
Complement Components in the Diagnosis and Treatment after Kidney Transplantation—Is There a Missing Link?

 , ,
, ,  ,
,
Abstract
:1. Introduction
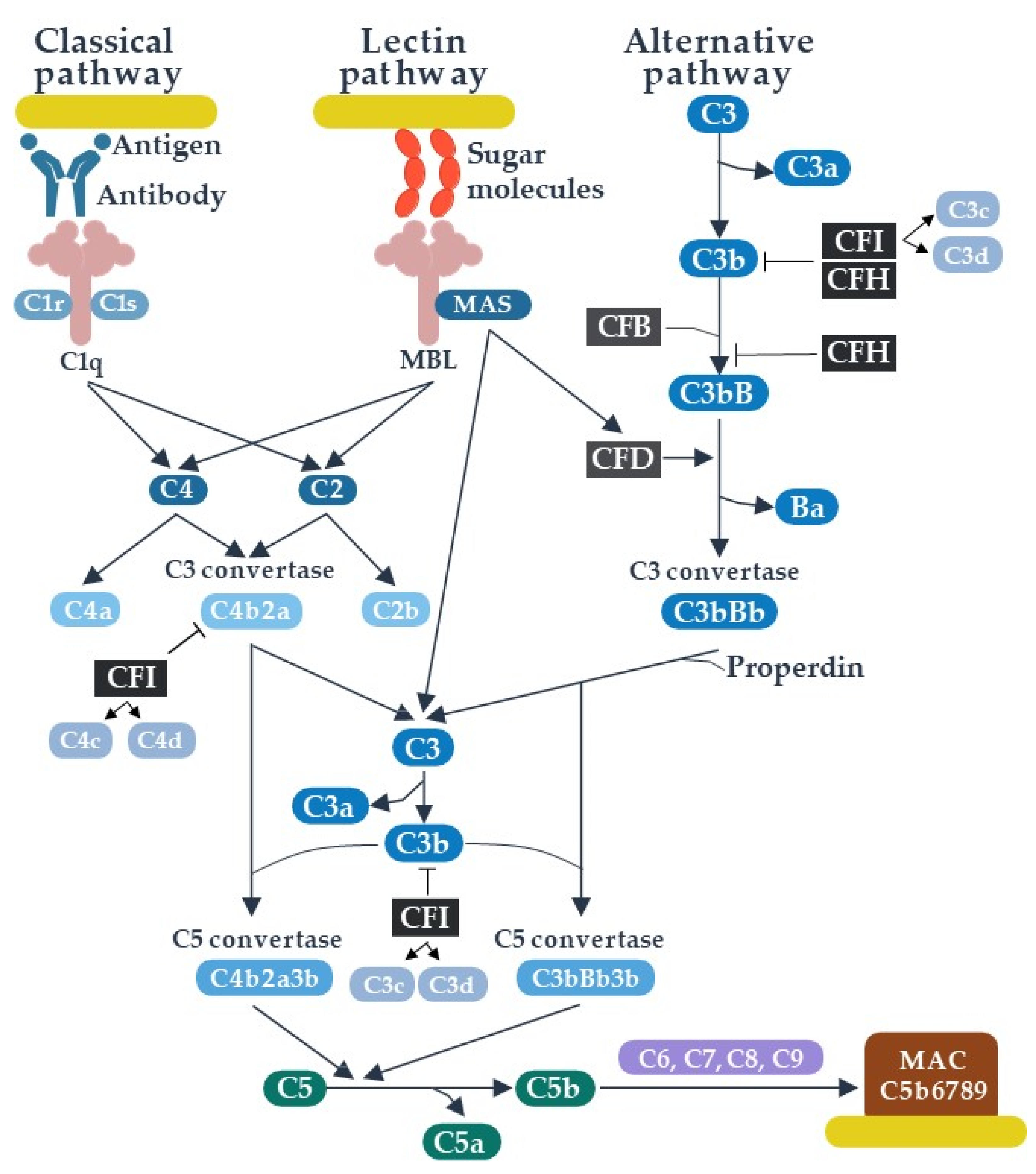
2. The Role of Complement in Human Immunity
2.1. Systemic Roles of Complement
2.2. Local and Intracellular Roles of Complement
3. The Role of Complement in Kidney Graft Injury
3.1. Ischemia-Reperfusion Injury of Kidney Graft
3.2. Humoral Rejection of Kidney Allograft
3.3. Cellular Rejection of Kidney Allograft
3.4. Post-Transplant Thrombotic Microangiopathy (TMA)
3.5. Recurrent Nephropathy in a Transplanted Kidney
3.6. Calcineurin Inhibitor-Induced Nephrotoxicity
4. Therapies Affecting Complement in Kidney Transplantation
5. The Role of Complement in Transplant Tolerance
6. The Relevance of Complement Gene Polymorphism for Kidney Transplant
7. Conclusions and Prospects for the Future
Author Contributions
Funding
Conflicts of Interest
References
- Mosquera Reboredo, J.M.; Vázquez Martul, E. Diagnostic criteria of antibody-mediated rejection in kidney transplants. Nefrologia 2011, 31, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Nankivell, B.J.; Alexander, S.I. Rejection of the Kidney Allograft. N. Engl. J. Med. 2010, 363, 1451–1462. [Google Scholar] [CrossRef] [Green Version]
- Kidney Disease: Improving Global Outcomes (KDIGO) Transplant Work Group. KDIGO Clinical Practice Guideline for the Care of Kidney Transplant Recipients. Am. J. Transplant. 2009, 9, S1–S155. [Google Scholar] [CrossRef]
- Heemann, U.; Abramowicz, D.; Spasovski, G.; Vanholder, R. Endorsement of the Kidney Disease Improving Global Outcomes (KDIGO) guidelines on kidney transplantation: A European Renal Best Practice (ERBP) position statement. Nephrol. Dial. Transplant. 2011, 26, 2099–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, M.; Loupy, A.; Lefaucheur, C.; Roufosse, C.; Glotz, D.; Seron, D.; Nankivell, B.J.; Halloran, P.F.; Colvin, R.B.; Akalin, E.; et al. The Banff 2017 Kidney Meeting Report: Revised diagnostic criteria for chronic active T cell–mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am. J. Transplant. 2018, 18, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef]
- Morris, A.B.; Ford, M.L. When rubber meets the road: How innate features of adaptive immune cells play critical roles in transplant alloimmunity. Curr. Opin. Organ Transplant. 2019, 24, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Luque, A.; Serrano, I.; Aran, J.M. Complement components as promoters of immunological tolerance in dendritic cells. Semin. Cell Dev. Biol. 2019, 85, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Cernoch, M.; Viklicky, O. Complement in kidney transplantation. Front. Med. 2017, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Damman, J.; Nijboer, W.N.; Schuurs, T.A.; Leuvenink, H.G.; Morariu, A.M.; Tullius, S.G.; Van Goor, H.; Ploeg, R.J.; Seelen, M.A. Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrol. Dial. Transplant. 2011, 26, 2345–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgari, E.; Zhou, W.; Sacks, S. Complement in organ transplantation. Curr. Opin. Organ Transplant. 2010, 15, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Nesargikar, P.; Spiller, B.; Chavez, R. The complement system: History, pathways, cascade and inhibitors. Eur. J. Microbiol. Immunol. 2012, 2, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ehrnthaller, C.; Ignatius, A.; Gebhard, F.; Huber-Lang, M. New insights of an old defense system: Structure, function, and clinical relevance of the complement system. Mol. Med. 2011, 17, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Grafals, M.; Thurman, J.M. The Role of Complement in Organ Transplantation. Front. Immunol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pratt, J.R.; Basheer, S.A.; Sacks, S.H. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat. Med. 2002, 8, 582–587. [Google Scholar] [CrossRef]
- Franzin, R.; Stasi, A.; Fiorentino, M.; Stallone, G.; Cantaluppi, V.; Gesualdo, L.; Castellano, G. Inflammaging and Complement System: A Link Between Acute Kidney Injury and Chronic Graft Damage. Front. Immunol. 2020, 11, 1–23. [Google Scholar] [CrossRef]
