Synergy between Auranofin and Celecoxib against Colon Cancer In Vitro and In Vivo through a Novel Redox-Mediated Mechanism

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
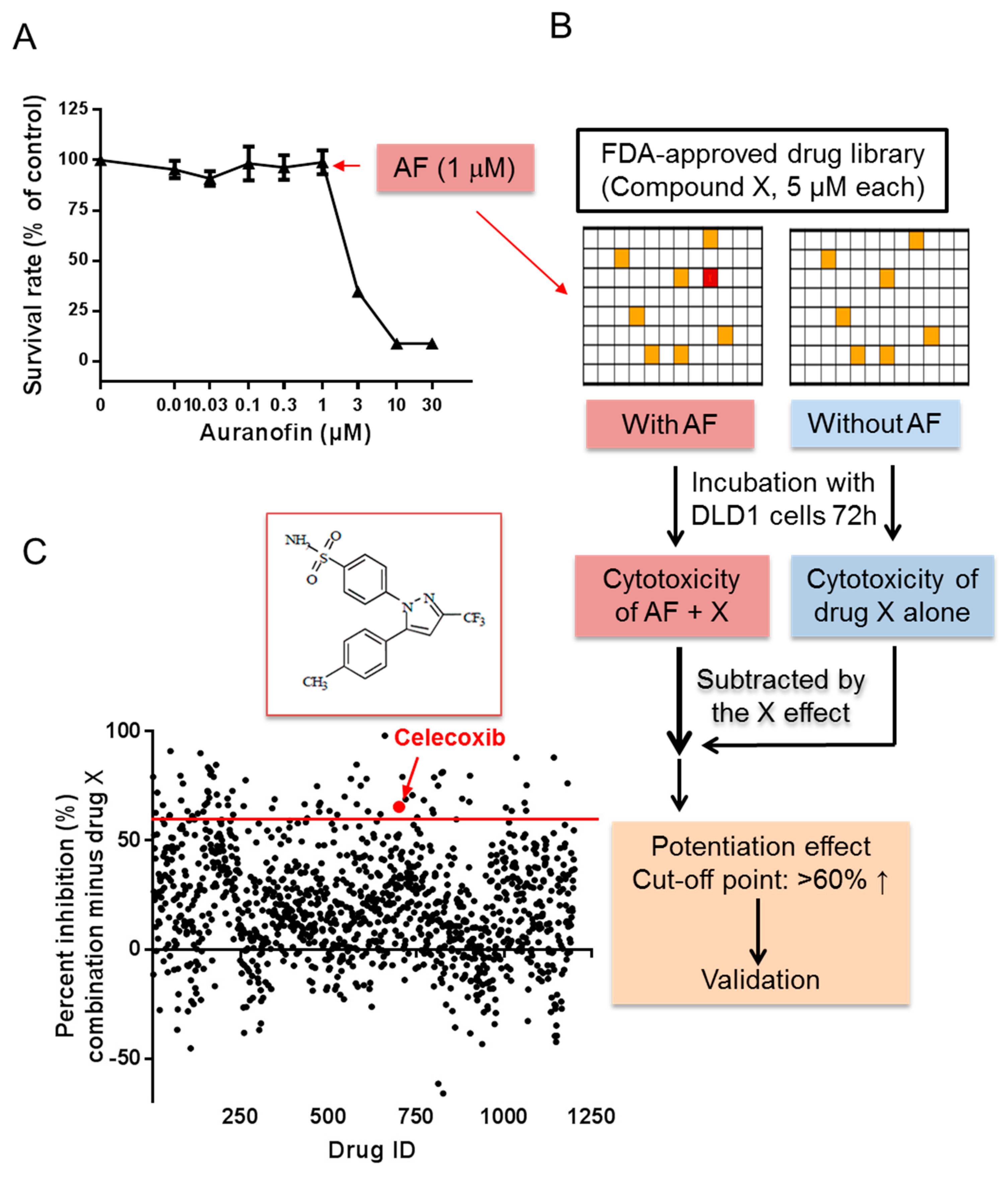
2.1. High-Throughput Screening for Drugs that Synergize with Auranofin against Cancer Cells In Vitro
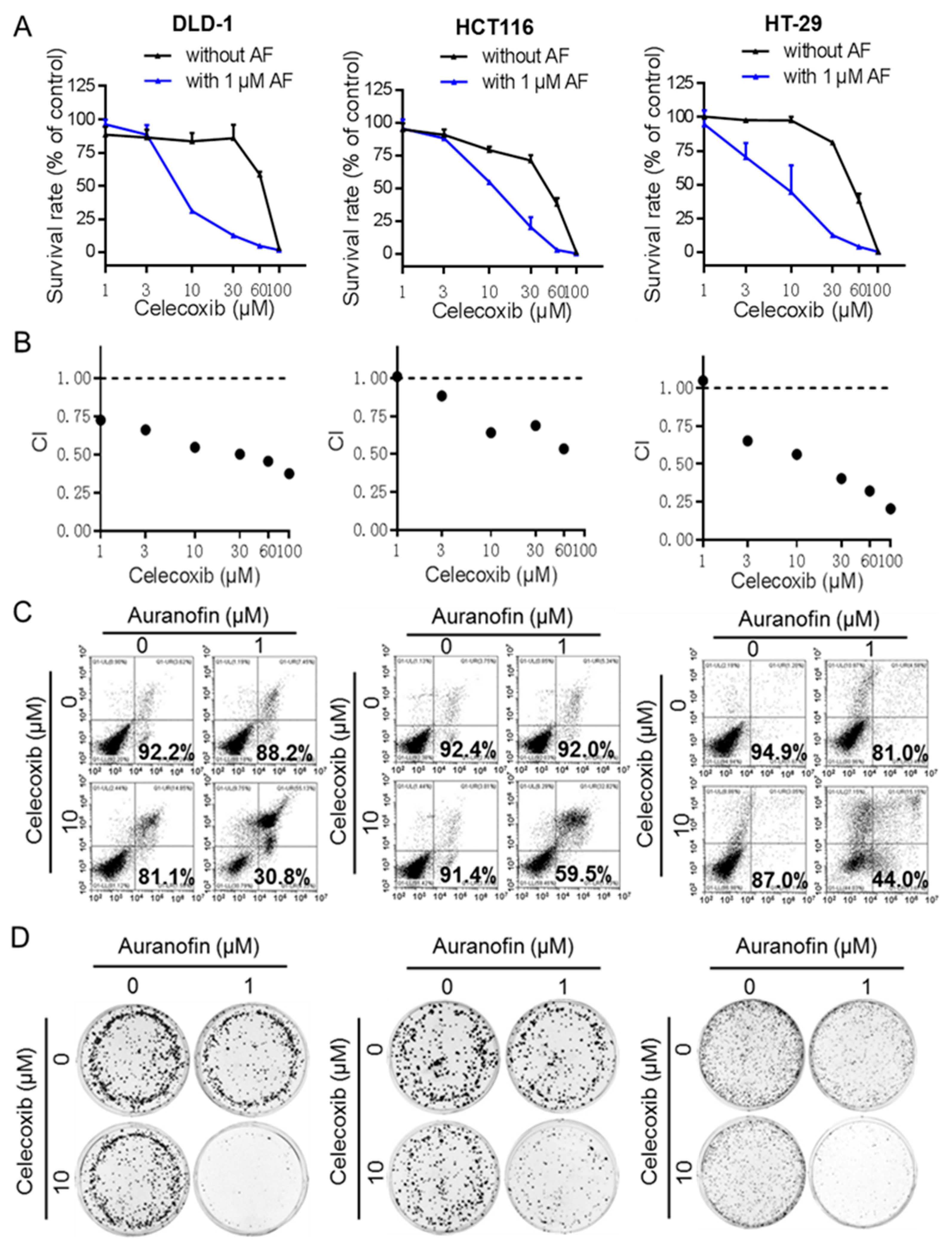
2.2. Auranofin and Celecoxib Synergistically Induce Colorectal Cancer Cell Death In Vitro
