
Upper Motor Neuron Disorders: Primary Lateral Sclerosis, Upper Motor Neuron Dominant Amyotrophic Lateral Sclerosis, and Hereditary Spastic Paraplegia


Abstract
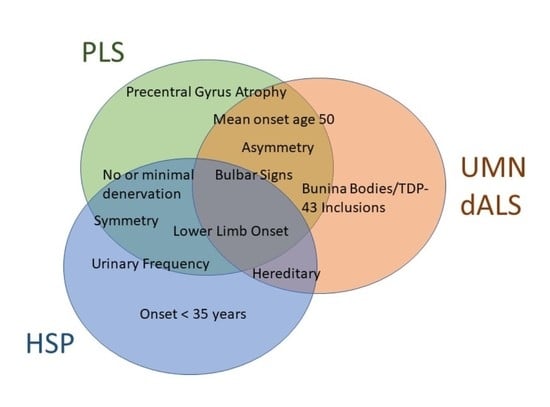
:
1. Introduction
2. Clinical Presentation of Select Upper Motor Neuron Disorders
2.1. Primary Lateral Sclerosis
2.2. Upper Motor Neuron Dominant ALS
2.3. Hereditary Spastic Paraparesis
3. Diagnostics
3.1. Electromyography
3.2. Genetic Testing
3.3. MRI
3.4. Positron Emission Tomography
3.5. Transcranial Magnetic Stimulation
3.6. Neurofilaments and Chitinases
3.7. Histopathological Findings
4. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Erb, W. Concerning Spastic and Syphilitic Spinal Paralysis. BMJ 1902, 2, 1114–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.R.; Talbot, K. Primary lateral sclerosis: Diagnosis and management. Pract. Neurol. 2020, 20, 262–269. [Google Scholar] [CrossRef]
- Pringle, C.E.; Hudson, A.J.; Munoz, D.G.; Kiernan, J.A.; Brown, W.F.; Ebers, G.C. Primary lateral sclerosis: Clinical features, neuropathology and diagnostic criteria. Brain 1992, 115, 495–520. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Barohn, R.J.; Corcia, P.; Fink, J.K.; Harms, M.B.; Kiernan, M.C.; Ravits, J.; Silani, V.; Simmons, Z.; Statland, J.; et al. Primary lateral sclerosis: Consensus diagnostic criteria. J. Neurol. Neurosurg. Psychiatry 2020, 91, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Stark, F.M.; Moersch, F.P. Primary lateral sclerosis: A distinct clinical entity. J. Nerv. Ment. Dis. 1945, 102, 332–337. [Google Scholar] [CrossRef]
- Statland, J.M.; Barohn, R.J.; Dimachkie, M.M.; Floeter, M.K.; Mitsumoto, H. Primary lateral sclerosis. Neurol. Clin. 2015, 33, 749–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, M.A.; Statland, J.M.; Wolfe, G.I.; Barohn, R.J. Primary lateral sclerosis. Muscle Nerve 2007, 35, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Gordon, P.H.; Cheng, B.; Katz, I.B.; Pinto, M.; Hays, A.P.; Mitsumoto, H.; Rowland, L.P. The natural history of primary lateral sclerosis. Neurology 2006, 66, 647–653. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Factor-Litvak, P.; Andrews, H.; Goetz, R.R.; Andrews, L.; Rabkin, J.G.; McElhiney, M.; Nieves, J.; Santella, R.M.; Murphy, J.; et al. ALS Multicenter Cohort Study of Oxidative Stress (ALS COSMOS): Study methodology, recruitment, and baseline demographic and disease characteristics. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 192–203. [Google Scholar] [CrossRef]
- D’Amico, E.; Ba, M.P.; Lee, Y.-W.; Weimer, L.; Mitsumoto, H. Clinical evolution of pure upper motor neuron disease/dysfunction (PUMMD). Muscle Nerve 2013, 47, 28–32. [Google Scholar] [CrossRef]