- West, E.E.; Kunz, N.; Kemper, C. Complement and human T cell metabolism: Location, location, location. Immunol. Rev. 2020, 295, 68–81. [Google Scholar] [CrossRef]
- Arbore, G.; Kemper, C.; Kolev, M. Intracellular complement—The complosome—In immune cell regulation. Mol. Immunol. 2017, 89, 2–9. [Google Scholar] [CrossRef]
- Kolev, M.; Kemper, C. Keeping it all going-complement meets metabolism. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Hilbrands, L.B. Latest developments in living kidney donation. Curr. Opin. Organ Transplant. 2020, 25, 74–79. [Google Scholar] [CrossRef]
- Damman, J.; Bloks, V.W.; Daha, M.R.J.; van der Most, P.; Sanjabi, B.; van der Vlies, P.; Snieder, H.; Ploeg, R.J.; Krikke, C.; Leuvenink, H.G.D.; et al. Hypoxia and complement-and-coagulation pathways in the deceased organ donor as the major target for intervention to improve renal allograft outcome. Transplantation 2015, 99, 1293–1300. [Google Scholar] [CrossRef]
- Danobeitia, J.S.; Djamali, A.; Fernandez, L.A. The role of complement in the pathogenesis of renal ischemia-reperfusion injury and fibrosis. Fibrogenesis Tissue Repair 2014, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Zhou, W.; Farrar, C.A.; Abe, K.; Pratt, J.R.; Marsh, J.E.; Wang, Y.; Stahl, G.L.; Sacks, S.H. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J. Clin. Investig. 2000, 105, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Medof, M.; Heeger, P.; Sacks, S. Graft-derived complement as a mediator of transplant injury. Curr. Opin. Immunol. 2007, 19, 569–576. [Google Scholar] [CrossRef]
- Sacks, S.H.; Chowdhury, P.; Zhou, W. Role of the complement system in rejection. Curr. Opin. Immunol. 2003, 15, 487–492. [Google Scholar] [CrossRef]
- Vandendriessche, S.; Cambier, S.; Proost, P.; Marques, P.E. Complement Receptors and Their Role in Leukocyte Recruitment and Phagocytosis. Front. Cell Dev. Biol. 2021, 9, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.A. Functions of C5a receptors. J. Mol. Med. 2009, 87, 375–378. [Google Scholar] [CrossRef]
- Martin, I.V.; Bohner, A.; Boor, P.; Shagdarsuren, E.; Raffetseder, U.; Lammert, F.; Floege, J.; Ostendorf, T.; Weber, S.N. Complement C5a receptors C5L2 and C5aR in renal fibrosis. Am. J. Physiol. Ren. Physiol. 2018, 314, F35–F46. [Google Scholar] [CrossRef]
- Nauser, C.L.; Farrar, C.A.; Sacks, S.H. Complement recognition pathways in renal transplantation. J. Am. Soc. Nephrol. 2017, 28, 2571–2578. [Google Scholar] [CrossRef]
- Bobka, S.; Ebert, N.; Koertvely, E.; Jacobi, J.; Wiesener, M.; Büttner-Herold, M.; Amann, K.; Daniel, C. Is early complement activation in renal transplantation associated with later graft outcome? Kidney Blood Press. Res. 2018, 43, 1488–1504. [Google Scholar] [CrossRef]
- Einecke, G.; Sis, B.; Reeve, J.; Mengel, M.; Campbell, P.M.; Hidalgo, L.G.; Kaplan, B.; Halloran, P.F. Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am. J. Transplant. 2009, 9, 2520–2531. [Google Scholar] [CrossRef]
- Stites, E.; Le Quintrec, M.; Thurman, J.M. The Complement System and Antibody-Mediated Transplant Rejection. J. Immunol. 2015, 195, 5525–5531. [Google Scholar] [CrossRef] [Green Version]
- Sellarés, J.; De Freitas, D.G.; Mengel, M.; Sis, B.; Hidalgo, L.G.; Matas, A.J.; Kaplan, B.; Halloran, P.F. Inflammation lesions in kidney transplant biopsies: Association with survival is due to the underlying diseases. Am. J. Transplant. 2011, 11, 489–499. [Google Scholar] [CrossRef]
- Sellarés, J.; De Freitas, D.G.; Mengel, M.; Reeve, J.; Einecke, G.; Sis, B.; Hidalgo, L.G.; Famulski, K.; Matas, A.; Halloran, P.F. Understanding the causes of kidney transplant failure: The dominant role of antibody-mediated rejection and nonadherence. Am. J. Transplant. 2012, 12, 388–399. [Google Scholar] [CrossRef]
- Loupy, A.; Lefaucheur, C.; Vernerey, D.; Prugger, C.; Van Huyen, J.P.D.; Mooney, N.; Suberbielle, C.; Frémeaux-Bacchi, V.; Méjean, A.; Desgrandchamps, F.; et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N. Engl. J. Med. 2013, 369, 1215–1226. [Google Scholar] [CrossRef] [Green Version]
