
Endoplasmic Reticulum Stress Promotes iNOS/NO and Influences Inflammation in the Development of Doxorubicin-Induced Cardiomyopathy


Abstract
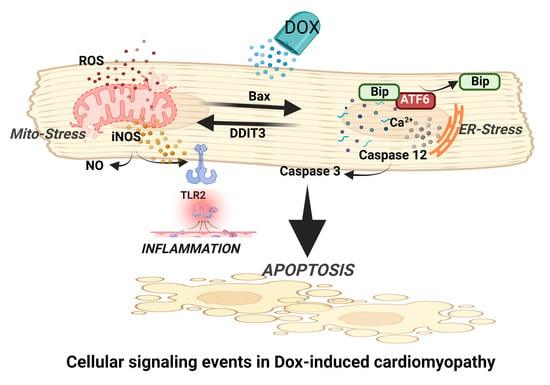
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Study Animals for In Vivo Dox Treatment
2.2. Cardiomyocyte Isolation and Treatments
2.3. NO Production Assay
2.4. Western Blot
2.5. Immunoprecipitation
2.6. Dual Immunofluorescence
2.6.1. Adherent Cells
2.6.2. Cardiac Tissue Sections
2.7. Statistical Analysis
3. Results
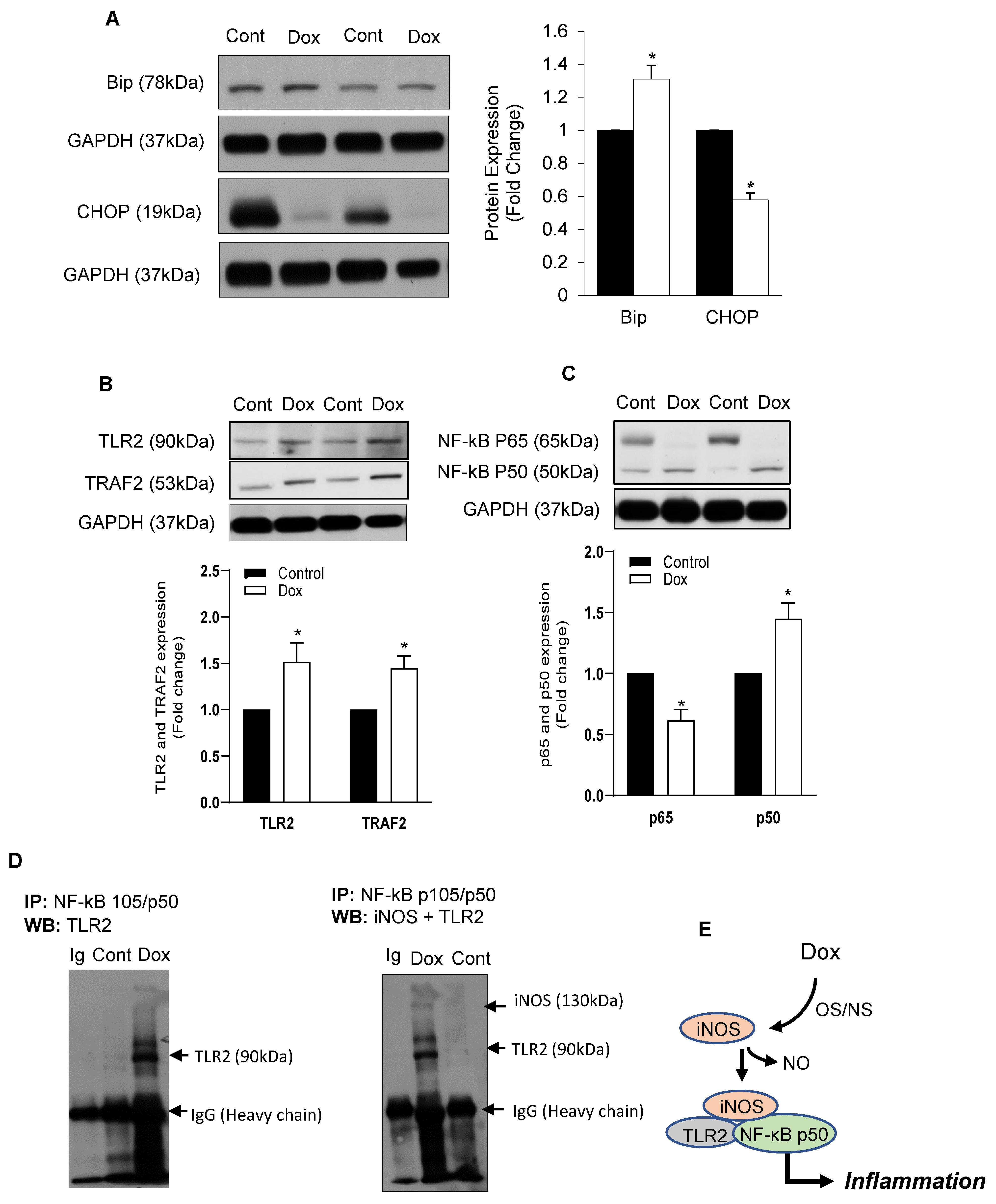
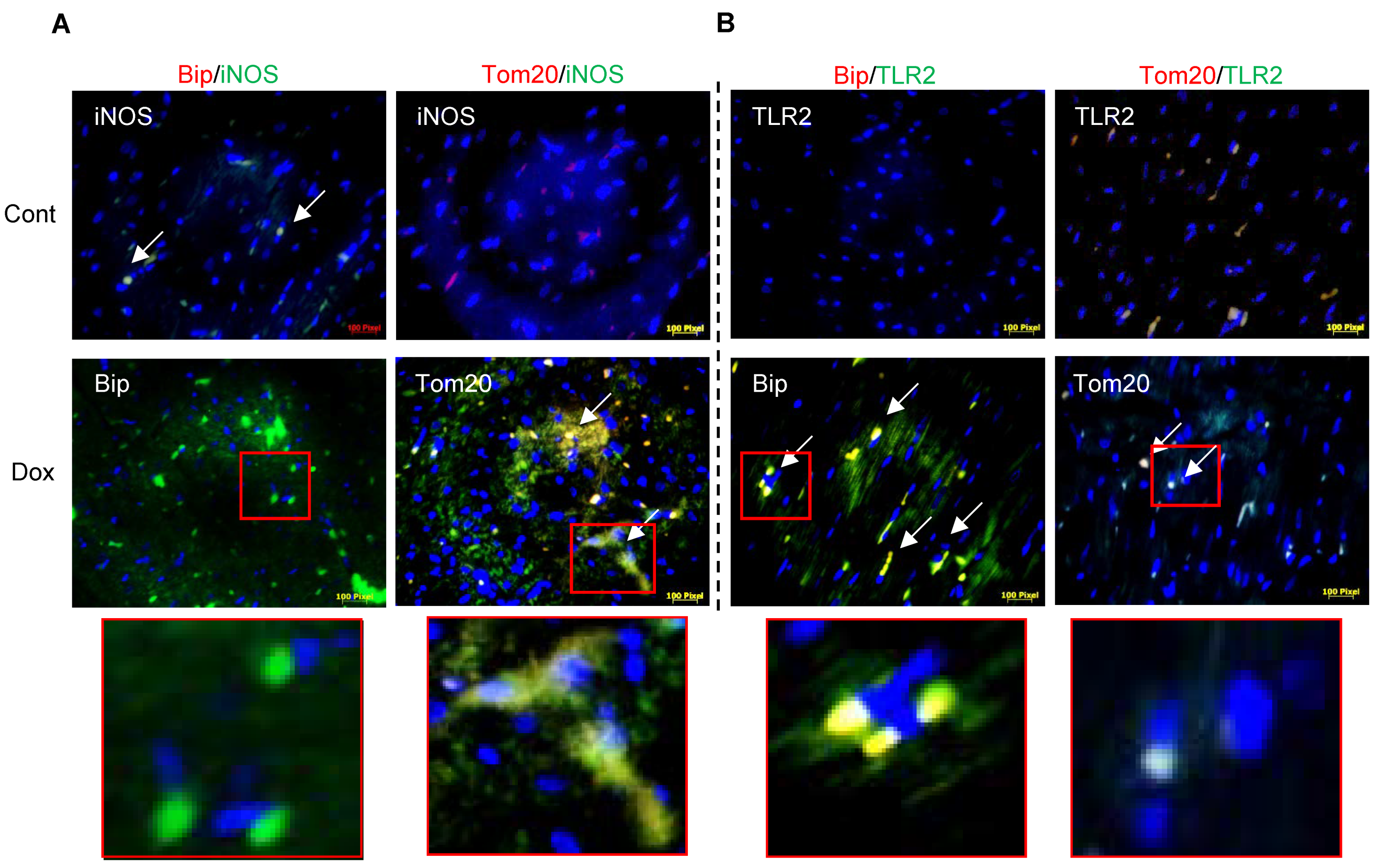
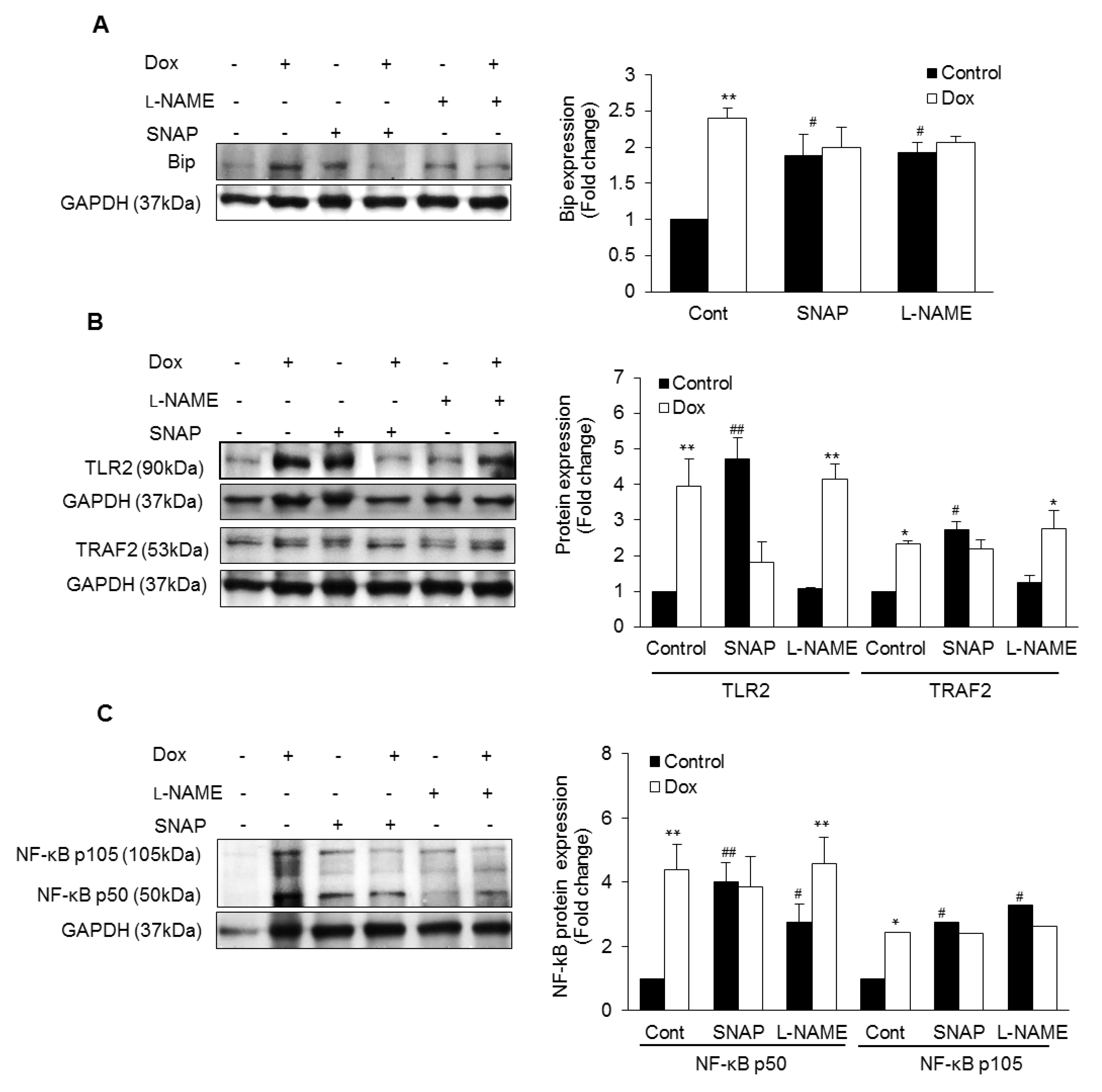
3.1. Dox-Induced ER Stress Mediates Inflammation and Mitochondrial iNOS
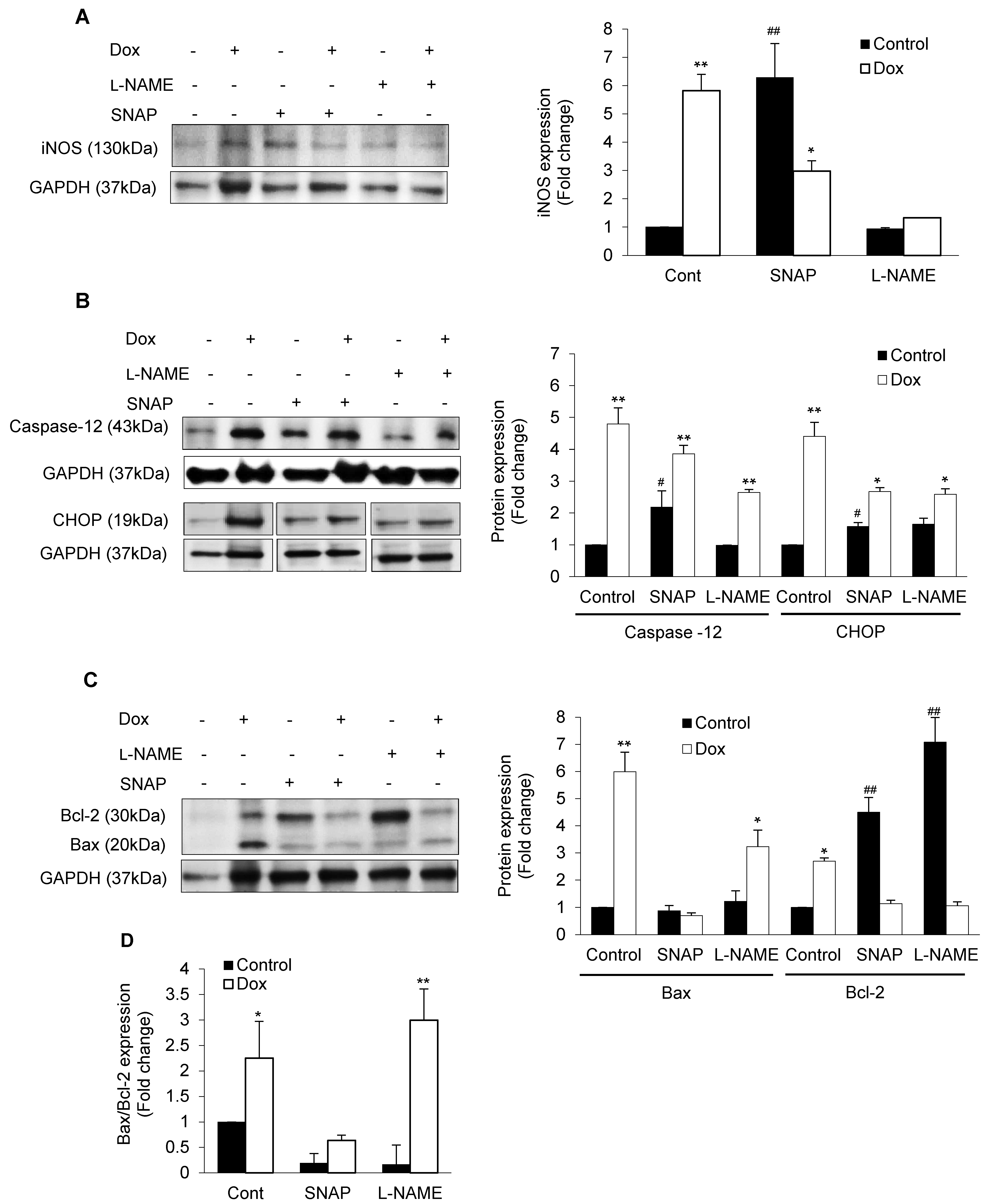
3.2. iNOS Synergistically Regulates Dox-ER Stress and Inflammatory Responses
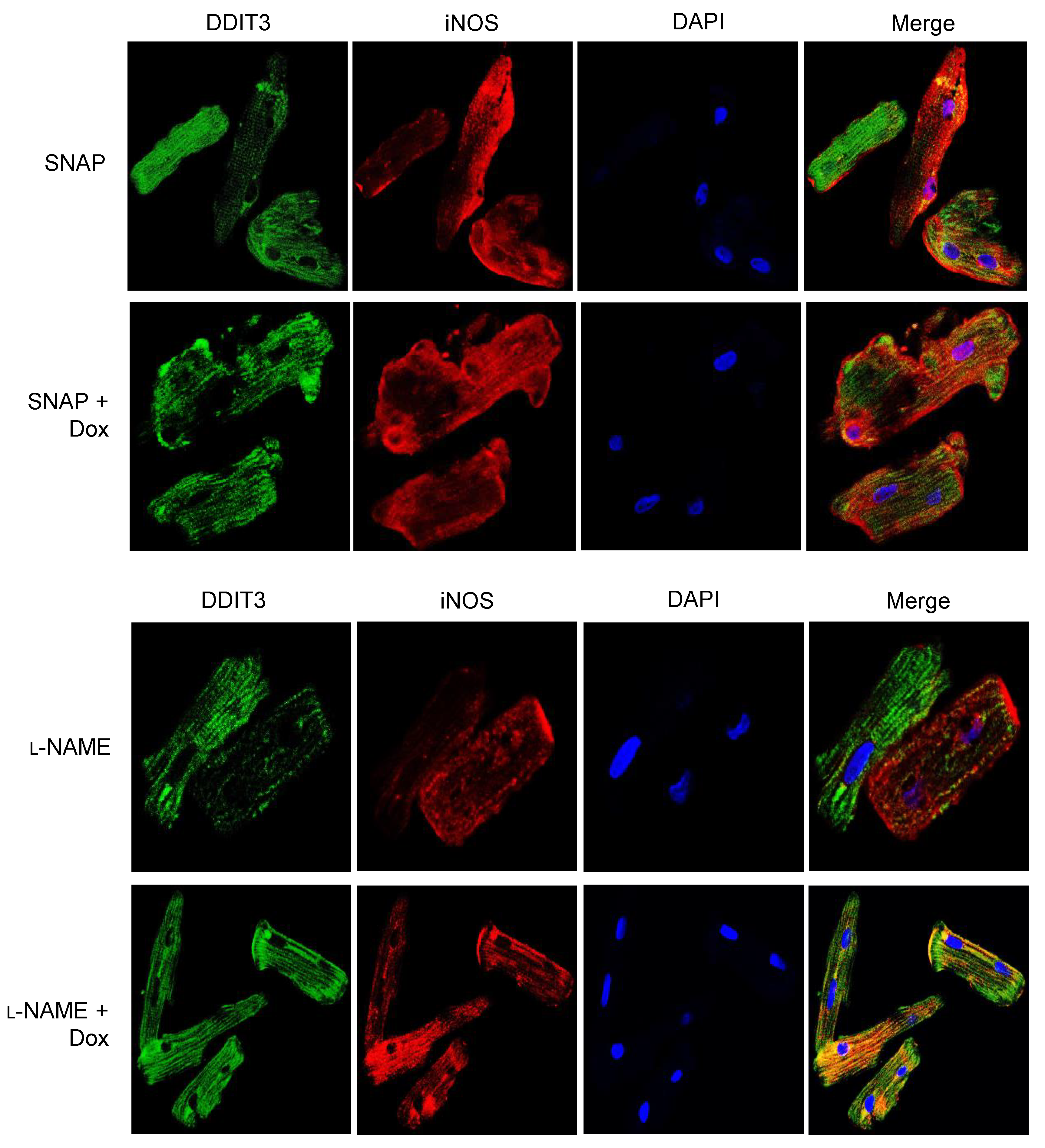
3.3. iNOS Regulates ER Stress-Induced Apoptosis
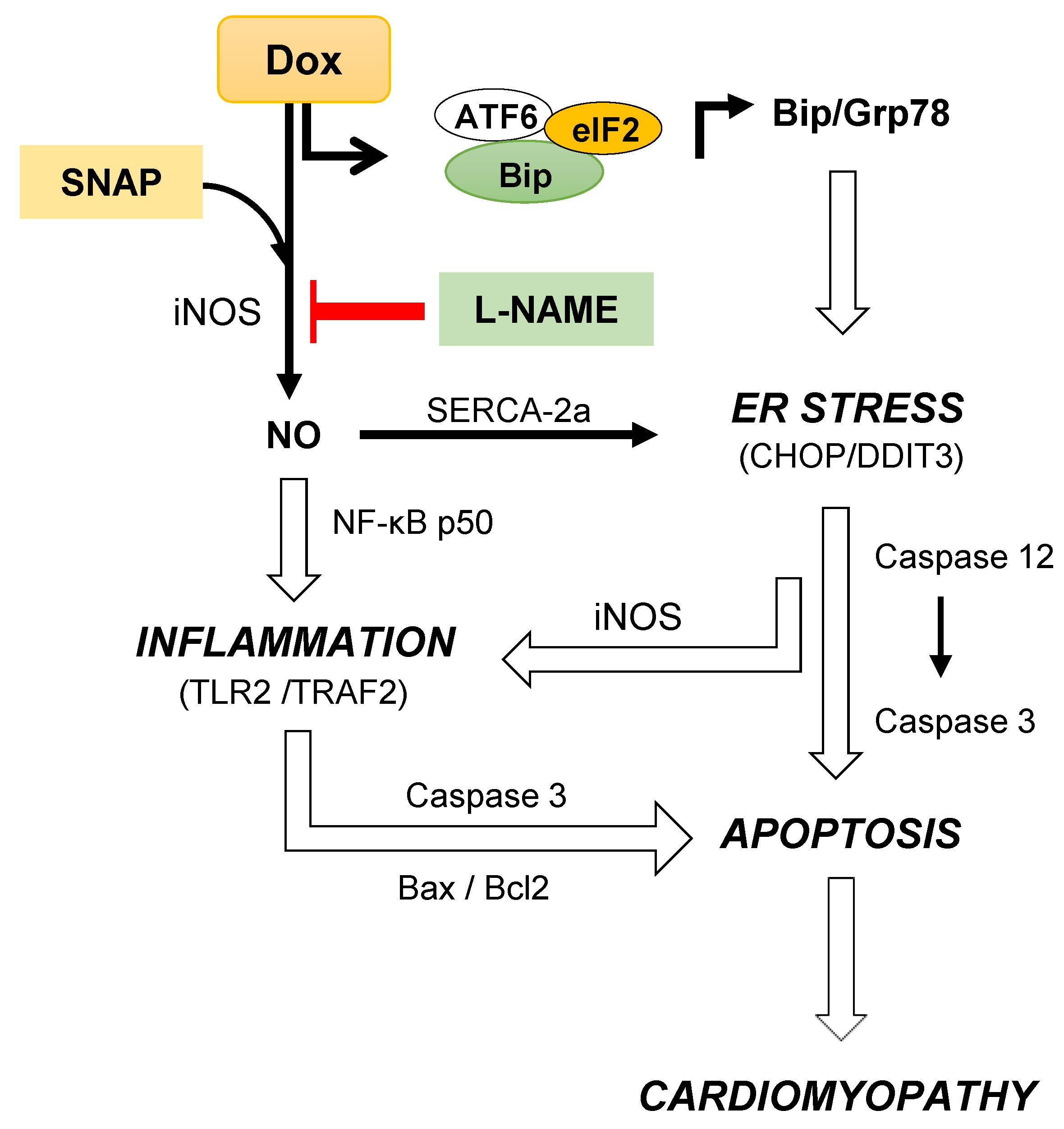
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lefrak, E.A.; Pitha, J.; Rosenheim, S.; Gottlieb, J.A. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer 1973, 32, 302–314. [Google Scholar] [CrossRef]