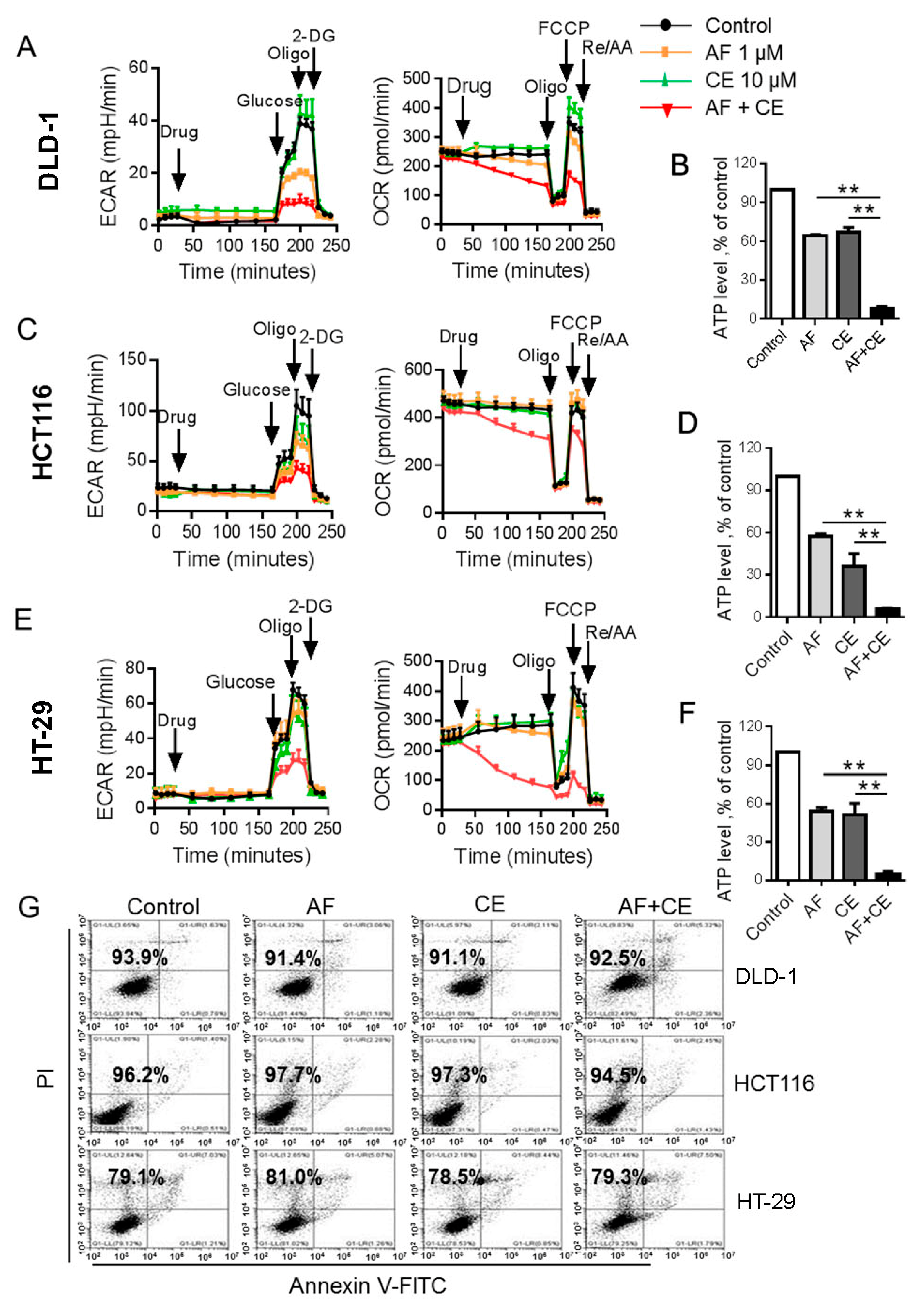
2.3. Combination of AF and CE Induced a Severe Energy Crisis in Cancer Cells
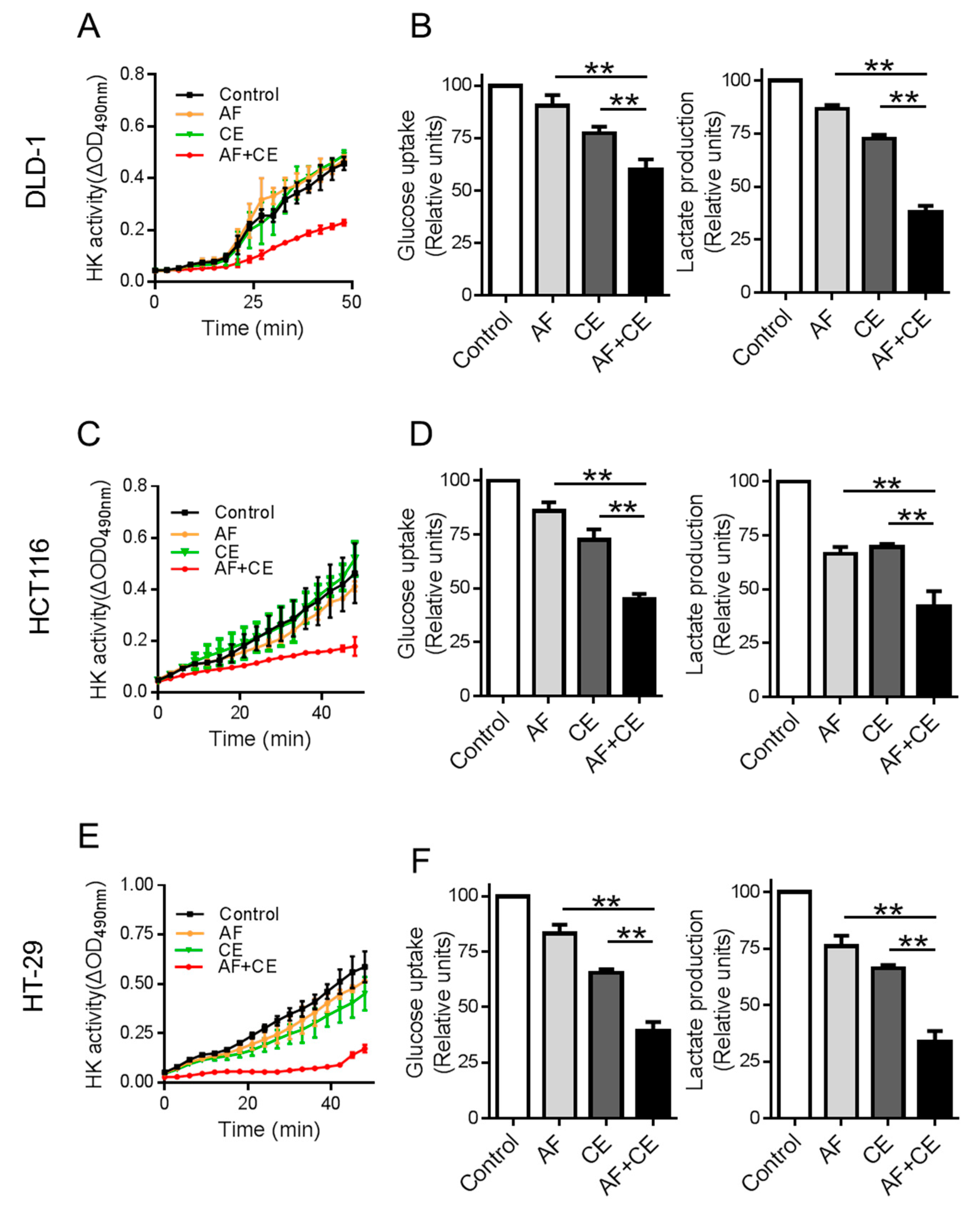
2.4. Combination of CE and AF Caused Inhibition of Hexokinase Enzyme Activity
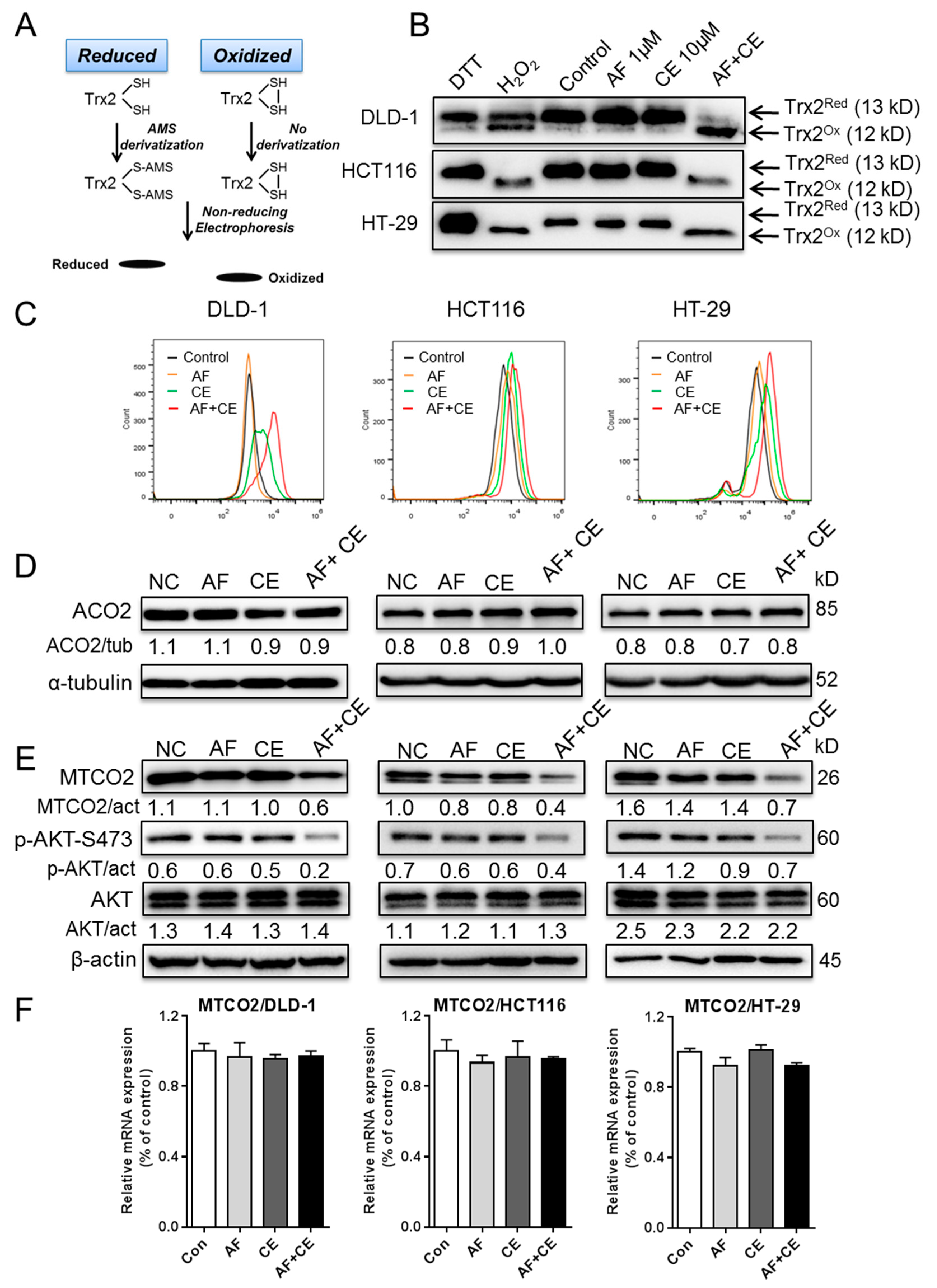
2.5. AF and CE Combination Induced Severe ROS Stress Leading to Protein Oxidation and Dysfunction of Mitochondrial Electron Transport Chain
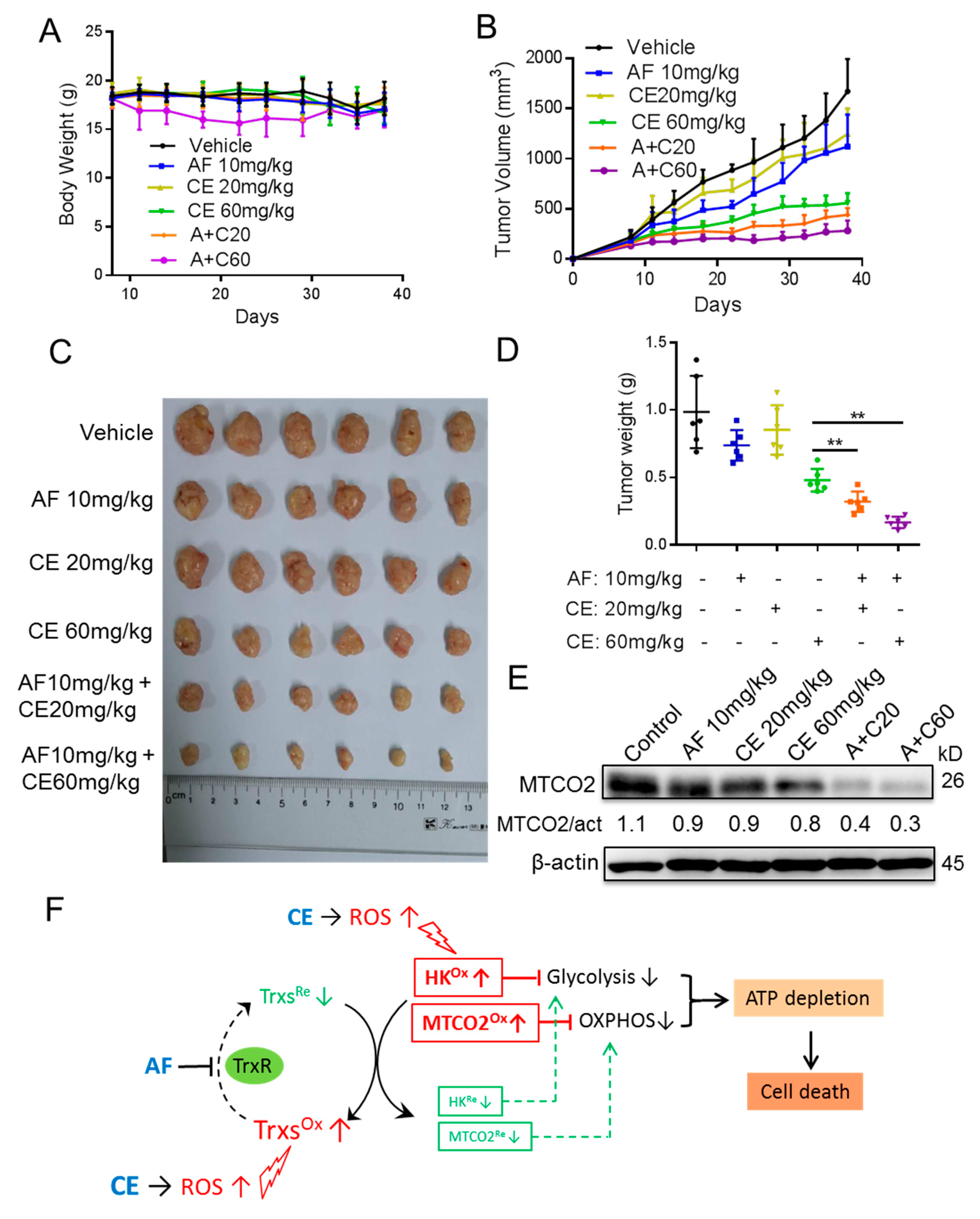
