen, X.; Longo, N.S.; Louder, M.; McKee, K.; et al. Focused Evolution of HIV-1 Neutralizing Antibodies Revealed by Structures and Deep Sequencing. Science 2011, 333, 1593–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.-X.; Chen, X.; Munshaw, S.; Zhang, R.; Marshall, D.J.; Vandergrift, N.; Whitesides, J.F.; Lu, X.; Yu, J.-S.; Hwang, K.-K.; et al. Initial Antibodies Binding to HIV-1 Gp41 in Acutely Infected Subjects Are Polyreactive and Highly Mutated. J. Exp. Med. 2011, 208, 2237–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kervevan, J.; Chakrabarti, L.A. Role of CD4+ T Cells in the Control of Viral Infections: Recent Advances and Open Questions. Int. J. Mol. Sci. 2021, 22, 523. [Google Scholar] [CrossRef] [PubMed]
- Kirpach, J.; Colone, A.; Bürckert, J.-P.; Faison, W.J.; Dubois, A.R.S.X.; Sinner, R.; Reye, A.L.; Muller, C.P. Detection of a Low Level and Heterogeneous B Cell Immune Response in Peripheral Blood of Acute Borreliosis Patients With High Throughput Sequencing. Front. Immunol. 2019, 10, 1105. [Google Scholar] [CrossRef]
- Briney, B.; Inderbitzin, A.; Joyce, C.; Burton, D.R. Commonality despite Exceptional Diversity in the Baseline Human Antibody Repertoire. Nature 2019, 566, 393–397. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, M.; Galperin, M.; Patgaonkar, M.; Vasan, S.; Ho, D.D.; Nouël, A.; Claireaux, M.; Benati, D.; Lambotte, O.; Huang, Y.; et al. DNA Vaccination by Electroporation Amplifies Broadly Cross-Restricted Public TCR Clonotypes Shared with HIV Controllers. J. Immunol. 2017, 199, 3437–3452. [Google Scholar] [CrossRef] [Green Version]
- Gantner, P.; Pagliuzza, A.; Pardons, M.; Ramgopal, M.; Routy, J.-P.; Fromentin, R.; Chomont, N. Single-Cell TCR Sequencing Reveals Phenotypically Diverse Clonally Expanded Cells Harboring Inducible HIV Proviruses during ART. Nat. Commun. 2020, 11, 4089. [Google Scholar] [CrossRef]
- Wanjalla, C.N.; McDonnell, W.J.; Ram, R.; Chopra, A.; Gangula, R.; Leary, S.; Mashayekhi, M.; Simmons, J.D.; Warren, C.M.; Bailin, S.; et al. Single-Cell Analysis Shows That Adipose Tissue of Persons with Both HIV and Diabetes Is Enriched for Clonal, Cytotoxic, and CMV-Specific CD4+ T Cells. Cell Rep. Med. 2021, 2, 100205. [Google Scholar] [CrossRef] [PubMed]
- Horton, H.; Vogel, T.; O’Connor, D.; Picker, L.; Watkins, D.I. Analysis of the Immune Response and Viral Evolution during the Acute Phase of SIV Infection. Vaccine 2002, 20, 1927–1932. [Google Scholar] [CrossRef]
- Zaunders, J.J.; Munier, M.L.; Seddiki, N.; Pett, S.; Ip, S.; Bailey, M.; Xu, Y.; Brown, K.; Dyer, W.B.; Kim, M.; et al. High Levels of Human Antigen-Specific CD4 + T Cells in Peripheral Blood Revealed by Stimulated Coexpression of CD25 and CD134 (OX40). J. Immunol. 2009, 183, 2827–2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, S.; Baxter, A.E.; Cirelli, K.M.; Dan, J.M.; Morou, A.; Daigneault, A.; Brassars, N.; Silvestri, G.; Routy, J.-P.; Havenar-Daughton, C.; et al. Comparative Analysis of Activation Induced Marker (AIM) Assays for Sensitive Identification of Antigen-Specific CD4 T Cells. PLoS ONE 2017, 12, e0186998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilkinton, M.A.; McDonnell, W.J.; Barnett, L.; Chopra, A.; Gangula, R.; White, K.D.; Leary, S.; Currenti, J.; Gaudieri, S.; Mallal, S.A.; et al. In Chronic Infection, HIV Gag-Specific CD4 + T Cell Receptor Diversity Is Higher than CD8 + T Cell Receptor Diversity and Is Associated with Less HIV Quasispecies Diversity. J. Virol. 2021, 95, e02380-20. [Google Scholar] [CrossRef] [PubMed]