- Singal, P.K.; Iliskovic, N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 1998, 339, 900–905. [Google Scholar] [CrossRef]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid. Med. Cell Longev. 2017, 2017, 1521020. [Google Scholar] [CrossRef]
- Ludke, A.R.; Al-Shudiefat, A.A.; Dhingra, S.; Jassal, D.S.; Singal, P.K. A concise description of cardioprotective strategies in doxorubicin-induced cardiotoxicity. Can. J. Physiol. Pharm. 2009, 87, 756–763. [Google Scholar]
- Akolkar, G.; Bagchi, A.K.; Ayyappan, P.; Jassal, D.S.; Singal, P.K. Doxorubicin-induced nitrosative stress is mitigated by vitamin C via the modulation of nitric oxide synthases. Am. J. Physiol. Cell Physiol. 2017, 312, C418–C427. [Google Scholar] [CrossRef] [Green Version]
- Akolkar, G.; da Silva Dias, D.; Ayyappan, P.; Bagchi, A.K.; Jassal, D.S.; Salemi, V.M.C.; Irigoyen, M.C.; De Angelis, K.; Singal, P.K. Vitamin C mitigates oxidative/nitrosative stress and inflammation in doxorubicin-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H795–H809. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Kashiwaya, Y.; Haskó, G.; Liaudet, L.; Szabó, C.; Pacher, P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1466–H1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neilan, T.G.; Blake, S.L.; Ichinose, F.; Raher, M.J.; Buys, E.S.; Jassal, D.S.; Furutani, E.; Perez-Sanz, T.M.; Graveline, A.; Janssens, S.P.; et al. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation 2007, 116, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farías, J.G.; Molina, V.M.; Carrasco, R.A.; Zepeda, A.B.; Figueroa, E.; Letelier, P.; Castillo, R.L. Antioxidant Therapeutic Strategies for Cardiovascular Conditions Associated with Oxidative Stress. Nutrients 2017, 9, 966. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, L.T.; Erthal, F.; Corte, C.L.; Muller, L.G.; Piovezan, C.M.; Nogueira, C.W.; Rocha, J.B. Involvement of oxidative stress in the pre-malignant and malignant states of cervical cancer in women. Clin. Biochem. 2005, 11, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Pizarro, M.; Troncoso, R.; Martínez, G.J.; Chiong, M.; Castro, P.F.; Lavandero, S. Basal autophagy protects cardiomyocytes from doxorubicin-induced toxicity. Toxicology 2016, 370, 41–48. [Google Scholar] [CrossRef]
- Singal, K.P.; Li, T.; Kumar, D.; Danelisen, I.; Iliskovic, N. Adriamycin-induced heart failure: Mechanism and modulation. Mol. Cell. Biochem. 2000, 207, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, X.; Bao, H. Toll-like receptor (TLR) 2 and TLR4 differentially regulate doxorubicin induced cardiomyopathy in mice. PLoS ONE 2012, 7, e40763. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Hendershot, L.M. The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. 2004, 71, 289–297. [Google Scholar]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, A.K.; Malik, A.; Akolkar, G.; Zimmer, A.; Belló-Klein, A.; De Angelis, K.; Jassal, D.S.; Fini, M.A.; Stenmark, K.R.; Singal, P.K. Study of ER stress and apoptotic proteins in the heart and tumor exposed to doxorubicin. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119039. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Su, I.J.; Lei, H.Y.; Lai, M.D.; Chang, W.W.; Huang, W. Differential endoplasmic reticulum stress signaling pathways mediated by iNOS. Biochem. Biophys. Res. Commun. 2007, 359, 643–648. [Google Scholar] [CrossRef]
- Yarmohammadi, F.; Rezaee, R.; Haye, A.W.; Karimi, G. Endoplasmic reticulum stress in doxorubicin-induced cardiotoxicity may be therapeutically targeted by natural and chemical compounds: A review. Pharmacol. Res. 2021, 164, 105383. [Google Scholar] [CrossRef]
- Bagchi, A.K.; Akolkar, G.; Mandal, S.; Ayyappan, P.; Yang, X.; Singal, P.K. Toll-like receptor 2 dominance over Toll-like receptor 4 in stressful conditions for its detrimental role in the heart. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1238–H1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagchi, A.K.; Sharma, A.; Dhingra, S.; Lehenbauer Ludke, A.R.; Al-Shudiefat, A.A.; Singal, P.K. Interleukin-10 activates Toll-like receptor 4 and requires MyD88 for cardiomyocyte survival. Cytokine 2013, 61, 304–314. [Google Scholar] [CrossRef]
- Pravdic, D.; Vladic, N.; Cavar, I.; Bosnjak, Z.J. Effect of nitric oxide donors S-nitroso-N-acetyl-DL-penicillamine, spermine NONOate and propylamine propylamine NONOate on intracellular pH in cardiomyocytes. Clin. Exp. Pharm. Physiol. 2012, 39, 772–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strijdom, H.; Muller, C.; Lochner, A. Direct intracellular nitric oxide detection in isolated adult cardiomyocytes: Flow cytometric analysis using the fluorescent probe, diaminofluorescein. J. Mol. Cell Cardiol. 2004, 37, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Nitric oxide and mitochondria. Front. Biosci. 2007, 12, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, L.; Charles, I.G.; Moncada, S. Nitric oxide induces coupling of mitochondrial signalling with the endoplasmic reticulum stress response. Nat. Cell Biol. 2004, 6, 1129–1134. [Google Scholar] [CrossRef]
