3.2.2. General Protocol for the Synthesis of Thioureas 10a–10k
Amine (2.0 mmol) was added to a heterogeneous solution of 3,6-diisothiocyanatoacridine (
9) (60 mg, 0.20 mmol) in methanol (1.50 mL) [
14]. The reaction mixture was stirred vigorously and the course of the reaction was monitored using TLC in which chloroform was used as an eluent. When the TLC result was negative in the presence of reactant
9, diethylether was added and the resultant mixture was stirred for an additional two hours to maximize the precipitation of the product. The heterogeneous mixture was then filtered off and the crude product was washed with ethylacetate then with
n-hexane in a funnel. The final product was crystalized from the DMF–methanol mixture.
N,N’-Acridine-3,6-diylbis(N’-cyclohexylthiourea) (10a, 92 mg, 91.3%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (600 MHz, DMSO–d6) δ 9.80 (s, 2 × NH, 2H) 8.84 (s, H9, 1H) 8.39 (s, H4, H5, 2H), 8.03 (d, J = 7.8 Hz, 2 × NH, 2H), 8.00 (d, J = 9.1 Hz, H1, H8, 2H), 7.60 (d, J = 9.1 Hz, H2, H7, 2H), 4.26–4.05 (m, 2 × NHCH, 2H) 2.08–1.92 (m, 4 × NHCHCHA, 4H), 1.84–1.65 (m, 4 × NHCHCH2CHA, 4H) 1.65–1.52 (m, 2 × NHCHCH2CH2CHA, 2H), 1.47–1.26 (m, 4 × NHCHCHB, 4H), 1.47–1.26 (m, 4 × NHCHCH2CHB, 4H), 1.26–1.11 (m, 2 × NHCHCH2CH2CHB, 2H). 13C NMR (150 MHz, DMSO–d6) δ 179.0 (2 × CS), 149.4 (C4a, C10a), 141.6 (C3, C6), 135.0 (C9), 128.5 (C1, C8), 122.6 (C8a, C9a), 122.5 (C2, C7), 115.6 (C4, C5), 52.3 (2 × NHCH), 31.8 (4 × NHCHCH2), 25.2 (2 × NHCHCH2CH2CH2), 24.5 (4 × NHCHCH2CH2). HRMS (ESI): m/z calculated for C27H33N5S2 [M + H]+ 492.22501, found 492.22650.
N,N’-Acridine-3,6-diylbis(N’-(azepan-1-yl)thiourea) (10b, 96 mg, 95.2%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystallized from the DMSO-diethyleter-methanol mixture. 1H NMR (400 MHz, DMSO–d6) δ 9.38 (s, 2 × NH, 2H), 8.88 (s, H9, 1H), 7.97 (s, H1, H8, 2H), 7.85 (d, J = 9.1 Hz, H4, H5, 2H), 7.70 (d, J = 9.1 Hz, H2, H7, 2H), 4.01–3.80 (m, 4 × NCH2, 8H), 1.86–1.77 (m, 4 × NCH2CH2, 8H), 1.63–1.51 (m, 4 × NCH2CH2CH2, 8H). 13C NMR (100 MHz, DMSO–d6) δ 180.6 (2 × CS), 149.1 (C4a, C10a), 143.2 (C3, C6), 134.8 (C9), 126.9 (C1, C8), 125.9 (C2, C7), 123.3 (C8a, C9a), 119.9 (C4, C5), 50.0 (4 × NCH2), 26.9 (4 × NCH2CH2), 26.3 (2 × NCH2CH2CH2). HRMS (ESI): m/z calculated for C27H33N5S2 [M + H]+ 492.22501, found 492.22750.