- Proserpio, V.; Piccolo, A.; Haim-Vilmovsky, L.; Kar, G.; Lönnberg, T.; Svensson, V.; Pramanik, J.; Natarajan, K.N.; Zhai, W.; Zhang, X.; et al. Single-Cell Analysis of CD4+ T-Cell Differentiation Reveals Three Major Cell States and Progressive Acceleration of Proliferation. Genome Biol. 2016, 17, 103. [Google Scholar] [CrossRef] [Green Version]
- Qi, Q.; Liu, Y.; Cheng, Y.; Glanville, J.; Zhang, D.; Lee, J.-Y.; Olshen, R.A.; Weyand, C.M.; Boyd, S.D.; Goronzy, J.J. Diversity and Clonal Selection in the Human T-Cell Repertoire. Proc. Natl. Acad. Sci. USA 2014, 111, 13139–13144. [Google Scholar] [CrossRef] [Green Version]
- Rolland, M.; Heckerman, D.; Deng, W.; Rousseau, C.M.; Coovadia, H.; Bishop, K.; Goulder, P.J.R.; Walker, B.D.; Brander, C.; Mullins, J.I. Broad and Gag-Biased HIV-1 Epitope Repertoires Are Associated with Lower Viral Loads. PLoS ONE 2008, 3, e1424. [Google Scholar] [CrossRef] [Green Version]
- Mothe, B.; Llano, A.; Ibarrondo, J.; Zamarreño, J.; Schiaulini, M.; Miranda, C.; Ruiz-Riol, M.; Berger, C.T.; Herrero, J.M.; Palou, E.; et al. CTL Responses of High Functional Avidity and Broad Variant Cross-Reactivity Are Associated with HIV Control. PLoS ONE 2012, 7, e29717. [Google Scholar] [CrossRef]
- Kunwar, P.; Hawkins, N.; Dinges, W.L.; Liu, Y.; Gabriel, E.E.; Swan, D.A.; Stevens, C.E.; Maenza, J.; Collier, A.C.; Mullins, J.I.; et al. Superior Control of HIV-1 Replication by CD8+ T Cells Targeting Conserved Epitopes: Implications for HIV Vaccine Design. PLoS ONE 2013, 8, e64405. [Google Scholar] [CrossRef] [Green Version]
- Sunshine, J.; Kim, M.; Carlson, J.M.; Heckerman, D.; Czartoski, J.; Migueles, S.A.; Maenza, J.; McElrath, M.J.; Mullins, J.I.; Frahm, N. Increased Sequence Coverage through Combined Targeting of Variant and Conserved Epitopes Correlates with Control of HIV Replication. J. Virol. 2014, 88, 1354–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndhlovu, Z.M.; Stampouloglou, E.; Cesa, K.; Mavrothalassitis, O.; Alvino, D.M.; Li, J.Z.; Wilton, S.; Karel, D.; Piechocka-Trocha, A.; Chen, H.; et al. The Breadth of Expandable Memory CD8 + T Cells Inversely Correlates with Residual Viral Loads in HIV Elite Controllers. J. Virol. 2015, 89, 10735–10747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Q.; Xu, J.-H.; Yu, Y.-Y.; Lo KK, E.; Felicianna; El-Nezami, H.; Zeng, Z. Human Immune Repertoire in Hepatitis B Virus Infection. World J. Gastroenterol. 2021, 27, 3790–3801. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Ye, J.; Wang, J.; Lu, H.; Yao, X. Characterization of the TCR β Chain Repertoire in Peripheral Blood from Hepatitis B Vaccine Responders and Non-Responders. J. Inflamm. Res. 2022, 15, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Osuch, S.; Laskus, T.; Perlejewski, K.; Berak, H.; Bukowska-Ośko, I.; Pollak, A.; Zielenkiewicz, M.; Radkowski, M.; Caraballo Cortés, K. CD8+ T-Cell Exhaustion Phenotype in Chronic Hepatitis C Virus Infection Is Associated With Epitope Sequence Variation. Front. Immunol. 2022, 13, 832206. [Google Scholar] [CrossRef]
- Silva, D.N.; Chrobok, M.; Rovesti, G.; Healy, K.; Wagner, A.K.; Maravelia, P.; Gatto, F.; Mazza, M.; Mazzotti, L.; Lohmann, V.; et al. Process Development for Adoptive Cell Therapy in Academia: A Pipeline for Clinical-Scale Manufacturing of Multiple TCR-T Cell Products. Front. Immunol. 2022, 13, 896242. [Google Scholar] [CrossRef]