- Kawahara, K.; Oyadomari, S.; Gotoh, T.; Kohsaka, S.; Nakayama, H.; Mori, M. Induction of CHOP and apoptosis by nitric oxide in p53-deficient microglial cells. FEBS Lett. 2001, 506, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, T.; Oyadomari, S.; Mori, K.; Mori, M. Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J. Biol. Chem. 2002, 277, 12343–12350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, T.; Mori, M. Nitric oxide and endoplasmic reticulum stress. Arter. Thromb. Vasc. Biol. 2006, 26, 1439–1446. [Google Scholar] [CrossRef]
- Oyadomari, S.; Araki, E.; Mori, M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 2002, 7, 335–345. [Google Scholar] [CrossRef]
- Messmer, U.K.; Brüne, B. Nitric oxide-induced apoptosis: p53-dependent and p53-independent signalling pathways. Biochem. J. 1996, 319, 299–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Fujii, R.; Toyofuku, Y.; Saito, T.; Koseki, H.; Hsu, V.W.; Aoe, T. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 2001, 20, 3082–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- East, J.M. Sarco(endo)plasmic reticulum calcium pumps: Recent advances in our understanding of structure/function and biology (review). Mol. Membr. Biol 2000, 17, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, K.; Shichiri, M.; Marumo, F.; Hirata, Y. NO inhibits cytokine-induced