- Loupy, A.; Haas, M.; Roufosse, C.; Naesens, M.; Adam, B.; Afrouzian, M.; Akalin, E.; Alachkar, N.; Bagnasco, S.; Becker, J.U.; et al. The Banff 2019 Kidney Meeting Report (I): Updates on and clarification of criteria for T cell– and antibody-mediated rejection. Am. J. Transplant. 2020, 1–14. [Google Scholar] [CrossRef]
- Cohen, D.; Colvin, R.B.; Daha, M.R.; Drachenberg, C.B.; Haas, M.; Nickeleit, V.; Salmon, J.E.; Sis, B.; Zhao, M.H.; Bruijn, J.A.; et al. Pros and cons for C4d as a biomarker. Kidney Int. 2012, 81, 628–639. [Google Scholar] [CrossRef] [Green Version]
- Sis, B.; Halloran, P.F. Endothelial transcripts uncover a previously unknown phenotype: C4d-negative antibody-mediated rejection. Curr. Opin. Organ Transplant. 2010, 15, 42–48. [Google Scholar] [CrossRef]
- Zhang, X.; Reed, E.F. Effect of antibodies on endothelium: Minireview. Am. J. Transplant. 2009, 9, 2459–2465. [Google Scholar] [CrossRef]
- Sutherland, S.M.; Chen, G.; Sequeira, F.A.; Lou, C.D.; Alexander, S.R.; Tyan, D.B. Complement-fixing donor-specific antibodies identified by a novel C1q assay are associated with allograft loss. Pediatr. Transplant. 2012, 16, 12–17. [Google Scholar] [CrossRef]
- Yell, M.; Muth, B.L.; Kaufman, D.B.; Djamali, A.; Ellis, T.M. C1q Binding Activity of De Novo Donor-specific HLA Antibodies in Renal Transplant Recipients With and Without Antibody-mediated Rejection. Transplantation 2015, 99, 1151–1155. [Google Scholar] [CrossRef]
- Viglietti, D.; Bouatou, Y.; Kheav, V.D.; Aubert, O.; Suberbielle-Boissel, C.; Glotz, D.; Legendre, C.; Taupin, J.L.; Zeevi, A.; Loupy, A.; et al. Complement-binding anti-HLA antibodies are independent predictors of response to treatment in kidney recipients with antibody-mediated rejection. Kidney Int. 2018, 94, 773–787. [Google Scholar] [CrossRef]
- Sicard, A.; Ducreux, S.; Rabeyrin, M.; Couzi, L.; McGregor, B.; Badet, L.; Scoazec, J.Y.; Bachelet, T.; Lepreux, S.; Visentin, J.; et al. Detection of C3d-binding donor-specific anti-HLA antibodies at diagnosis of humoral rejection predicts renal graft loss. J. Am. Soc. Nephrol. 2015, 26, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Von Moos, S.; Schalk, G.; Mueller, T.F.; Laube, G. Age-associated decrease in de novo donor-specific antibodies in renal transplant recipients reflects changing humoral immunity. Immun. Ageing 2019, 16, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Terasaki, P.I. Deduction of the fraction of immunologic and nonimmunologic failure in cadaver donor transplants. Clin. Transpl. 2003, 1, 449–452. [Google Scholar]
- Delville, M.; Charreau, B.; Rabant, M.; Legendre, C.; Anglicheau, D. Pathogenesis of non-HLA antibodies in solid organ transplantation: Where do we stand? Hum. Immunol. 2016, 77, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Siu, J.H.Y.; Surendrakumar, V.; Richards, J.A.; Pettigrew, G.J. T cell allorecognition pathways in solid organ transplantation. Front. Immunol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kusztal, M.; Jezior, D.; Weyde, W.; Krajewska, M.; Klinger, M. The immune response to kidney allograft. Part I: The role of MHC antigens, antigen-presenting cells, and lymphocytes in alloantigen recognition; two-signal activation of T cells. Postepy Hig. Med. Dosw. 2007, 61, 13–20. [Google Scholar]
- Kerstan, A.; Hünig, T. Cutting Edge: Distinct TCR- and CD28-Derived Signals Regulate CD95L, Bcl-x L, and the Survival of Primary T Cells. J. Immunol. 2004, 172, 1341–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusztal, M.; Jezior, D.; Weyde, W.; Krajewska, M.; Klinger, M. The immune response to kidney allograft. Part II: The role of costimulatory and accessory molecules in T-cell activation; The effector phase of response. Postepy Hig. Med. Dosw. 2007, 61, 21–27. [Google Scholar]
- Strainic, M.G.; Liu, J.; Huang, D.; An, F.; Lalli, P.N.; Muqim, N.; Shapiro, V.S.; Dubyak, G.R.R.; Heeger, P.S.; Medof, M.E. Locally Produced Complement Fragments C5a and C3a Provide Both Costimulatory and Survival Signals to Naive CD4+ T Cells. Immunity 2008, 28, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Li, K.; Anderson, K.; Farrar, C.A.; Lu, B.; Smith, R.A.G.; Sacks, S.H.; Zhou, W. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood 2008, 111, 2452–2461. [Google Scholar] [CrossRef]