N,N’-Acridine-3,6-diylbis(N’-phenylthiourea) (10c, 92 mg, 93.5%). Yellow crystalline solid, mp > 200°C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (600 MHz, DMSO–d6) δ 10.28 (bs, 2 × NH, 2H), 10.15 (bs, 2 × NH, 2H), 8.91 (s, H9, 1H), 8.28 (s, H4, H5, 2H), 8.06 (d, J = 9.1 Hz, H1, H8, 2H), 7.72 (d, J = 9.1 Hz, H2, H7, 2H), 7.58–7.53 (m, 2 × (H2′, H6′), 4H), 7.41–7.34 (m, 2 × (H3’, H5’), 4H), 7.20–7.13 (m, 2 × H4’, 2H). 13C NMR (150 MHz, DMSO–d6) δ 179.5 (2 × CS), 149.3 (C4a, C10a), 141.5 (C3, C6), 139.3 (2 × C1’), 135.2 (C9), 128.6 (2 × (C3’, C5’), 128.4 (C1, C8), 124.7 (2 × C4’), 123.7 (2 × (C2’, C6’)), 123.2 (C2, C7), 123.1 (C8a, C9a), 117.5 (C4, C5). HRMS (ESI): m/z calculated for C27H21N5S2 [M + H]+ 480.13111, found 480.13252.
N,N’-Acridine-3,6-diylbis(N’-benzylthiourea) (10d, 100 mg, 96.1%). Yellow crystalline solid, mp > 200°C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (600 MHz, DMSO–d6) δ 10.08 (s, 2 × NH, 2H), 8.88 (s, H9, 1H), 8.58 (t, J = 5.7 Hz, 2 × NH, 2H), 8.35 (s, H4, H5, 2H), 8.04 (d, J = 9.1 Hz, H1, H8, 2H), 7.61 (d, J = 9.1 Hz, H2, H7, 2H), 7.40 (d, J = 7.3 Hz, 2 × (H2’, H6’), 4H), 7.44–7.34 (m, 2 × (H3’, H5’), 4H), 7.32–7.26 (m, 2 × H4’, 2H), 4.81 (d, J = 5.7 Hz, 2 × NHCH2, 4H). 13C NMR (150 MHz, DMSO–d6) δ 180.6 (CS), 149.4 (C4a, C10a), 141.3 (C3, C6), 138.7 (2 × C1’), 135.1 (C9), 128.7 (C1, C8), 128.4 (2 × (C3’, C5’)), 127.6 (2 × (C2’, C6’)), 127.0 (2 × C4’), 125.9 (C8a, C9a), 122.8 (C2, C7), 116.6 (C4, C5), 47.3 (2 × NHCH2). HRMS (ESI): m/z calculated for C29H25N5S2 [M + H]+ 508.16241, found 508.16480.
N,N’-Acridine-3,6-diylbis(N’-(4-methylphenyl)thiourea) (10e, 92 mg, 88.4%). Yellow crystalline solid, mp > 120 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture.1H NMR (400 MHz, DMSO–d6) δ 10.19 (s, 2 × NH, 2H), 10.04 (s, 2 × NH, 2H), 8.89 (s, H9, 1H), 8.26 (s, H4, H5, 2H), 8.04 (d, J = 9.1 Hz, H1, H8, 2H), 7.71 (d, J = 9.1 Hz, H2, H7, 2H), 7.39 (d, J = 7.3 Hz, 2 × (H2’, H6’), 4H), 7.17 (d, J = 7.3 Hz, 2 × (H3’, H5’), 4H), 2.29 (s, 2 × CH3, 6H). 13C NMR (100 MHz, DMSO–d6) δ 179.7 (2 × CS), 149.4 (C4a, C10a), 141.8 (C3, C6), 136.8 (2 × C1’), 135.4 (2 × C4’), 134.3 (C9), 129.2 (C1, C8), 128.5 (2 × (C3’, C5’)), 124.1 (2 × (C2’, C6’)), 123.4 (C2, C7), 123.2 (C8a, C9a), 117.6 (C4, C5), 20.7 (2 × CH3). HRMS (ESI): m/z calculated for C29H25N5S2 [M + H]+ 508.16241, found 508.16475.