- La Gruta, N.L.; Gras, S.; Daley, S.R.; Thomas, P.G.; Rossjohn, J. Understanding the Drivers of MHC Restriction of T Cell Receptors. Nat. Rev. Immunol 2018, 18, 467–478. [Google Scholar] [CrossRef]
- Chen, X.; Ling, J.; Mo, P.; Zhang, Y.; Jiang, Q.; Ma, Z.; Cao, Q.; Hu, W.; Zou, S.; Chen, L.; et al. Restoration of Leukomonocyte Counts Is Associated with Viral Clearance in COVID-19 Hospitalized Patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. SARS-CoV-2-Reactive T Cells in Healthy Donors and Patients with COVID-19. Nature 2020, 587, 270–274. [Google Scholar] [CrossRef]
- García, L.F. Immune Response, Inflammation, and the Clinical Spectrum of COVID-19. Front. Immunol. 2020, 11, 1441. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Meng, M.; Kumar, R.; Wu, Y.; Huang, J.; Deng, Y.; Weng, Z.; Yang, L. Lymphopenia Is Associated with Severe Coronavirus Disease 2019 (COVID-19) Infections: A Systemic Review and Meta-Analysis. Int. J. Infect. Dis. 2020, 96, 131–135. [Google Scholar] [CrossRef]
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.-Q.; Wang, Q.; Miao, H. Lymphopenia Predicts Disease Severity of COVID-19: A Descriptive and Predictive Study. Signal Transduct. Target. Ther. 2020, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- de Candia, P.; Prattichizzo, F.; Garavelli, S.; Matarese, G. T Cells: Warriors of SARS-CoV-2 Infection. Trends Immunol. 2021, 42, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A Single-Cell Atlas of the Peripheral Immune Response in Patients with Severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thurmann, L.; Kurth, F.; Volker, M.T.; et al. COVID-19 Severity Correlates with Airway Epithelium–Immune Cell Interactions Identified by Single-Cell Analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Su, W.; Tang, H.; Le, W.; Zhang, X.; Zheng, Y.; Liu, X.; Xie, L.; Li, J.; Ye, J.; et al. Immune Cell Profiling of COVID-19 Patients in the Recovery Stageby Single-Cell Sequencing. Cell Discov. 2020, 6, 31. [Google Scholar] [CrossRef]
- Ong, E.Z.; Chan, Y.F.Z.; Leong, W.Y.; Lee, N.M.Y.; Kalimuddin, S.; Haja Mohideen, S.M.; Chan, K.S.; Tanoto Tan, A.; Bertoletti, A.; Ooi, E.E.; et al. A Dynamic Immune Response Shapes COVID-19 Progression. Cell Host Microbe 2020, 27, 879–882.e2. [Google Scholar] [CrossRef]
- Ouyang, Y.; Yin, J.; Wang, W.; Shi, H.; Shi, Y.; Xu, B.; Qiao, L.; Feng, Y.; Pang, L.; Wei, F.; et al. Downregulated Gene Expression Spectrum and Immune Responses Changed During the Disease Progression in Patients With COVID-19. Clin. Infect. Dis. 2020, 71, 2052–2060. [Google Scholar] [CrossRef]
- Wang, P.; Jin, X.; Zhou, W.; Luo, M.; Xu, Z.; Xu, C.; Li, Y.; Ma, K.; Cao, H.; Huang, Y.; et al. Comprehensive Analysis of TCR Repertoire in COVID-19 Using Single Cell Sequencing. Genomics 2021, 113, 456–462. [Google Scholar] [CrossRef]
- Niu, X.; Li, S.; Li, P.; Pan, W.; Wang, Q.; Feng, Y.; Mo, X.; Yan, Q.; Ye, X.; Luo, J.; et al. Longitudinal Analysis of T and B Cell Receptor Repertoire Transcripts Reveal Dynamic Immune Response in COVID-19 Patients. Front. Immunol. 2020, 11, 582010. [Google Scholar] [CrossRef]
- Snyder, T.M.; Gittelman, R.M.; Klinger, M.; May, D.H.; Osborne, E.J.; Taniguchi, R.; Zahid, H.J.; Kaplan, I.M.; Dines, J.N.; Noakes, M.T.; et al. Magnitude and Dynamics of the T-Cell Response to SARS-CoV-2 Infection at Both Individual and Population Levels. MedRxiv 2020. [Google Scholar] [CrossRef]