- Le Forestier, N.; Maisonobe, T.; Piquard, A.; Rivaud, S.; Crevier-Buchman, L.; Salachas, F.; Pradat, P.-F.; Lacomblez, L.; Meininger, V. Does primary lateral sclerosis exist? A study of 20 patients and a review of the literature. Brain 2001, 124, 1989–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, M.A.; Kojan, S.; Barohn, R.J.; Herbelin, L.; Nations, S.P.; Trivedi, J.R.; Jackson, C.E.; Burns, D.K.; Boyer, P.J.; Wolfe, G. Primary Lateral Sclerosis: Clinical and laboratory features in 25 patients. J. Neuromuscul. Dis. 2005, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Panzeri, C.; De Palma, C.; Martinuzzi, A.; Daga, A.; De Polo, G.; Bresolin, N.; Miller, C.C.; Tudor, E.L.; Clementi, E.; Bassi, M.T. The first ALS2 missense mutation associated with JPLS reveals new aspects of alsin biological function. Brain 2006, 129 (Pt 7), 1710–1719. [Google Scholar] [CrossRef]
- Floeter, M.K.; Mills, R. Progression in primary lateral sclerosis: A prospective analysis. Amyotroph. Lateral Scler. 2009, 10, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, P.H.; Cheng, B.; Katz, I.B.; Mitsumoto, H.; Rowland, L.P. Clinical features that distinguish PLS, upper motor neuron-dominant ALS, and typical ALS. Neurology 2009, 72, 1948–1952. [Google Scholar] [CrossRef]
- Finegan, E.; Chipika, R.H.; Shing, S.L.H.; Doherty, M.A.; Hengeveld, J.C.; Vajda, A.; Donaghy, C.; McLaughlin, R.L.; Pender, N.; Hardiman, O.; et al. The clinical and radiological profile of primary lateral sclerosis: A population-based study. J. Neurol. 2019, 266, 2718–2733. [Google Scholar] [CrossRef]
- Zhai, P.; Pagan, F.; Statland, J.; Butman, J.A.; Floeter, M.K. Primary lateral sclerosis: A heterogeneous disorder composed of different subtypes? Neurology 2003, 60, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.C.; Rowe, A.; Findlater, K.; Orange, J.B.; Grace, G.; Strong, M.J. Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis. Arch. Neurol. 2007, 64, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.; Mittal, S.O.; Hu, W.T.; Josephs, K.A.; Sorenson, E.J.; Ahlskog, J.E. Natural History of Pure Primary Lateral Sclerosis. Neurology 2021, 96, e2231–e2238. [Google Scholar] [CrossRef]
- Gastaut, J.L.; Bartolomei, F. Mills’ syndrome: Ascending (or descending) progressive hemiplegia: A hemiplegic form of primary lateral sclerosis? J. Neurol. Neurosurg. Psychiatry 1994, 57, 1280–1281. [Google Scholar] [CrossRef] [Green Version]
- Eisen, A.; Kuwabara, S. The split hand syndrome in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 399–403. [Google Scholar] [CrossRef]
- Norlinah, I.M.; Bhatia, K.P.; Østergaard, K.; Howard, R.; Arabia, G.; Quinn, N.P. Primary lateral sclerosis mimicking atypical parkinsonism. Mov. Disord. 2007, 22, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, N.; Watanabe, H.; Atsuta, N.; Hirayama, M.; Ito, H.; Fukatsu, H.; Kato, T.; Ito, K.; Sobue, G. Primary lateral sclerosis presenting parkinsonian symptoms without nigrostriatal involvement. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Kuipers-Upmeijer, J.; Jager, A.E.J.D.; Hew, J.M.; Snoek, J.W.; Van Weerden, T.W. Primary lateral sclerosis: Clinical, neurophysiological, and magnetic resonance findings. J. Neurol. Neurosurg. Psychiatry 2001, 71, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, B.S.; Rustemeijer, L.M.M.; Bakker, L.A.; Schroder, C.D.; Veldink, J.H.; van den Berg, L.H.; Nijboer, T.C.W.; van Es, M.A. Cognitive and behavioural changes in PLS and PMA: Challenging the concept of restricted phenotypes. J. Neurol. Neurosurg. Psychiatry 2019, 90, 141–147. [Google Scholar] [CrossRef] [Green Version]