N,N’-Acridine-3,6-diylbis(N’-(2-phenylethyl)thiourea) (10f, 100 mg, 91.1%). Yellow crystalline solid, mp > 180 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (400 MHz, DMSO–d6) δ 10.02 (bs, 2 × NH, 2H), 8.86 (s, H9, 1H), 8.29 (s, H4, H5, 2H), 8.25–8.14 (m, 2 × NH, 2H), 8.01 (d, J = 9.1 Hz, H1, H8, 2H), 7.55 (d, J = 9.1 Hz, H2, H7, 2H), 7.39–7.28 (m, 2 × (H2’, H6’), 2 × (H3’, H5’), 8H), 7.27–7.21 (m, 2 × H4’, 2H), 3.84–3.74 (m, 2 × NHCH2, 4H), 2.95 (t, J = 6.4 Hz, 2 × NHCH2CH2, 4H). 13C NMR (100 MHz, DMSO–d6) δ 180.2 (2 × CS), 149.4 (C4a, C10a), 141.2 (C3, C6), 139.3 (2 × C1’), 135.0 (C9), 128.7 (2 × (C2’, C6’), (C1, C8)), 128.5 (2 × (C3’, C5’), 126.2 (2 × C4’), 122.8 (C8a, C9a), 122.6 (C2, C7), 116.4 (C4, C5), 45.5 (2 × NHCH2CH2), 34.3 (2 × NHCH2CH2). HRMS (ESI): m/z calculated for C31H29N5S2 [M + H]+ 536.19371, found 536.19362.
N,N’-Acridine-3,6-diylbis(N’-(2-methylbenzyl)thiourea) (10g, 104 mg, 94.7%). Yellow crystalline solid, mp > 180 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (400 MHz, DMSO–d6) δ 10.04 (bs, 2 × NH, 2H), 8.87 (s, H9, 1H), 8.60–8.46 (m, 2 × NH, 2H), 8.35 (s, H4, H5, 2H), 8.03 (d, J = 9.1 Hz, H1, H8, 2H), 7.61 (d, J = 9.1 Hz, H2, H7, 2H), 7.29 (d, J = 7.3 Hz, 2 × (H2’, H6’), 4H), 7.18 (d, J = 7.3 Hz, 2 × (H3’, H5’), 4H), 4.75 (d, J = 5.7 Hz, 2 × NHCH2, 4H), 2.30 (s, 2 × CH3, 6H). 13C NMR (100 MHz, DMSO–d6) δ 180.5 (2 × CS), 149.4 (C4a, C10a), 141.3 (C3, C6), 136.1 (2 × C1’), 135.5 (2 × C4’), 135.1 (C9), 128.9 (2 × (C3’, C5’), 128.7 (C1, C8), 127.6 (2 × (C2’, C6’), 122.9 (C8a, C9a), 122.8 (C2, C7), 116.5 (C4, C5), 47.0 (2 × NHCH2), 20.7 (2 × CH3). HRMS (ESI): m/z calculated for C31H29N5S2 [M + H]+ 536.19371, found 536.19574.
N,N’-Acridine-3,6-diylbis(N’-[(1S)-1-phenylethyl]thiourea) (10h, 40 mg, 36.4%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the diethyleter-n-hexane mixture. 1H NMR (600 MHz, DMSO–d6) δ 9.89 (s, 2 × NH, 2H), 8.85 (s, H9, 1H), 8.62–8.51 (m, 2 × NH, 2H), 8.41 (s, H4, H5, 2H), 8.01 (d, J = 9.1 Hz, H1, H8, 2H), 7.61 (d, J = 9.1 Hz, H2, H7, 2H), 7.42 (d, J = 7.3 Hz, 2 × (H2’, H6’), 4H), 7.46–7.33 (m, 2 × (H3’, H5’), 4H), 7.31–7.24 (m, 2 × H4’, 2H), 5.67–5.51 (m, 2 × NHCH, 2H), 1.52 (d, J = 7.0 Hz, 2 × CH3, 6H). 13C NMR (150 MHz, DMSO–d6) δ 179.5 (2 × CS), 149.4 (C4a, C10a), 143.7 (2 × C1’), 141.6 (C3, C6), 135.0 (C9), 128.5 (C1, C8), 128.4 (2 × (C3’, C5’)), 126.9 (2 × C4’), 126.3 (2 × (C2’, C6’)), 122.7 (C8a, C9a), 122.6 (C2, C7), 116.1 (C4, C5), 52.7 (2 × NHCH), 21.9 (2 × CH3). [α]D20 = +33.3 (c 0.06, Methanol). HRMS (ESI): m/z calculated for C31H29N5S2 [M + H]+ 536.19371, found 536.19405.