- Wang, Y.; Duan, F.; Zhu, Z.; Yu, M.; Jia, X.; Dai, H.; Wang, P.; Qiu, X.; Liu, Y.; Huang, J. Analysis of TCR Repertoire by High-Throughput Sequencing Indicates the Feature of T Cell Immune Response after SARS-CoV-2 Infection. Cells 2021, 11, 68. [Google Scholar] [CrossRef]
- Shomuradova, A.S.; Vagida, M.S.; Sheetikov, S.A.; Zornikova, K.V.; Kiryukhin, D.; Titov, A.; Peshkova, I.O.; Khmelevskaya, A.; Dianov, D.V.; Malasheva, M.; et al. SARS-CoV-2 Epitopes Are Recognized by a Public and Diverse Repertoire of Human T Cell Receptors. Immunity 2020, 53, 1245–1257.e5. [Google Scholar] [CrossRef] [PubMed]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.-B.; Olsson, A.; Llwellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef] [PubMed]
- Nolan, S.; Vignali, M.; Klinger, M.; Dines, J.N.; Kaplan, I.M.; Svejnoha, E.; Craft, T.; Boland, K.; Pesesky, M.; Gittelman, R.M.; et al. A Large-Scale Database of T-Cell Receptor Beta (TCRβ) Sequences and Binding Associations from Natural and Synthetic Exposure to SARS-CoV-2. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Pasetto, A.; Lu, Y.-C. Single-Cell TCR and Transcriptome Analysis: An Indispensable Tool for Studying T-Cell Biology and Cancer Immunotherapy. Front. Immunol. 2021, 12, 689091. [Google Scholar] [CrossRef]
- Ambrosini-Spaltro, A.; Farnedi, A.; Calistri, D.; Rengucci, C.; Prisinzano, G.; Chiadini, E.; Capelli, L.; Angeli, D.; Bennati, C.; Valli, M.; et al. The Role of next-Generation Sequencing in Detecting Gene Fusions with Known and Unknown Partners: A Single-Center Experience with Methodologies’ Integration. Hum. Pathol. 2022, 123, 20–30. [Google Scholar] [CrossRef]
- Volpi, A.; Bravaccini, S.; Medri, L.; Cerasoli, S.; Gaudio, M.; Amadori, D. Usefulness of Immunological Detection of the Human Telomerase Reverse Transcriptase. Anal. Cell. Pathol. 2005, 27, 347–353. [Google Scholar] [CrossRef]
- Liu, J.; Qu, S.; Zhang, T.; Gao, Y.; Shi, H.; Song, K.; Chen, W.; Yin, W. Applications of Single-Cell Omics in Tumor Immunology. Front. Immunol. 2021, 12, 697412. [Google Scholar] [CrossRef]
- Rempala, G.A.; Seweryn, M. Methods for Diversity and Overlap Analysis in T-Cell Receptor Populations. J. Math. Biol. 2013, 67, 1339–1368. [Google Scholar] [CrossRef] [Green Version]
- Gohil, S.H.; Iorgulescu, J.B.; Braun, D.A.; Keskin, D.B.; Livak, K.J. Applying High-Dimensional Single-Cell Technologies to the Analysis of Cancer Immunotherapy. Nat. Rev. Clin. Oncol. 2020, 18, 244–256. [Google Scholar] [CrossRef]
- Nam, A.S.; Chaligne, R.; Landau, D.A. Integrating Genetic and Non-Genetic Determinants of Cancer Evolution by Single-Cell Multi-Omics. Nat. Rev. Genet. 2021, 22, 3–18. [Google Scholar] [CrossRef]
- Stuart, T.; Satija, R. Integrative Single-Cell Analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef]
- Jin, M.Z.; Jin, W.L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.-A.; Wright, G.S.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [Green Version]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [Green Version]