iNOS expression and NF-kappaB activation by interfering with phosphorylation and degradation of IkappaB-alpha. Arter. Thromb. Vasc. Biol. 1998, 18, 1796–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, K.; Okazaki, T.; Sakon, S.; Kobarai, T.; Kurosawa, K.; Yamaoka, S.; Hashimoto, H.; Mak, T.W.; Yagita, H.; Okumura, K.; et al. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-kappa B activation and protection from cell death. J. Biol. Chem. 2001, 276, 36530–36534. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Mancilla, T.R.; Iskra, B.; Aune, G.J. Doxorubicin-Induced Cardiomyopathy in Children. Compr. Physiol. 2019, 9, 905–931. [Google Scholar] [CrossRef]
- Taylor, B.S.; Kim, Y.M.; Wang, Q.; Shapiro, R.A.; Billiar, T.R.; Geller, D.A. Nitric oxide down-regulates hepatocyte-inducible nitric oxide synthase gene expression. Arch. Surg. 1997, 132, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D. Cytokine-induced modulation of cardiac function. Circ. Res. 2004, 95, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.E.; Stamler, J.S. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry 2001, 40, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, A.K.; Surendran, A.; Malik, A.; Jassal, D.S.; Ravandi, A.; Singal, P.K. IL-10 attenuates OxPCs-mediated lipid metabolic responses in ischemia reperfusion injury. Sci. Rep. 2020, 10, 12120. [Google Scholar] [CrossRef]