N,N’-Acridine-3,6-diylbis(N’-[(1R)-1-phenylethyl]thiourea) (10i, 37 mg, 33.7%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the diethyleter-n-hexane mixture. 1H NMR (400 MHz, DMSO–d6) δ 9.89 (s, 2 × NH, 2H), 8.85 (s, H9, 1H), 8.62–8.51 (m, 2 × NH, 2H), 8.41 (s, H4, H5, 2H), 8.01 (d, J = 9.1 Hz, H1, H8, 2H), 7.61 (d, J = 9.1 Hz, H2, H7, 2H), 7.42 (d, J = 7.3 Hz, 2 × (H2’, H6’), 4H), 7.46–7.33 (m, 2 × (H3’, H5’), 4H), 7.31–7.24 (m, 2 × H4’, 2H), 5.67–5.51 (m, 2 × NHCH, 2H), 1.52 (d, J = 7.0 Hz, 2 × CH3, 6H). 13C NMR (100 MHz, DMSO–d6) δ 179.5 (2 × CS), 149.4 (C4a, C10a), 143.7 (2 × C1’), 141.6 (C3, C6), 135.0 (C9), 128.5 (C1, C8), 128.4 (2 × (C3’, C5’)), 126.9 (2 × C4’), 126.3 (2 × (C2’, C6’)), 122.7 (C8a, C9a), 122.6 (C2, C7), 116.1 (C4, C5), 52.7 (2 × NHCH), 21.9 (2 × CH3). [α]D20 = −33.3 (c 0.06, Methanol). HRMS (ESI): m/z calculated for C31H29N5S2 [M + H]+ 536.19371, found 536.19574.
N,N’-Acridine-3,6-diylbis(N’-hexylthiourea) (
10j, 90 mg, 90.8%). All physicochemical properties were in accordance with previously published data [
14].
3.2.3. General Protocol for the Synthesis of the Ureas 11a–11l
MNO (2.30 mol%) was added to a heterogeneous mixture of urea (50 mg) in methanol (3 mL) [
14]. The reaction mixture was stirred vigorously in dark conditions for 3–5 h. The course of the reaction was monitored using TLC with a methanol–ammonium hydroxide mixture at a volume ratio of 1:10 as an eluent. The crude product was then filtered off and washed with methanol (1 mL), diethyleter (1 mL) and ethylacetate (1 mL).
N,N’-Acridine-3,6-diylbis(N’-cyclohexylurea) (11a, 35 mg, 74.7%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (600 MHz, DMSO–d6) δ 8.75 (s, 2 × NH, 2H), 8.69 (s, H9, 1H), 8.12 (s, H4, H5, 2H), 7.91 (d, J = 9.1 Hz, H1, H8, 2H), 7.43 (d, J = 9.1 Hz, H2, H7, 2H), 6.28 (d, J = 7.8 Hz, 2 × NH, 2H), 3.60–3.47 (m, 2 × NHCH, 2H), 1.91–1.76 (m, 4 × NHCHCHA, 4H), 1.76–1.62 (m, 4 × NHCHCH2CHA, 4H), 1.62–1.45 (m, 2 × NHCHCH2CH2CHA, 2H), 1.43–1.27 (m, 4 × NHCHCH2CHB, 4H), 1.27–1.05 (m, 2 × NHCHCH2CH2CHB, 2H), 1.27–1.05 (m, 4 × NHCHCHB, 4H). 13C NMR (150 MHz, DMSO–d6) δ 154.2 (2 × CO), 150.1 (C4a, C10a), 142.1 (C3, C6), 134.9 (C9), 129.0 (C1, C8), 121.2 (C8a, C9a), 119.4 (C2, C7), 110.8 (C4, C5), 47.7 (2 × NHCH), 32.9 (4 × NHCHCH2), 25.3 (2 × NHCHCH2CH2CH2), 24.4 (4 × NHCHCH2CH2). HRMS (ESI): m/z calculated for C27H33N5O2 [M + H]+ 460.27070, found 460.27032.