- Massard, C.; Gordon, M.S.; Sharma, S.; Rafii, S.; Wainberg, Z.A.; Luke, J.; Curiel, T.J.; Colon-Otero, G.; Hamid, O.; Sanborn, R.E.; et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti–Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J. Clin. Oncol. 2016, 34, 3119–3125. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [Green Version]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Gueguen, P.; Metoikidou, C.; Dupic, T.; Lawand, M.; Goudot, C.; Baulande, S.; Lameiras, S.; Lantz, O.; Girard, N.; Seguin-Givelet, A.; et al. Contribution of Resident and Circulating Precursors to Tumor-Infiltrating CD8+ T Cell Populations in Lung Cancer. Sci. Immunol. 2021, 6, eabd5778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, X.; Zheng, L.; Zhang, Y.; Li, Y.; Fang, Q.; Gao, R.; Kang, B.; Zhang, Q.; Huang, J.Y.; et al. Lineage Tracking Reveals Dynamic Relationships of T Cells in Colorectal Cancer. Nature 2018, 564, 268–272. [Google Scholar] [CrossRef]
- Rudqvist, N.-P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.-W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; Demaria, S. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol. Res. 2018, 6, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Zhang, Q.; Wu, S.; Zhang, X.; Wang, M.; He, F.; Wei, T.; Yang, J.; Lou, Y.; Cai, Z.; et al. Characteristics of Tumor Infiltrating Lymphocyte and Circulating Lymphocyte Repertoires in Pancreatic Cancer by the Sequencing of T Cell Receptors. Sci. Rep. 2015, 5, 13664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.K.; et al. PD-1 Identifies the Patient-Specific CD8+ Tumor-Reactive Repertoire Infiltrating Human Tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Han, A.; Glanville, J.; Hansmann, L.; Davis, M.M. Linking T-Cell Receptor Sequence to Functional Phenotype at the Single-Cell Level. Nat. Biotechnol. 2014, 32, 684–692. [Google Scholar] [CrossRef]
- Valpione, S.; Mundra, P.A.; Galvani, E.; Campana, L.G.; Lorigan, P.; De Rosa, F.; Gupta, A.; Weightman, J.; Mills, S.; Dhomen, N.; et al. The T Cell Receptor Repertoire of Tumor Infiltrating T Cells Is Predictive and Prognostic for Cancer Survival. Nat. Commun. 2021, 12, 4098. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Creaney, J.; Redwood, A.; Robinson, B. The Current Lung Cancer Neoantigen Landscape and Implications for Therapy. J. Thorac. Oncol. 2021, 16, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.; McNeil, L.K.; Starobinets, H.; DeVault, V.L.; Cohen, R.B.; Twardowski, P.; Johnson, M.L.; Gillison, M.L.; Stein, M.N.; Vaishampayan, U.N.; et al. An Empirical Antigen Selection Method Identifies Neoantigens That Either Elicit Broad Antitumor T-Cell Responses or Drive Tumor Growth. Cancer Discov. 2021, 11, 696–713. [Google Scholar] [CrossRef]
- Wang, C.; Ding, Y.; Liu, Y.; Zhang, Q.; Xu, S.; Xia, L.; Duan, H.; Wang, S.; Ji, P.; Huang, W.; et al. Identification of Mutated Peptides in Bladder Cancer From Exomic Sequencing Data Reveals Negative Correlation Between Mutation-Specific Immunoreactivity and Inflammation. Front. Immunol. 2020, 11, 576603. [Google Scholar] [CrossRef]
- Wijetunga, N.A.; Yu, Y.; Morris, L.G.; Lee, N.; Riaz, N. The Head and Neck Cancer Genome in the Era of Immunotherapy. Oral Oncol. 2021, 112, 105040. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.-M.; Ren, M.-D.; Liu, N.; Gao, H.; Wang, J.-J.; Zheng, X.-Q.; Fu, X.; Liang, X.; Ruan, Z.-P.; Tian, T.; et al. Tumor Mutation Burden, Immune Cell Infiltration, and Construction of Immune-Related Genes Prognostic Model in Head and Neck Cancer. Int. J. Med. Sci. 2021, 18, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Ji, C.; Jiang, L.; Zhu, Y.; Zhou, Y.; Meng, J.; Gao, J.; Lu, T.; Ye, J.; Yan, F. Tumour Microenvironment-based Molecular Profiling Reveals Ideal Candidates for High-grade Serous Ovarian Cancer Immunotherapy. Cell Prolif. 