- Dixon, K.O.; O’Flynn, J.; Klar-Mohamad, N.; Daha, M.R.; van Kooten, C. Properdin and factor H production by human dendritic cells modulates their T-cell stimulatory capacity and is regulated by IFN-γ. Eur. J. Immunol. 2017, 47, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Ponticelli, C. De novo thrombotic microangiopathy. An underrated complication of renal transplantation. Clin. Nephrol. 2007, 67, 335–340. [Google Scholar] [CrossRef]
- Garg, N.; Rennke, H.G.; Pavlakis, M.; Zandi-Nejad, K. De novo thrombotic microangiopathy after kidney transplantation. Transplant. Rev. 2018, 32, 58–68. [Google Scholar] [CrossRef]
- Benz, K.; Amann, K. Thrombotic microangiopathy: New insights. Curr. Opin. Nephrol. Hypertens. 2010, 19, 242–247. [Google Scholar] [CrossRef]
- Fortin, M.C.; Raymond, M.A.; Madore, F.; Fugère, J.A.; Pâquet, M.; St-Louis, G.; Hébert, M.J. Increased risk of thrombotic microangiopathy in patients receiving a cyclosporin-sirolimus combination. Am. J. Transplant. 2004, 4, 946–952. [Google Scholar] [CrossRef]
- Nava, F.; Cappelli, G.; Mori, G.; Granito, M.; Magnoni, G.; Botta, C.; Solazzo, A.; Fontana, F.; Baisi, A.; Bonucchi, D. Everolimus, cyclosporine, and thrombotic microangiopathy: Clinical role and preventive tools in renal transplantation. Transplant. Proc. 2014, 46, 2263–2268. [Google Scholar] [CrossRef]
- Le Quintrec, M.; Lionet, A.; Kamar, N.; Karras, A.; Barbier, S.; Buchler, M.; Fakhouri, F.; Provost, F.; Fridman, W.H.; Thervet, E.; et al. Complement mutation-associated de novo thrombotic microangiopathy following kidney transplantation. Am. J. Transplant. 2008, 8, 1694–1701. [Google Scholar] [CrossRef]
- Broecker, V.; Bardsley, V.; Torpey, N.; Perera, R.; Montero, R.; Dorling, A.; Bentall, A.; Neil, D.; Willicombe, M.; Berry, M.; et al. Clinical–pathological correlations in post-transplant thrombotic microangiopathy. Histopathology 2019, 75, 88–103. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Glomerular Diseases Dependent on Complement Activation, Including Atypical Hemolytic Uremic Syndrome, Membranoproliferative Glomerulonephritis, and C3 Glomerulopathy: Core Curriculum 2015. Am. J. Kidney Dis. 2015, 66, 359–375. [Google Scholar] [CrossRef] [Green Version]
- Pickering, M.C.; D’agati, V.D.; Nester, C.M.; Smith, R.J.; Haas, M.; Appel, G.B.; Alpers, C.E.; Bajema, I.M.; Bedrosian, C.; Braun, M.; et al. C3 glomerulopathy: Consensus report. Kidney Int. 2013, 84, 1079–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhouri, F.; Le Quintrec, M.; Frémeaux-Bacchi, V. Practical management of C3 glomerulopathy and Ig-mediated MPGN: Facts and uncertainties. Kidney Int. 2020, 98, 1135–1148. [Google Scholar] [CrossRef]
- Cosio, F.G.; Cattran, D.C. Recent advances in our understanding of recurrent primary glomerulonephritis after kidney transplantation. Kidney Int. 2017, 91, 304–314. [Google Scholar] [CrossRef]
- Sethi, S.; Fervenza, F.C. Membranoproliferative Glomerulonephritis—A New Look at an Old Entity. N. Engl. J. Med. 2012, 366, 1119–1131. [Google Scholar] [CrossRef] [Green Version]
- Salvadori, M.; Bertoni, E. Complement related kidney diseases: Recurrence after transplantation. World J. Transplant. 2016, 6, 632. [Google Scholar] [CrossRef]
- Bentata, Y. Tacrolimus: 20 years of use in adult kidney transplantation. What we should know about its nephrotoxicity. Artif. Organs 2020, 44, 140–152. [Google Scholar] [CrossRef]
- Kim, Y.O.; Lim, S.W.; Li, C.; Kang, H.J.; Ahn, K.O.; Yang, H.J.; Ghee, J.Y.; Kim, S.H.; Kim, J.Y.; Choi, B.S.; et al. Activation of intrarenal complement system in mouse model for chronic cyclosporine nephrotoxicity. Yonsei Med. J. 2007, 48, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Renner, B.; Klawitter, J.; Goldberg, R.; McCullough, J.W.; Ferreira, V.P.; Cooper, J.E.; Christians, U.; Thurman, J.M. Cyclosporine induces endothelial cell release of complement-activating microparticles. J. Am. Soc. Nephrol. 2013, 24, 1849–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuwirt, H.; Loeschenberger, B.; Eder, I.; Puhr, M.; Rudnicki, M. Complement System and MAPK Signaling in Calcineurin-Inhibitor Induced Nephrotoxicity. Transplantation 2014, 98, 661. [Google Scholar] [CrossRef]