- De Vries, B.S.; Spreij, L.A.; Rustemeijer, L.M.M.; Bakker, L.A.; Veldink, J.A.; van den Berg, L.H.; Nijboer, T.C.W.; van Es, M.A. A neuropsychological and behavioral study of PLS. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 376–384. [Google Scholar] [CrossRef]
- Agarwal, S.; Highton-Williamson, E.; Caga, J.; Matamala, J.M.; Dharmadasa, T.; Howells, J.; Zoing, M.C.; Shibuya, K.; Geevasinga, N.; Vucic, S.; et al. Primary lateral sclerosis and the amyotrophic lateral sclerosis–frontotemporal dementia spectrum. J. Neurol. 2018, 265, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Abrahams, S.; Goldstein, L.H.; Woolley, S.; Mclaughlin, P.; Snowden, J.; Mioshi, E.; Roberts-South, A.; Benatar, M.; Hortobágyi, T.; et al. Amyotrophic lateral sclerosis—Frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 153–174. [Google Scholar] [CrossRef]
- Bruyn, R.P.; Koelman, J.H.; Troost, D.; De Jong, J.M. Motor neuron disease (amyotrophic lateral sclerosis) arising from longstanding primary lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 1995, 58, 742–744. [Google Scholar] [CrossRef]
- Bunina, T.L. On intracellular inclusions in familial amyotrophic lateral sclerosis. Zhurnal Nevropatol. Psikhiatrii Im. S.S. Korsakova 1962, 62, 1293–1299. (In Russian) [Google Scholar]
- Hart, M.N.; Cancilla, P.A.; Frommes, S.; Hirano, A. Anterior horn cell degeneration and Bunina-type inclusions associated with dementia. Acta Neuropathol. 1977, 38, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Leigh, P.; Anderton, B.; Dodson, A.; Gallo, J.-M.; Swash, M.; Power, D. Ubiquitin deposits in anterior horn cells in motor neurone disease. Neurosci. Lett. 1988, 93, 197–203. [Google Scholar] [CrossRef]
- Lowe, J.; Lennox, G.; Jefferson, D.; Morrell, K.; McQuire, D.; Gray, T.; Landon, M.; Doherty, F.; Mayer, R. A filamentous inclusion body within anterior horn neurones in motor neurone disease defined by immunocytochemical localisation of ubiquitin. Neurosci. Lett. 1988, 94, 203–210. [Google Scholar] [CrossRef]
- Kato, T.; Katagiri, T.; Hirano, A.; Sasaki, H.; Arai, S. Sporadic lower motor neuron disease with Lewy body-like inclusions: A new subgroup? Acta Neuropathol. 1988, 76, 208–211. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Klebe, S.; Stevanin, G.; Depienne, C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: From SPG1 to SPG72 and still counting. Rev. Neurol. 2015, 171, 505–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Giudice, T.; Lombardi, F.; Santorelli, F.M.; Kawarai, T.; Orlacchio, A. Hereditary spastic paraplegia: Clinical-genetic characteristics and evolving molecular mechanisms. Exp. Neurol. 2014, 261, 518–539. [Google Scholar] [CrossRef]
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [Green Version]
- De Souza, P.V.S.; de Rezende Pinto, W.B.V.; de Rezende Batistella, G.N.; Bortholin, T.; Oliviera, A.C.B. Hereditary Spastic Paraplegia: Clinical and Genetic Hallmarks. Cerebellum 2017, 16, 525–551. [Google Scholar]