2021, 54, e12979. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen Screening Identifies Broad TP53 Mutant Immunogenicity in Patients with Epithelial Cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-Cell Responses to TP53 “Hotspot” Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Matsuzaki, J.; Wei, L.; Tsuji, T.; Battaglia, S.; Hu, Q.; Cortes, E.; Wong, L.; Yan, L.; Long, M.; et al. Efficient Identification of Neoantigen-Specific T-Cell Responses in Advanced Human Ovarian Cancer. J. Immunother. Cancer 2019, 7, 156. [Google Scholar] [CrossRef]
- Burrack, A.L.; Spartz, E.J.; Raynor, J.F.; Wang, I.; Olson, M.; Stromnes, I.M. Combination PD-1 and PD-L1 Blockade Promotes Durable Neoantigen-Specific T Cell-Mediated Immunity in Pancreatic Ductal Adenocarcinoma. Cell Rep. 2019, 28, 2140–2155.e6. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.C.; Denroche, R.; Jang, G.H.; Nowak, K.M.; Zhang, A.; Borgida, A.; Holter, S.; Topham, J.T.; Wilson, J.; Dodd, A.; et al. Clinical and Genomic Characterisation of Mismatch Repair Deficient Pancreatic Adenocarcinoma. Gut 2020, 7, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Zheng, Z.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Li, Y.F.; Ray, S.; Franco, Z.; Bliskovsky, V.; et al. An Efficient Single-Cell RNA-Seq Approach to Identify Neoantigen-Specific T Cell Receptors. Mol. Ther. 2018, 26, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, M.R.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Ivey, G.; Li, Y.F.; El-Gamil, M.; Lalani, A.; et al. Unique Neoantigens Arise from Somatic Mutations in Patients with Gastrointestinal Cancers. Cancer Discov. 2019, 9, 1022–1035. [Google Scholar] [CrossRef]
- Gros, A.; Tran, E.; Parkhurst, M.R.; Ilyas, S.; Pasetto, A.; Groh, E.M.; Robbins, P.F.; Yossef, R.; Garcia-Garijo, A.; Fajardo, C.A.; et al. Recognition of Human Gastrointestinal Cancer Neoantigens by Circulating PD-1+ Lymphocytes. J. Clin. Investig. 2019, 129, 4992–5004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.-C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.; Malekzadeh, P.; et al. Immunologic Recognition of a Shared P53 Mutated Neoantigen in a Patient with Metastatic Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Chasov, V.; Zaripov, M.; Mirgayazova, R.; Khadiullina, R.; Zmievskaya, E.; Ganeeva, I.; Valiullina, A.; Rizvanov, A.; Bulatov, E. Promising New Tools for Targeting P53 Mutant Cancers: Humoral and Cell-Based Immunotherapies. Front. Immunol. 2021, 12, 707734. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, F.; Sonnemann, H.; Jackson, K.R.; Talukder, A.H.; Katailiha, A.S.; Lizee, G. Evolution of CD8+ T Cell Receptor (TCR) Engineered Therapies for the Treatment of Cancer. Cells 2021, 10, 2379. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, X.; Zhang, F.; Zhang, X.; Tang, F.; Han, Z.; Li, Y. TCR-T Immunotherapy: The Challenges and Solutions. Front. Oncol. 2022, 11, 794183. [Google Scholar] [CrossRef] [PubMed]
- Sheih, A.; Voillet, V.; Hanafi, L.-A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.; Riddell, S.R.; Maloney, D.G.; Wohlfhart, M.E.; et al. Clonal Kinetics and Single-Cell Transcriptional Profiling of CAR-T Cells in Patients Undergoing CD19 CAR-T Immunotherapy. Nat. Commun. 2020, 11, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [Green Version]