- Loeschenberger, B.; Niess, L.; Würzner, R.; Schwelberger, H.; Eder, I.E.; Puhr, M.; Guenther, J.; Troppmair, J.; Rudnicki, M.; Neuwirt, H. Calcineurin inhibitor-induced complement system activation via ERK1/2 signalling is inhibited by SOCS-3 in human renal tubule cells. Eur. J. Immunol. 2018, 48, 330–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinstock, C.A.; Mannon, R.B.; Budde, K.; Chong, A.S.; Haas, M.; Knechtle, S.; Lefaucheur, C.; Montgomery, R.A.; Nickerson, P.; Tullius, S.G.; et al. Recommended Treatment for Antibody-mediated Rejection After Kidney Transplantation. Transplantation 2020, 104, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Viglietti, D.; Gosset, C.; Loupy, A.; Deville, L.; Verine, J.; Zeevi, A.; Glotz, D.; Lefaucheur, C. C1 Inhibitor in Acute Antibody-Mediated Rejection Nonresponsive to Conventional Therapy in Kidney Transplant Recipients: A Pilot Study. Am. J. Transplant. 2016, 16, 1596–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, A.A.; Zeevi, A.; Choi, J.; Cisneros, K.; Toyoda, M.; Kahwaji, J.; Peng, A.; Villicana, R.; Puliyanda, D.; Reinsmoen, N.; et al. A Phase I/II Placebo-Controlled Trial of C1-Inhibitor for Prevention of Antibody-Mediated Rejection in HLA Sensitized Patients. Transplantation 2015, 99, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.S.; Ying, T.D.; Wyburn, K.; Roberts, D.M.; Wyld, M.; Chadban, S.J. The treatment of antibody-mediated rejection in kidney transplantation: An updated systematic review and meta-analysis. Transplantation 2018, 102, 557–568. [Google Scholar] [CrossRef]
- Montgomery, R.A.; Orandi, B.J.; Racusen, L.; Jackson, A.M.; Garonzik-Wang, J.M.; Shah, T.; Woodle, E.S.; Sommerer, C.; Fitts, D.; Rockich, K.; et al. Plasma-Derived C1 Esterase Inhibitor for Acute Antibody-Mediated Rejection Following Kidney Transplantation: Results of a Randomized Double-Blind Placebo-Controlled Pilot Study. Am. J. Transplant. 2016, 16, 3468–3478. [Google Scholar] [CrossRef] [Green Version]
- Okumi, M.; Kakuta, Y.; Unagami, K.; Takagi, T.; Iizuka, J.; Inui, M.; Ishida, H.; Tanabe, K. Current protocols and outcomes of ABO-incompatible kidney transplantation based on a single-center experience. Transl. Androl. Urol. 2019, 8, 126–133. [Google Scholar] [CrossRef]
- Baek, C.H.; Kim, H.; Yu, H.; Shin, E.; Cho, H.; Yang, W.S.; Han, D.J.; Park, S.K. Low dose of mycophenolate mofetil is enough in desensitized kidney transplantation using rituximab. BMC Nephrol. 2015, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, K.; Mehta, A.A. Rituximab in kidney disease and transplant. Anim. Model. Exp. Med. 2019, 76–82. [Google Scholar] [CrossRef]
- Eskandary, F.; Regele, H.; Baumann, L.; Bond, G.; Kozakowski, N.; Wahrmann, M.; Hidalgo, L.G.; Haslacher, H.; Kaltenecker, C.C.; Aretin, M.B.; et al. A Randomized Trial of Bortezomib in Late Antibody-Mediated Kidney Transplant Rejection. J. Am. Soc. Nephrol. 2018, 29, 591–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parajuli, S.; Mandelbrot, D.A.; Muth, B.; Mohamed, M.; Garg, N.; Aziz, F.; Redfield, R.R.; Zhong, W.; Astor, B.C.; Djamali, A. Rituximab and Monitoring Strategies for Late Antibody-Mediated Rejection After Kidney Transplantation. Transplant. Direct 2017, 3, e227. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Iwadoh, K.; Ogawa, Y.; Miki, K.; Kato, Y.; Kai, K.; Sannomiya, A.; Koyama, I.; Kitajima, K.; Nakajima, I.; et al. Single fixed low-dose rituximab as induction therapy suppresses de novo donor-specific anti-HLA antibody production in ABO compatible living kidney transplant recipients. PLoS ONE 2019, 14, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, V.; Madhavan, D.; Narayanasamy, K.; Kumar, S.; Ramalingam, V.; Sengodagounder, B.; Bodonyi-Kovacs, G. Low-dose Rituximab and Thymoglobulin Induction with Steroid-free Maintenance Immunosuppression and Protocol Biopsies Improves Long-term Patient and Graft Survival after Kidney Transplantation: Survival and Safety Outcomes in More Than 1100 Patients from a. Transplant. Direct 2019, 5, 1–9. [Google Scholar] [CrossRef]