- Dupré, N.; Valdmanis, P.N.; Bouchard, J.-P.; Rouleau, G.A. Autosomal dominant primary lateral sclerosis. Neurology 2007, 68, 1156–1157. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.K. Progressive Spastic Paraparesis: Hereditary Spastic Paraplegia and Its Relation to Primary and Amyotrophic Lateral Sclerosis. Semin. Neurol. 2001, 21, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Burgman, F.; Veldink, J.H.; Franssen, H.; de Visser, M.; de Jong, J.M.B.V.; Faber, C.G.; Kremer, B.H.P.; Schelhaas, H.J.; van Doorn, P.A.; Verschuuren, J.J.G.M.; et al. Differentiation of hereditary spastic paraparesis from primary lateral sclerosis in sporadic adult-onset upper motor neuron syndroms. Arch. Neurol. 2009, 66, 509–514. [Google Scholar]
- Mitsumoto, H.; Nagy, P.L.; Gennings, C.; Murphy, J.; Andrews, H.; Goetz, R.; Floeter, M.K.; Hupf, J.; Singleton, J.; Barohn, R.J.; et al. Phenotypic and molecular analyses of primary lateral sclerosis. Neurol. Genet. 2015, 1, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, C.N.; Murphy, A.; Loci, L.; Mitsumoto, H.; Lomen-Hoerth, C.; Kisanuki, Y.; Simmons, Z.; Maragakis, N.J.; McVey, A.L.; Al-Lahham, T.; et al. Primary Lateral Sclerosis and Early Upper Motor Neuron Disease: Characteristics of a Cross-Sectional Population. J. Clin. Neuromuscul. Dis. 2016, 17, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooch, C.L.; Pullman, S.L.; Shungu, D.C.; Uluğ, A.M.; Chan, S.; Gordon, P.H.; Tang, M.-X.; Mao, X.; Rowland, L.P.; Mitsumoto, H. Motor unit number estimation (MUNE) in diseases of the motor neuron: Utility and comparative analysis in a multimodal biomarker study. Suppl. Clin. Neurophysiol. 2009, 60, 153–162. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Ulug, A.M.; Pullman, S.L.; Gooch, C.L.; Chan, S.; Tang, M.-X.; Mao, X.; Hays, A.P.; Floyd, A.G.; Battista, V.; et al. Quantitative objective markers for upper and lower motor neuron dysfunction in ALS. Neurology 2007, 68, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Craciun, L.; Floeter, M.K. Motor Unit Number Estimation in Primary Lateral Sclerosis; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- DeDe, H.O.; Sirin, N.G.; Kocasoy-Orhan, E.; Idrisoglu, H.A.; Baslo, M.B. Electrophysiological Findings of Subclinical Lower Motor Neuron Involvement in Degenerative Upper Motor Neuron Diseases. Noropsikiyatr. Ars. 2020, 57, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Gazulla, J.; Ferrer, I.; Izquierdo-Alvarez, S.; Alvarez, S.; Sánchez-Alcudia, R.; Bestué-Cardiel, M.; Seral, M.; Benavente, I.; Sierra-Martínez, E.; Berciano, J. Hereditary primary lateral sclerosis and progressive nonfluent aphasia. J. Neurol. 2019, 266, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Van Rheenen, W.; van Blitterswijk, M.; Huisman, M.H.B.; Vlam, L.; van Doormaal, P.T.C.; Seelen, M.; Medic, J.; Dooijes, D.; de Visser, M.; van der Kooi, A.J.; et al. Hexanucleotide repeat expansions in C9ORF72 int he spectrum of motor neuron diseases. Neurology 2012, 79, 878–882. [Google Scholar] [CrossRef]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef] [Green Version]