- Martino, M.; Canale, F.A.; Alati, C.; Vincelli, I.D.; Moscato, T.; Porto, G.; Loteta, B.; Naso, V.; Mazza, M.; Nicolini, F.; et al. CART-Cell Therapy: Recent Advances and New Evidence in Multiple Myeloma. Cancers 2021, 13, 2639. [Google Scholar] [CrossRef]
- Nagarsheth, N.B.; Norberg, S.M.; Sinkoe, A.L.; Adhikary, S.; Meyer, T.J.; Lack, J.B.; Warner, A.C.; Schweitzer, C.; Doran, S.L.; Korrapati, S.; et al. TCR-Engineered T Cells Targeting E7 for Patients with Metastatic HPV-Associated Epithelial Cancers. Nat. Med. 2021, 27, 419–425. [Google Scholar] [CrossRef]
- Wei, F.; Cheng, X.-X.; Xue, J.Z.; Xue, S.-A. Emerging Strategies in TCR-Engineered T Cells. Front. Immunol. 2022, 13, 850358. [Google Scholar] [CrossRef]
- Tan, E.; Gakhar, N.; Kirtane, K. TCR Gene-Engineered Cell Therapy for Solid Tumors. Best Pract. Res. Clin. Haematol. 2021, 34, 101285. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor Regression in Patients With Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive With NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1–Reactive T-Cell Receptor: Long-Term Follow-up and Correlates with Response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 C259T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langerak, A.W. (Ed.) Immunogenetics: Methods and Protocols. In Methods in Molecular Biology; Springer: New York, NY, USA, 2022; Volume 2453, ISBN 978-1-07-162114-1. [Google Scholar]
- Hou, X.; Lu, C.; Chen, S.; Xie, Q.; Cui, G.; Chen, J.; Chen, Z.; Wu, Z.; Ding, Y.; Ye, P.; et al. High Throughput Sequencing of T Cell Antigen Receptors Reveals a Conserved TCR Repertoire. Medicine 2016, 95, e2839. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Tunkey, C.D.; Pugh, T.J.; Ross, T.; Shah, M.; Lee, C.C.; Harkins, T.T.; Wells, W.A.; Tafe, L.J.; Amos, C.I.; et al. A Comprehensive Assay for CFTR Mutational Analysis Using Next-Generation Sequencing. Clin. Chem. 2013, 59, 1481–1488. [Google Scholar] [CrossRef] [Green Version]
- Howie, B.; Sherwood, A.M.; Berkebile, A.D.; Berka, J.; Emerson, R.O.; Williamson, D.W.; Kirsch, I.; Vignali, M.; Rieder, M.J.; Carlson, C.S.; et al. High-Throughput Pairing of T Cell Receptor α and β Sequences. Sci. Transl. Med. 2015, 7, ra131–ra301. [Google Scholar] [CrossRef]
- Lee, E.S.; Thomas, P.G.; Mold, J.E.; Yates, A.J. Identifying T Cell Receptors from High-Throughput Sequencing: Dealing with Promiscuity in TCRα and TCRβ Pairing. PLoS Comput. Biol. 2017, 13, e1005313. [Google Scholar] [CrossRef] [Green Version]
- Chronister, W.D.; Crinklaw, A.; Mahajan, S.; Vita, R.; Koşaloğlu-Yalçın, Z.; Yan, Z.; Greenbaum, J.S.; Jessen, L.E.; Nielsen, M.; Christley, S.; et al. TCRMatch: Predicting T-Cell Receptor Specificity Based on Sequence Similarity to Previously Characterized Receptors. Front. Immunol. 2021, 12, 640725. [Google Scholar] [CrossRef]
- Ma, K.-Y.; Schonnesen, A.A.; He, C.; Xia, A.Y.; Sun, E.; Chen, E.; Sebastian, K.R.; Guo, Y.-W.; Balderas, R.; Kulkarni-Date, M.; et al. High-Throughput and High-Dimensional Single-Cell Analysis of Antigen-Specific CD8+ T Cells. Nat. Immunol. 2021, 22, 1590–1598. [Google Scholar] [CrossRef]