- Stegall, M.D.; Diwan, T.; Raghavaiah, S.; Cornell, L.D.; Burns, J.; Dean, P.G.; Cosio, F.G.; Gandhi, M.J.; Kremers, W.; Gloor, J.M. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am. J. Transplant. 2011, 11, 2405–2413. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Suarez, M.L.; Thongprayoon, C.; Mao, M.A.; Leeaphorn, N.; Bathini, T.; Cheungpasitporn, W. Outcomes of Kidney Transplant Patients with Atypical Hemolytic Uremic Syndrome Treated with Eculizumab: A Systematic Review and Meta-Analysis. J. Clin. Med. 2019, 8, 919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, W.H.; Mamode, N.; Montgomery, R.A.; Stegall, M.D.; Ratner, L.E.; Cornell, L.D.; Rowshani, A.T.; Colvin, R.B.; Dain, B.; Boice, J.A.; et al. Safety and efficacy of eculizumab in the prevention of antibody-mediated rejection in living-donor kidney transplant recipients requiring desensitization therapy: A randomized trial. Am. J. Transplant. 2019, 19, 2876–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licht, C.; Greenbaum, L.A.; Muus, P.; Babu, S.; Bedrosian, C.L.; Cohen, D.J.; Delmas, Y.; Douglas, K.; Furman, R.R.; Gaber, O.A.; et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015, 87, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian C Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; Eitner, F.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef] [Green Version]
- Legendre, C.M.; Campistol, J.M.; Feldkamp, T.; Remuzzi, G.; Kincaid, J.F.; Lommelé, Å.; Wang, J.; Weekers, L.E.; Sheerin, N.S. Outcomes of patients with atypical haemolytic uraemic syndrome with native and transplanted kidneys treated with eculizumab: A pooled post hoc analysis. Transpl. Int. 2017, 30, 1275–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, S.; Kirkiles-Smith, N.C.; Deng, Y.H.; Formica, R.N.; Moeckel, G.; Broecker, V.; Bow, L.; Tomlin, R.; Pober, J.S. Eculizumab Therapy for Chronic Antibody-Mediated Injury in Kidney Transplant Recipients: A Pilot Randomized Controlled Trial. Am. J. Transplant. 2017, 17, 682–691. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.E.; Mejia, P.; Lu, F. Biological activities of C1 inhibitor. Mol. Immunol. 2008, 45, 4057–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paréj, K.; Dobó, J.; Závodszky, P.; Gál, P. The control of the complement lectin pathway activation revisited: Both C1-inhibitor and antithrombin are likely physiological inhibitors, While α2-macroglobulin is not. Mol. Immunol. 2013, 54, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Baldwin, W.M.; Jordan, S.C. Potential roles for C1 inhibitor in transplantation. Transplantation 2016, 100, 1415–1424. [Google Scholar] [CrossRef]
- Eskandary, F.; Jilma, B.; Mühlbacher, J.; Wahrmann, M.; Regele, H.; Kozakowski, N.; Firbas, C.; Panicker, S.; Parry, G.C.; Gilbert, J.C.; et al. Anti-C1s monoclonal antibody BIVV009 in late antibody-mediated kidney allograft rejection—Results from a first-in-patient phase 1 trial. Am. J. Transplant. 2018, 18, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.C.; Lorant, T.; Choi, J.; Kjellman, C.; Winstedt, L.; Bengtsson, M.; Zhang, X.; Eich, T.; Toyoda, M.; Eriksson, B.M.; et al. IgG endopeptidase in highly sensitized patients undergoing transplantation. N. Engl. J. Med. 2017, 377, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Kassimatis, T.; Qasem, A.; Douiri, A.; Ryan, E.G.; Rebollo-Mesa, I.; Nichols, L.L.; Greenlaw, R.; Olsburgh, J.; Smith, R.A.; Sacks, S.H.; et al. A double-blind randomised controlled investigation into the efficacy of Mirococept (APT070) for preventing ischaemia reperfusion injury in the kidney allograft (EMPIRIKAL): Study protocol for a randomised controlled trial. Trials 2017, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Ma, L.; Zhao, M.; Smith, R.A.; Huang, G.; Jones, P.M.; Persaud, S.; Pingitore, A.; Dorling, A.; Lechler, R.; et al. APT070 (mirococept), a membrane-localizing C3 convertase inhibitor, attenuates early human islet allograft damage in vitro and in vivo in a humanized mouse model. Br. J. Pharmacol. 2016, 173, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.L.; Heurich, M.; de Cordoba, S.R.; Morgan, B.P. The complotype: Dictating risk for inflammation and infection. Trends Immunol. 2012, 33, 513–521. [Google Scholar] [CrossRef]