- Kwan, J.Y.; Meoded, A.; Danielian, L.E.; Wu, T.; Floeter, M.K. Structural imaging differences and longitudinal changes in primary lateral sclerosis and amyotrophic lateral sclerosis. NeuroImage Clin. 2013, 2, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, V.; Pioro, E.P. Unbiased MRI Analyses Identify Micropathologic Differences Between Upper Motor Neuron-Predominant ALS Phenotypes. Front. Neurosci. 2019, 13, 704. [Google Scholar] [CrossRef]
- Ngai, S.; Tang, Y.M.; Du, L.; Stuckey, S. Hyperintensity of the precentral gyral subcortical white matter and hypointensity of hte precentral gyrus on fluid-attenuated inversion recovery: Variation with age and implications for the diagnosis of amyotrophic lateral sclerosis. Am. J. Neuroradiol. 2007, 28, 250–254. [Google Scholar] [PubMed]
- Finegan, E.; Shing, S.L.H.; Siah, W.F.; Chipika, R.H.; Chang, K.M.; McKenna, M.C.; Doherty, M.A.; Hengeveld, J.C.; Vajda, A.; Donaghy, C.; et al. Evolving diagnostic criteria in primary lateral sclerosis: The clinical and radiological basis of “probable PLS”. J. Neurol. Sci. 2020, 417, 117052. [Google Scholar] [CrossRef]
- Agosta, F.; Galantucci, S.; Riva, N.; Chiò, A.; Messina, S.; Iannaccone, S.; Calvo, A.; Silani, V.; Copetti, M.; Falini, A.; et al. Intrahemispheric and interhemispheric structural network abnormalities in PLS and ALS. Hum. Brain Mapp. 2014, 35, 1710–1722. [Google Scholar] [CrossRef]
- Bede, P.; Hardiman, O. Longitudinal structural changes in ALS: A three time-point imaging study of white and gray matter degeneration. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Menke, R.A.; Körner, S.; Filippini, N.; Douaud, G.; Knight, S.; Talbot, K.; Turner, M.R. Widespread grey matter pathology dominates the longitudinal cerebral MRI and clinical landscape of amyotrophic lateral sclerosis. Brain 2014, 137 Pt 9, 2546–2555. [Google Scholar] [CrossRef] [Green Version]
- Menke, R.A.L.; Proudfoot, M.; Talbot, K.; Turner, M. The two-year progression of structural and functional cerebral MRI in amyotrophic lateral sclerosis. NeuroImage Clin. 2018, 17, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Chipika, R.H.; Siah, W.F.; Shing, S.L.H.; Finegan, E.; McKenna, M.C.; Christidi, F.; Chang, K.M.; Karavasilis, E.; Vajda, A.; Hengeveld, J.C.; et al. MRI data confirm the selective involvement of thalamic and amygdalar nuclei in amyotrophic lateral sclerosis and primary lateral sclerosis. Data Brief 2020, 32, 106246. [Google Scholar] [CrossRef]
- Turner, M.R.; Hammers, A.; Al-Chalabi, A.; Shaw, C.E.; Andersen, P.M.; Brooks, D.J.; Leigh, P.N. Cortical involvement in four cases of primary lateral sclerosis using [11C]-flumazenil PET. J. Neurol. 2007, 254, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Claassen, D.O.; Josephs, K.A.; Peller, P.J. The stripe of primary lateral sclerosis: Focal primary motor cortex hypometabolism seen on fluorodeoxyglucose F18 positron emission tomography. Arch. Neurol. 2010, 67, 122–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, P.; Geevasinga, N.; Yiannikas, C.; Howells, J.; Kiernan, M.C.; Vucic, S. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: A prospective study. Lancet Neurol. 2015, 14, 478–484. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Sue, C.M.; Kumar, K.R.; Ng, K.; Yiannikas, C.; Kiernan, M.C.; Vucic, S. Cortical excitability changes distinguish the motor neuron disease phenotypes from hereditary spastic paraplegia. Eur. J. Neurol. 2015, 22, 826-e58. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Stewart, H.; Hirota, N.; Eisen, A. Corticomotoneuronal connections in primary lateral sclerosis (PLS). Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2002, 3, 190–198. [Google Scholar] [CrossRef]
- Falzone, Y.M.; Domi, T.; Agosta, F.; Pozzi, L.; Schito, P.; Fazio, R.; Del Carro, U.; Barbieri, A.; Comola, M.; Leocani, L.; et al. Serum phosphorylated neurofilament heavy-chain levels reflect phenotypic heterogeneity and are an independent predictor of survival in motor neuron disease. J. Neurol. 2020, 267, 2272–2280. [Google Scholar] [CrossRef]
- Gaiani, A.; Martinelli, I.; Bello, L.; Querin, G.; Puthenparampil, M.; Ruggero, S.; Toffani, E.; Cagnin, A.; Briani, C.; Pegoraro, E.; et al. Diagnostic and Prognostic Biomarkers in Amyotrophic Lateral Sclerosis: Neurofilament Light Chain Levels in Definite Subtypes of Disease. JAMA Neurol. 2017, 74, 525–532. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Vacchiano, V.; Zenesini, C.; Polischi, B.; De Pasqua, S.; Fileccia, E.; Mammana, A.; Di Stasi, V.; Capellari, S.; Salvi, F.; et al. Diagnostic-prognostic value and electrophysiological correlates of CSF biomarkers of neurodegeneration and neuroinflammation in amyotrophic lateral sclerosis. J. Neurol. 2020, 267, 1699–1708. [Google Scholar] [CrossRef]
- Gille, B.; De Schaepdryver, M.; Goossens, J.; Dedeene, L.; De Vocht, J.; Oldoni, E.; Goris, A.; Bosch, L.V.D.; Depreitere, B.; Claeys, K.G.; et al. Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with Amyotrophic Lateral Sclerosis. Neuropathol. Appl. Neurobiol. 2019, 45, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.; Rattay, T.W.; Hengel, H.; Zimmermann, M.; Brockmann, K.; Schöls, L.; Kuhle, J.; Schüle, R.; Synofzik, M. Serum neurofilament light chain is increased in hereditary spastic paraplegias. Ann. Clin. Transl. Neurol. 2018, 5, 876–882. [Google Scholar] [CrossRef]
- Steinacker, P.; Feneberg, E.; Weishaupt, J.; Brettschneider, J.; Tumani, H.; Andersen, P.M.; Arnim, C.A.F.V.; Böhm, S.; Kassubek, J.; Kubisch, C.; et al. Neurofilaments in the diagnosis of motoneuron diseases: A prospective study on 455 patients. J. Neurol. Neurosurg. Psychiatry 2016, 87, 12–20. [Google Scholar] [CrossRef]
- Zucchi, E.; Bedin, R.; Fasano, A.; Fini, N.; Gessani, A.; Vinceti, M.; Mandrioli, J. Cerebrospinal Fluid Neurofilaments May Discriminate Upper Motor Neuron Syndromes: A Pilot Study. Neurodegener. Dis. 2018, 18, 255–261. [Google Scholar] [CrossRef]
- Zucchi, E.; Bonetto, V.; Sorarù, G.; Martinelli, I.; Parchi, P.; Liguori, R.; Mandrioli, J. Neurofilaments in motor neuron disorders: Towards promising diagnostic and prognostic biomarkers. Mol. Neurodegener. 2020, 15, 58. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Bampton, A.; Raciborska, D.; Talbot, K.; Turner, M.R. CSF chitinase proteins in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1215–1220. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Thézénas, M.-L.; Charles, P.D.; Evetts, S.; Hu, M.T.; Talbot, K.; Fischer, R.; Bm, B.M.K.; Turner, M.R. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann. Neurol. 2018, 83, 258–268. [Google Scholar] [CrossRef]