- Francis, J.M.; Leistritz-Edwards, D.; Dunn, A.; Tarr, C.; Lehman, J.; Dempsey, C.; Hamel, A.; Rayon, V.; Liu, G.; Wang, Y.; et al. Allelic Variation in Class I HLA Determines CD8+ T Cell Repertoire Shape and Cross-Reactive Memory Responses to SARS-CoV-2. Sci. Immunol. 2021, 3070, eabk3070. [Google Scholar] [CrossRef]
- Bentzen, A.K.; Marquard, A.M.; Lyngaa, R.; Saini, S.K.; Ramskov, S.; Donia, M.; Such, L.; Furness, A.J.S.; McGranahan, N.; Rosenthal, R.; et al. Large-Scale Detection of Antigen-Specific T Cells Using Peptide-MHC-I Multimers Labeled with DNA Barcodes. Nat. Biotechnol. 2016, 34, 1037–1045. [Google Scholar] [CrossRef]
- Wang, L.; Lan, X. Rapid Screening of TCR-PMHC Interactions by the YAMTAD System. Cell Discov. 2022, 8, 30. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Sources of α/β TCR Diversity | |
|---|---|
| Recombination of the T-cell α-genes on chromosome 14 and T-cell β-genes on chromosome 7 by RAG1/2 enzymes. | Total α-genes combination: 2392 Total β-genes combination: 1248 Total αβ-genes combination: 2,985,216 [6]. |
| Theoretical diversity by pairing of different in-frame α- and β-chains plus junctional diversity by terminal deoxynucleotidyl transferase activity [6]. | 1015–1061 [14] |
| Experimental diversity evaluation by deep sequencing. | 104–106, based on the amount of the sample. |
| Type of HLA Alleles Association | HLA Typing Future Opportunities | Example |
|---|---|---|
| With specific infectious diseases or the severity of infection | To provide insight into differences in T-cell repertoires in infectious disease and patterns of T-cell targeting | Heterozygous individuals progress less rapidly to AIDS than HLA homozygous individuals after HIV infection [31]. Kaslow et al. found that HLA-B27 and B57 were strongly associated with slow progression to AIDS [36]. |
| With increased risk of or protection from various autoimmune disorders | To clarify a subject’s disease state and potentially stratify patients for treatment studies. | Association of the HLA class I region has been detected for several autoimmune diseases (AIDs); some examples are: |
| With cancer therapy outcomes | To understand and infer the efficacy of immunotherapy in specific individuals | Higher heterozygosity in HLA has been linked to a better response to anti-cancer treatments [41]. |
| Advantages | |
| gDNA | mRNA |
|
|
| Disadvantages | |
| gDNA | mRNA |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzotti, L.; Gaimari, A.; Bravaccini, S.; Maltoni, R.; Cerchione, C.; Juan, M.; Navarro, E.A.-G.; Pasetto, A.; Nascimento Silva, D.; Ancarani, V.; et al. T-Cell Receptor Repertoire Sequencing and Its Applications: Focus on Infectious Diseases and Cancer. Int. J. Mol. Sci. 2022, 23, 8590. https://doi.org/10.3390/ijms23158590
Mazzotti L, Gaimari A, Bravaccini S, Maltoni R, Cerchione C, Juan M, Navarro EA-G, Pasetto A, Nascimento Silva D, Ancarani V, et al. T-Cell Receptor Repertoire Sequencing and Its Applications: Focus on Infectious Diseases and Cancer. International Journal of Molecular Sciences. 2022; 23(15):8590. https://doi.org/10.3390/ijms23158590
Chicago/Turabian StyleMazzotti, Lucia, Anna Gaimari, Sara Bravaccini, Roberta Maltoni, Claudio Cerchione, Manel Juan, Europa Azucena-Gonzalez Navarro, Anna Pasetto, Daniela Nascimento Silva, Valentina Ancarani, and et al. 2022. "T-Cell Receptor Repertoire Sequencing and Its Applications: Focus on Infectious Diseases and Cancer" International Journal of Molecular Sciences 23, no. 15: 8590. https://doi.org/10.3390/ijms23158590