- Montero, R.M.; Sacks, S.H.; Smith, R.A. Complement—Here, there and everywhere, but what about the transplanted organ? Semin. Immunol. 2016, 28, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Jordan, S.C.; Choi, J.; Aubert, O.; Haas, M.; Loupy, A.; Huang, E.; Peng, A.; Kim, I.; Louie, S.; Ammerman, N.; et al. A phase I/II, double-blind, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Am. J. Transplant. 2018, 18, 2955–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, E.; Vo, A.; Choi, J.; Ammerman, N.; Lim, K.; Sethi, S.; Kim, I.; Kumar, S.; Najjar, R.; Peng, A.; et al. Three-Year Outcomes of a Randomized, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Clin. J. Am. Soc. Nephrol. 2020, 15, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.M.; Kondeatis, E.; Vaughan, R.W.; Kon, S.P.; Farmer, C.K.T.; Taylor, J.D.; He, X.; Johnston, A.; Horsfield, C.; Janssen, B.J.C.; et al. Influence of donor C3 allotype on late renal-transplantation outcome. N. Engl. J. Med. 2006, 354, 2014–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Touw, W.; Cravedi, P.; Kwan, W.; Paz-Artal, E.; Merad, M.; Heeger, P.S. Cutting Edge: Receptors for C3a and C5a Modulate Stability of Alloantigen-Reactive Induced Regulatory T Cells. J. Immunol. 2013, 190, 5921–5925. [Google Scholar] [CrossRef] [Green Version]
- Golshayan, D.; Wójtowicz, A.; Bibert, S.; Pyndiah, N.; Manuel, O.; Binet, I.; Buhler, L.H.; Huynh-Do, U.; Mueller, T.; Steiger, J.; et al. Polymorphisms in the lectin pathway of complement activation influence the incidence of acute rejection and graft outcome after kidney transplantation. Kidney Int. 2016, 89, 927–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heurich, M.; Martínez-Barricarte, R.; Francis, N.J.; Roberts, D.L.; Rodríguez De Córdoba, S.; Morgan, B.P.; Harris, C.L. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc. Natl. Acad. Sci. USA 2011, 108, 8761–8766. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Reference | Study Characteristics | Studied Group | Results |
|---|---|---|---|
| Parajuli et al., 2017 [82] | Observational single center study | Kidney transplant recipients with late AMR treated with steroids plus IVIG with (n = 40) or without (n = 38) rituximab | Rituximab treatment was associated with less frequent graft loss after 1-year follow-up (15% versus 32%). |
| Tomita et al., 2019 [83] | Propensity-matched retrospective analysis of single center data | Non-sensitized recipients of kidney transplant from AB0-compatible living donors; patients who received rituximab in induction (n = 115) were compared with non-rituximab controls (n = 115) | Rituximab was associated with lower incidence of biopsy proven acute rejection, lower de novo DSA generation, less frequent CMV infection in 5-year follow-up. No difference in patient and graft survival between groups. |
| Pathak et al., 2019 [84] | Retrospective single center observational study | Kidney transplant patients given thymoglobulin and low-dose rituximab induction followed by steroid-free immunosuppression (n = 1111) | Good patient and graft survival were observed (92.4% and 86.1%, respectively at 12 years), along with low percentage of biopsy-proven acute rejection (12.7%). |
| Reference | Study Characteristics | Studied Group | Results |
|---|---|---|---|
| Stegall et al., 2011 [85] | Prospectively enrolled patients treated with eculizumab were compared with historical controls | Highly sensitized recipients of kidney graft from living donors with a positive crossmatch; patients treated with plasma exchange protocol plus eculizumab (n = 26) were compared with non-eculizumab controls (n = 51). | Incidence of AMR during the first 3 months after transplantation was 7.7% in the eculizumab group versus 41.2% in controls. |
| Kulkarni et al., 2017 [91] | Pilot single center randomized trial | Kidney transplant recipients with chronic persistent DSA and deteriorating renal function, treated with eculizumab for 6 months and then observed for 6 months (n = 10) and non-treated (n = 5). | During treatment with eculizumab eGFR decrease was slowed as compared to controls. Long-term eculizumab was safe. |