N,N’-Acridine-3,6-diylbis(N’-(azepan-1-yl)urea) (11b, 30 mg, 53.5%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (600 MHz, DMSO–d6) δ 8.72 (s, H9, 1H), 8.57 (s, 2 × NH, 2H), 8.19 (s, H4, H5, 2H), 7.92 (d, J = 9.1 Hz, H1, H8, 2H), 7.71 (d, J = 9.1 Hz, H2, H7, 2H), 3.64–3.45 (m, 4 × NCH2, 8H), 1.81–1.62 (m, 4 × NCH2CH2, 8H), 1.62–1.42 (m, 4 × NCH2CH2CH2, 8H). 13C NMR (150 MHz, DMSO–d6) δ 154.8 (2 × CO), 149.8 (C4a, C10a), 142.4 (C3, C6), 134.6 (C9), 128.1 (C1, C8), 121.6 (C8a, C9a), 121.1 (C2, C7), 113.4 (C4, C5), 46.3 (4 × NCH2), 28.1 (4 × NCH2CH2), 26.6 (4 × NCH2CH2CH2). HRMS (ESI): m/z calculated for C27H33N5O2 [M + H]+ 460.27070, found 460.27074.
N,N’-Acridine-3,6-diylbis(N’-phenylurea) (11c, 34 mg, 73.1%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (600 MHz, DMSO–d6) δ 9.36 (s, 2 × NH, 2H), 9.00 (s, 2 × NH, 2H), 8.89 (s, H9, 1H), 8.31 (s, H4, H5, 2H), 8.05 (d, J = 9.1 Hz, H1, H8, 2H), 7.60–7.50 (m, H2, H7, 2 × (H2’, H6’), 6H), 7.40–7.30 (m, 2 × (H3’, H5’), 4H), 7.03–6.97 (m, 2 × H4’, 2H). 13C NMR (150 MHz, DMSO–d6) δ 152.4 (2 × CO), 148.9 (C4a, C10a), 142.3 (C3, C6), 139.4 (2 × C1’), 136.4 (C9), 129.6 (C1, C8), 128.9 (2 × (C3’, C5’)), 122.2 (2 × C4’), 121.5 (C8a, C9a), 119.8 (C2, C7), 118.4 (2 × (C2’, C6’)), 110.6 (C4, C5). HRMS (ESI): m/z calculated for C27H21N5O2 [M + H]+ 448.17680, found 448.17961.
N,N’-Acridine-3,6-diylbis(N’-benzylthiourea) (11d, 30 mg, 64.0%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (600 MHz, DMSO–d6) δ 9.04 (s, 2 × NH, 2H), 8.72 (s, H9, 1H), 8.17 (s, H4, H5, 2H), 7.93 (d, J = 9.1 Hz, H1, H8, 2H), 7.49 (d, J = 9.1 Hz, H2, H7, 2H), 7.38–7.31 (m, 2 × (H2’, H6’), 4H), 7.38–7.31 (m, 2 × (H3’, H5’), 4H), 7.30–7.21 (m, 2 × H4’, 2H), 6.84 (t, J = 5.7 Hz, 2 × NH, 2H), 4.38 (d, J = 5.7 Hz, 2 × NHCH2, 4H). 13C NMR (150 MHz, DMSO–d6) δ 155.1 (2 × CO), 150.1 (C4a, C10a), 142.0 (C3, C6), 140.1 (2 × C1’), 134.9 (C9), 129.0 (C1, C8), 128.4 (2 × (C3’, C5’)), 127.2 (2 × (C2’, C6’)), 126.8 (2 × C4’), 121.3 (C8a, C9a), 119.5 (C2, C7), 111.1 (C4, C5), 42.9 (2 × NHCH2). HRMS (ESI): m/z calculated for C29H25N5O2 [M + H]+ 476.20810, found 476.20822.