- Beal, M.F.; Richardson, E.P., Jr. Primary lateral sclerosis: A case report. Arch. Neurol. 1981, 38, 630–633. [Google Scholar] [CrossRef]
- Fisher, C.M. Pure spastic paralysis of corticospinal origin. Can. J. Neurol. Sci. 1977, 4, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Younger, D.S.; Chou, S.; Hays, A.P.; Lange, D.L.; Emerson, R.; Brin, M.; Thompson, H., Jr.; Rowland, L.P. Primary lateral sclerosis. A clinical diagnosis reemerges. Arch. Neurol. 1988, 45, 1304–1307. [Google Scholar] [CrossRef]
- Tan, C.F.; Kakita, A.; Piao, Y.-S.; Kikugawa, K.; Endo, K.; Tanaka, M.; Okamoto, K.; Takahashi, H. Primary lateral sclerosis: A rare upper-motor-predominant form of amyotrophic lateral sclerosis often accompanied by frontotemporal lobar degeneration with ubiquitinated neuronal inclusions? Report of an autopsy case and a review of the literature. Acta Neuropathol. 2003, 105, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Bigio, E.H.; Budka, H.; Dickson, D.W.; Ferrer, I.; Ghetti, B.; Giaccone, G.; Hatanpaa, K.J.; Holton, J.L.; Josephs, K.A.; et al. Globular glial tauopathies (GGT): Consensus recommendations. Acta Neuropathol. 2013, 126, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Z.; Tsuchiya, K.; Arai, T.; Yokota, O.; Yoshida, M.; Shimomura, Y.; Kondo, H.; Haga, C.; Asaoka, T.; Onaya, M.; et al. Clinicopathological characteristics of FTLD-TDP showing corticospinal tract degeneration but lacking lower motor neuron loss. J. Neurol. Sci. 2010, 298, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Yokota, O.; Tsuchiya, K.; Arai, T.; Yagishita, S.; Matsubara, O.; Mochizuki, A.; Tamaoka, A.; Kawamura, M.; Yoshida, H.; Terada, S.; et al. Clinicopathological characterization of Pick’s disease versus frontotemporal lobar degeneration with ubiquitin/TDP-43-positive inclusions. Acta Neuropathol. 2009, 117, 429–444. [Google Scholar] [CrossRef]
- Hainfellner, J.A.; Pilz, P.; Lassmann, H.; Ladurner, G.; Budka, H. Diffuse Lewy body disease as substrate of primary lateral sclerosis. J. Neurol. 1995, 242, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Feldman, H. Neurofilament inclusion body disease with early onset frontotemporal dementia and primary lateral sclerosis. Clin. Neuropathol. 2004, 23, 183–193. [Google Scholar]
- Nagao, S.; Yokota, O.; Nanba, R.; Takata, H.; Haraguchi, T.; Ishizu, H.; Ikeda, C.; Takeda, N.; Oshima, E.; Sakane, K.; et al. Progressive supranuclear palsy presenting as primary lateral sclerosis but lacking parkinsonism, gaze palsy, aphasia, or dementia. J. Neurol. Sci. 2012, 323, 147–153. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Briemberg, H. TDP-43 pathology in primary lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21 (Suppl. 1), 52–58. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Josephs, K.A.; Amador-Ortiz, C. TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol. 2007, 114, 71–79. [Google Scholar] [CrossRef]