| Marks et al., 2019 [87] | Phase 2 multicenter randomized open-label trial | Sensitized recipients of living-donor kidney transplant requiring desensitization (n = 102): half were treated with eculizumab for 9 weeks post transplantation (n = 51); the other half received standard of care. | A composite of biopsy-proven acute AMR, graft loss, death, or loss to follow-up at 9 weeks was observed in 11.8% of the eculizumab group and 29.4% controls. Eculizumab treatment was safe. |
| Gonzalez Suarez et al., 2019 [86] | Systematic review of observational studies and case series | Adult kidney transplant recipients with aHUS who received eculizumab in treatment or prevention of aHUS recurrence (n = 380). | Prophylactic eculizumab was associated with 6.3% (95%CI: 2.8–13.4%) recurrence of TMA and 5.5% (95%CI: 2.9–10.0%) graft loss due to TMA. Treatment with eculizumab was associated with 22.5% (95%CI: 13.6–34.8%) graft loss due to TMA. |
| Reference | Study Characteristics | Studied Group | Result |
|---|---|---|---|
| Vo et al., 2015 [75] | Phase 1/2 randomized placebo-controlled | Highly sensitized kidney transplant recipients desensitized with IVIG, rituximab with or without plasma exchange (n = 20); half received plasma-derived human C1-INH intraoperatively and 7 doses twice weekly (n = 10). | Blinded assessment showed reduction in C1q+ HLA antibodies in treatment group, no AMR was observed during the study. Treatment was safe. |
| Montgomery et al., 2016 [77] | Phase 2b, multicenter double-blind randomized placebo-controlled pilot study | Kidney transplant recipients with biopsy-proved DSA-positive AMR (n = 18); half received plasmapheresis and IVIG plus C1-INH every other day for two weeks (n = 9), the other half plasmapheresis and IVIG plus placebo. | No difference in renal biopsy on day 20 (primary endpoint). Transplant glomerulopathy was not observed in treatment arm and was observed in 3/7 patients in placebo arm after 6 months. Treatment was safe. |
| Viglietti et al., 2016 [74] | Prospective, single-arm pilot study | Kidney transplant patients with AMR and acute graft dysfunction nonresponsive to standard therapy; patients were treated with high-dose IVIG plus C1-INH for 6 months (n = 6) | eGFR improved during 6-month treatment from mean 38.7 to 45.2 mL/min/1.73 m2. C4d deposition in tubular capillaries was less frequent after treatment. Treatment was safe. |
| Jordan et al., 2018 [101] | Phase 1/2 randomized placebo-controlled single center trial | Recipients of deceased-donor kidney transplant at increased risk for delayed graft function (n = 70); half received C1-INH (n = 35). | C1-INH did not reduced the need for dialysis at 1-week posttransplant (primary endpoint) but reduced the need for dialysis sessions after 2 to 4 weeks and was associated with better renal function 1 year posttransplant. |
| Huang et al., 2020 [102] | Post-hoc analysis of randomized placebo-controlled trial of Jordan et al. [101] | Recipients of deceased-donor kidney transplant at increased risk for delayed graft function (n = 70); half received C1-INH (n = 35). | Cumulative incidence of graft failure was lower over 3.5-year follow-up among patients treated with C1 esterase inhibitor (50 U/kg perioperatively and after 24 h) compared with placebo. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kielar, M.; Gala-Błądzińska, A.; Dumnicka, P.; Ceranowicz, P.; Kapusta, M.; Naumnik, B.; Kubiak, G.; Kuźniewski, M.; Kuśnierz-Cabala, B. Complement Components in the Diagnosis and Treatment after Kidney Transplantation—Is There a Missing Link? Biomolecules 2021, 11, 773. https://doi.org/10.3390/biom11060773
Kielar M, Gala-Błądzińska A, Dumnicka P, Ceranowicz P, Kapusta M, Naumnik B, Kubiak G, Kuźniewski M, Kuśnierz-Cabala B. Complement Components in the Diagnosis and Treatment after Kidney Transplantation—Is There a Missing Link? Biomolecules. 2021; 11(6):773. https://doi.org/10.3390/biom11060773
Chicago/Turabian StyleKielar, Małgorzata, Agnieszka Gala-Błądzińska, Paulina Dumnicka, Piotr Ceranowicz, Maria Kapusta, Beata Naumnik, Grzegorz Kubiak, Marek Kuźniewski, and Beata Kuśnierz-Cabala. 2021. "Complement Components in the Diagnosis and Treatment after Kidney Transplantation—Is There a Missing Link?" Biomolecules 11, no. 6: 773. https://doi.org/10.3390/biom11060773