N,N’-Acridine-3,6-diylbis(N’-(4-methylphenyl)urea) (11e, 30 mg, 64.0%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (400 MHz, DMSO–d6) δ 9.12 (s, 2 × NH, 2H), 8.81–8.76 (m, H9, NH, 2H), 8.24 (s, H4, H5, 2H), 8.00 (d, J = 9.1 Hz, H1, H8, 2H), 7.53 (d, J = 9.1 Hz, H2, H7, 2H), 7.41 (d, J = 7.3 Hz, 2 × (H2’, H6’), 4H), 7.13 (d, J = 7.3 Hz, 2 × (H3’, H5’), 4H), 2.26 (s, 2 × CH3, 6H). 13C NMR (100 MHz, DMSO–d6) δ 152.5 (2 × CO), 150.0 (C4a, C10a), 141.5 (C3, C6), 136.9 (2 × C1’), 135.5 (C9), 131.0 (2 × C4’), 129.3 (2 × (C3’, C5’)), 129.2 (C1, C8), 121.6 (C8a, C9a), 119.7 (C2, C7), 118.6 (2 × (C2’, C6’)), 111.8 (C4, C5), 20.4 (2 × CH3). HRMS (ESI): m/z calculated for C29H25N5O2 [M + H]+ 476.20810, found 476.20803.
N,N’-Acridine-3,6-diylbis(N’-(2-phenylethyl)urea) (11f, 35 mg, 74.5%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (600 MHz, DMSO–d6) δ 8.95 (s, 2 × NH, 2H), 8.70 (s, H9, 1H), 8.15 (s, H4, H5, 2H), 7.92 (d, J = 9.1 Hz, H1, H8, 2H), 7.45 (d, J = 9.1 Hz, H2, H7, 2H), 7.37–7.30 (m, 2 × (H3’, H5’), 4H), 7.28 (d, J = 7.3 Hz, 2 × (H2’, H6’), 4H), 7.23–7.21 (m, 2 × H4’, 2H), 6.33 (t, J = 5.7 Hz, 2 × NH, 2H), 3.42 (m, 2 × NHCH2, 4H), 2.81 (t, J = 6.4 Hz, 2 × NHCH2CH2, 4H). 13C NMR (150 MHz, DMSO–d6) δ 155.0 (2 × CO), 150.1 (C4a, C10a), 142.0 (C3, C6), 139.5 (2 × C1’), 134.9 (C9), 129.0 (C1, C8), 128.7 (2 × (C2’, C6’)), 128.4 (2 × (C3’, C5’)), 126.1 (2 × C4’), 121.3 (C8a, C9a), 119.4 (C2, C7), 111.0 (C4, C5), 40.7 (2 × NHCH2CH2), 35.7 (2 × NHCH2CH2). HRMS (ESI): m/z calculated for C31H29N5O2 [M + H]+ 504.23940, found 504.24187.
N,N’-Acridine-3,6-diylbis(N’-(2-methylbenzyl)thiourea) (11g, 35 mg, 74.5%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the DMSO-diethyleter-methanol mixture. 1H NMR (400 MHz, DMSO–d6) δ 9.00 (s, 2 × NH, 2H), 8.71 (s, H9, 1H), 8.16 (s, H4, H5, 2H), 7.93 (d, J = 9.1 Hz, H1, H8, 2H), 7.48 (d, J = 9.1 Hz, H2, H7, 2H), 7.24 (d, J = 7.3 Hz, 2 × (H2’, H6’), 4H), 7.16 (t, J = 7.5 Hz, 2 × (H3’, H5’), 4H), 6.77 (t, J = 5.7 Hz, 2 × NH, 2H), 4.32 (d, J = 5.7 Hz, NHCH2, 4H), 2.28 (s, 2 × CH3, 6H). 13C NMR (100 MHz, DMSO–d6) δ 155.0 (2 × CO), 150.1 (C4a, C10a), 142.0 (C3, C6), 137.0 (2 × C1’), 135.9 (2 × C4’), 135.0 (C9), 129.0 (C1, C8), 128.9 (2 × (C3’, C5’)), 127.2 (2 × (C2’, C6’)), 121.3 (C8a, C9a), 119.5 (C2, C7), 111.1 (C4, C5), 42.6 (2 × NHCH2), 20.7 (2 × CH3). HRMS (ESI): m/z calculated for C31H29N5O2 [M + H]+ 504.23940, found 504.24140.