- Behan, W.M.H.; Maia, M. Strumpell’s familial spastic paraplegia: Genetics and neuropathology. J. Neurol. Neurosurg. Psychiatry 1974, 37, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Mitsumoto, H.; Chiuzan, C.; Gilmore, M.; Zhang, Y.; Simmons, Z.; Paganoni, S.; Kisanuki, Y.Y.; Zinman, L.; Jawdat, O.; Sorenson, E.; et al. Primary lateral sclerosis (PLS) functional rating scale: PLS-specific clinimetric scale. Muscle Nerve 2020, 61, 163–172. [Google Scholar] [CrossRef] [PubMed]

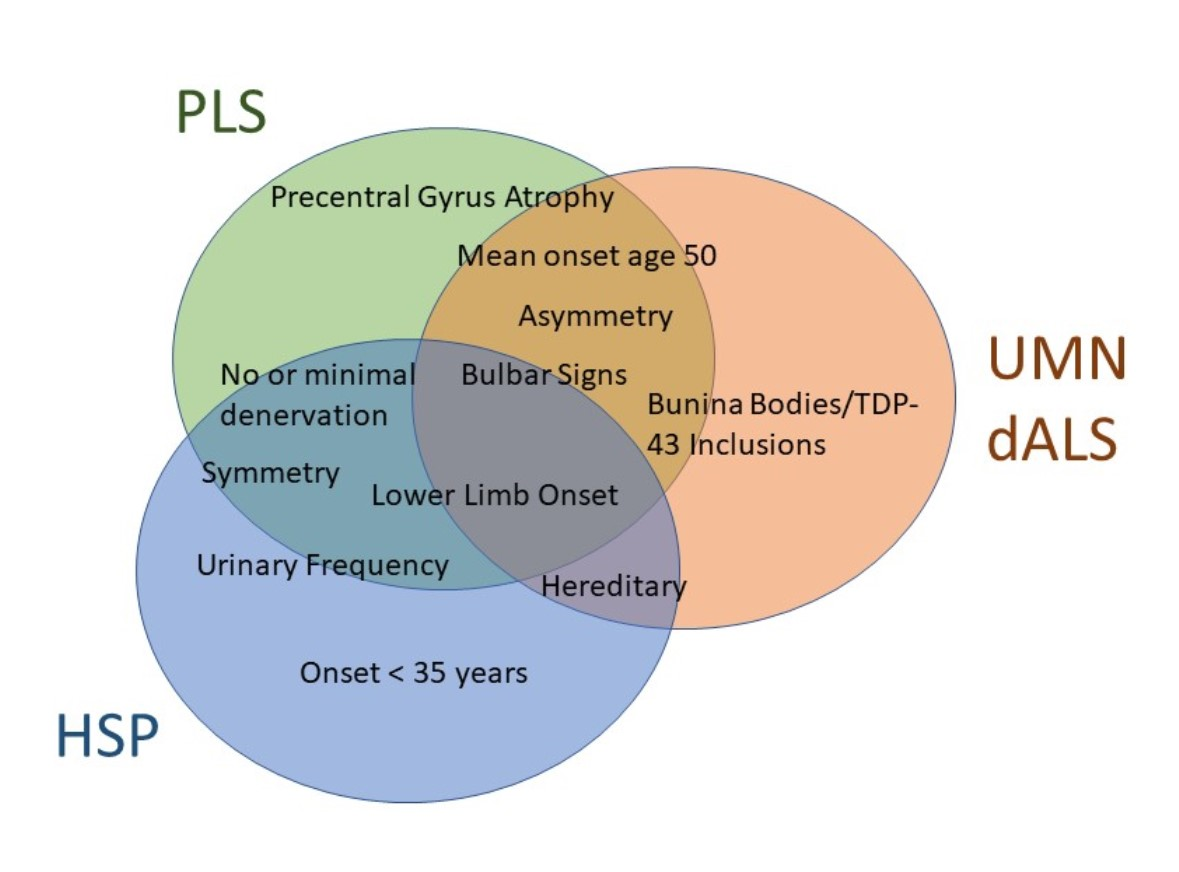{kind=link}
| Feature | PLS | UMNdALS | ALS | HSP |
|---|---|---|---|---|
| Incidence (per 100,000/year) | <0.1 | 1–3 | 1–3 | 4.3–9.6 1 |
| Age of onset (mean) | 50 | 50 | 65 | 30–40 |
| Gender Predominance | 2–4:1 2 | 1.6:1 | 1.6:1 | 1:1 |
| Prognosis | Slowly progressive, decade or greater survival | Typical survival beyond a decade | 3–5 years | Very slowly progressive, variable |
| Site of first symptoms | Lower limb 90%/Bulbar 10% | Lower Limb 30%/Bulbar 30%/Upper limb 30% | Lower Limb 30%/Bulbar 30%/Upper limb 30% | Lower Limb |
| Lowest MRC on initial evaluation ≤4? | - | +/- | + | - |
| Symmetry | Symmetric 3 | Asymmetric | Asymmetric | Symmetric |
| Weight Loss at Diagnosis? | - | + | ++ | - |
| Urinary Frequency | ++ (late) | +/- | - | ++ |
| Cognitive Impairment | + | + | ++ | +/- |
| Family History | None | 10% | 10% | 40–60% |
| Spasticity | +++ | +++ | +/- | +++ |
| PEG/Ventilator needs? | +/- | + | ++ | - |
| Frontotemporal Dementia | + | + | ++ | +/- |
| Pseudobulbar Affect | ++ | + | + | - |
| Diagnostic Test | PLS | ALS | HSP |
|---|---|---|---|
| Denervation Potentials on EMG | +/- 1 | +++ | +/- |
| Abnormal SSEPs | +/- | - | ++ |
| Precentral Gyrus Atrophy (MRI) | ++ | + | - |
| Corticospinal Tract Hyperintensity on T2-weighted imaging | + (late) | + (early) 2 | + |
| FDG-PET Findings | Focal motor cortex hypometabolism (‘stripe sign’) | Motor cortex + diffuse frontal cortex hypometabolism | Heterogenous areas of cortical hypometabolism |
| TMS MEPs prolonged? (early/late) | +++/+++ | +/++ | -/- |
| Neurofilament light chain levels | ++ | +++ | + |
| Bunina Bodies | +/- | ++ | - |
| TDP-43 Inclusions | + 3 | ++ | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fullam, T.; Statland, J. Upper Motor Neuron Disorders: Primary Lateral Sclerosis, Upper Motor Neuron Dominant Amyotrophic Lateral Sclerosis, and Hereditary Spastic Paraplegia. Brain Sci. 2021, 11, 611. https://doi.org/10.3390/brainsci11050611
Fullam T, Statland J. Upper Motor Neuron Disorders: Primary Lateral Sclerosis, Upper Motor Neuron Dominant Amyotrophic Lateral Sclerosis, and Hereditary Spastic Paraplegia. Brain Sciences. 2021; 11(5):611. https://doi.org/10.3390/brainsci11050611
Chicago/Turabian StyleFullam, Timothy, and Jeffrey Statland. 2021. "Upper Motor Neuron Disorders: Primary Lateral Sclerosis, Upper Motor Neuron Dominant Amyotrophic Lateral Sclerosis, and Hereditary Spastic Paraplegia" Brain Sciences 11, no. 5: 611. https://doi.org/10.3390/brainsci11050611