- Cole, M.P.; Chaiswing, L.; Oberley, T.D.; Edelmann, S.E.; Piascik, M.T.; Lin, S.M.; Kiningham, K.K.; St Clair, D.K. The protective roles of nitric oxide and superoxide dismutase in adriamycin-induced cardiotoxicity. Cardiovasc. Res. 2006, 69, 186–197. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagchi, A.K.; Malik, A.; Akolkar, G.; Jassal, D.S.; Singal, P.K. Endoplasmic Reticulum Stress Promotes iNOS/NO and Influences Inflammation in the Development of Doxorubicin-Induced Cardiomyopathy. Antioxidants 2021, 10, 1897. https://doi.org/10.3390/antiox10121897
Bagchi AK, Malik A, Akolkar G, Jassal DS, Singal PK. Endoplasmic Reticulum Stress Promotes iNOS/NO and Influences Inflammation in the Development of Doxorubicin-Induced Cardiomyopathy. Antioxidants. 2021; 10(12):1897. https://doi.org/10.3390/antiox10121897
Chicago/Turabian StyleBagchi, Ashim K., Akshi Malik, Gauri Akolkar, Davinder S. Jassal, and Pawan K. Singal. 2021. "Endoplasmic Reticulum Stress Promotes iNOS/NO and Influences Inflammation in the Development of Doxorubicin-Induced Cardiomyopathy" Antioxidants 10, no. 12: 1897. https://doi.org/10.3390/antiox10121897