N,N’-Acridine-3,6-diylbis(N’-[(1S)-1-phenylethyl]urea) (11h, 30 mg, 63.8%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the diethyleter-n-hexane mixture. 1H NMR (600 MHz, DMSO–d6) δ 8.85 (s, 2 × NH, 2H), 8.70 (s, H9, 1H), 8.10 (s, H4, H5, 2H), 7.92 (d, J = 9.1 Hz, H1, H8, 2H), 7.44 (d, J = 9.1 Hz, H2, H7, 2H), 7.38 (d, J = 7.3 Hz, 2 × (H2’, H6’), 4H), 7.36–7.32 (m, 2 × (H3’, H5’), 4H), 7.28–7.21 (m, 2 × H4’, 2H), 6.83 (d, J = 5.7 Hz, 2 × NH, 2H), 4.95–4.84 (m, 2 × NHCH, 2H), 1.43 (d, J = 7.0 Hz, 2 × CH3, 6H). 13C NMR (150 MHz, DMSO–d6) δ 154.2 (2 × CO), 150.0 (C4a, C10a), 145.0 (2 × C1’), 141.9 (C3, C6), 135.0 (C9), 129.1 (C1, C8), 128.4 (2 × (C3’, C5’)), 126.8 (2 × C4’), 125.9 (2 × (C2’, C6’)), 121.3 (C8a, C9a), 119.4 (C2, C7), 110.9 (C4, C5), 48.7 (2 × NHCH), 23.0 (2 × CH3). [α]D20 = +326.6 (c 0.06, DMSO). HRMS (ESI): m/z calculated for C31H29N5O2 [M + H]+ 504.23940, found 504.24143.
N,N’-Acridine-3,6-diylbis(N’-[(1R)-1-phenylethyl]urea) (11i, 35 mg, 74.5%). Yellow crystalline solid, mp > 200 °C with decomposition. Thiourea was crystalized from the diethyleter-n-hexane mixture. 1H NMR (400 MHz, DMSO–d6) δ 8.85 (s, 2 × NH, 2H), 8.70 (s, H9, 1H), 8.11 (s, H4, H5, 2H), 7.93 (d, J = 9.1 Hz, H1, H8, 2H), 7.44 (d, J = 9.1 Hz, H2, H7, 2H), 7.38 (d, J = 7.3 Hz, 2 × (H2’, H6’), 4H), 7.36–7.32 (m, 2 × (H3’, H5’), 4H), 7.28–7.21 (m, 2 × H4’, 2H), 6.83 (d, J = 5.7 Hz, 2 × NH, 2H), 4.95–4.84 (m, 2 × NHCH, 2H), 1.43 (d, J = 7.0 Hz, 2 × CH3, 6H). 13C NMR (100 MHz, DMSO–d6) δ 154.2 (2 × CO), 150.0 (C4a, C10a), 145.0 (2 × C1’), 141.9 (C3, C6), 135.0 (C9), 129.1 (C1, C8), 128.4 (2 × (C3’, C5’)), 126.8 (2 × C4’), 125.9 (2 × (C2’, C6’)), 121.3 (C8a, C9a), 119.4 (C2, C7), 110.9 (C4, C5), 48.7 (2 × NHCH), 23.0 (2 × CH3). [α]D20 = −326.6 (c 0.06, DMSO). HRMS (ESI): m/z calculated for C31H29N5O2 [M + H]+ 504.23940, found 504.24133.
N,N’-Acridine-3,6-diylbis(N’-hexylurea) (
11j, 35 mg, 31.2%). All physicochemical properties were in accordance with previously published data [
14].


 ,
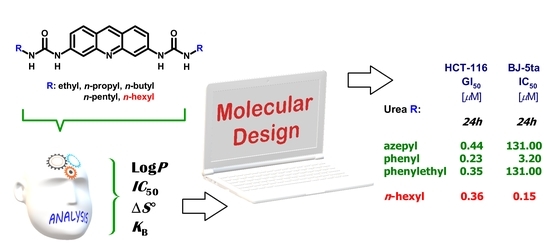
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}