2.6. Auranofin and Celecoxib Exhibit Synergistic Therapeutic Effect In Vivo
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. Cell Viability Assay
4.3. Apoptosis Assay
4.4. Real-Time Cell Metabolism Assay
4.5. Measurement of Cellular ATP
4.6. Hexokinase Enzymatic Activity Assay
4.7. Measurement of Cellular Glucose Uptake and Lactate Production
4.8. Thioredoxin-2 Redox Analysis
4.9. Measurement of Cellular ROS
4.10. Western Blotting
4.11. High-Throughput Screening Assay
4.12. Mouse Experiments
4.13. Determination of mRNA Expression
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, I.S.; Jung, Y.H. Metronomic chemotherapy in metastatic colorectal cancer. Cancer Lett. 2017, 400, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Buzzelli, J.N.; Ouaret, D.; Brown, G.; Allen, P.D.; Muschel, R.J. Colorectal cancer liver metastases organoids retain characteristics of original tumor and acquire chemotherapy resistance. Stem Cell Res. 2018, 27, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Huang, D.; Peng, Y.; Yao, Y.; Zhao, Y.; Yang, Y.; Wang, H.; Cao, L.; Zhu, W.G.; Gu, J. 5-Fluorouracil targets histone acetyltransferases p300/CBP in the treatment of colorectal cancer. Cancer Lett. 2017, 400, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Tong, M.; Liang, Q.; Guo, Y.; Sun, H.Q.; Zheng, W.; Ao, L.; Guo, Z.; She, F. Epigenomics alternations and dynamic transcriptional changes in responses to 5-fluorouracil stimulation reveal mechanisms of acquired drug resistance of colorectal cancer cells. Pharmacogenom. J. 2017, 18, 23–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41. [Google Scholar] [CrossRef]
- Hatem, E.; Azzi, S.; El Banna, N.; He, T.; Heneman-Masurel, A.; Vernis, L.; Baille, D.; Masson, V.; Dingli, F.; Loew, D.; et al. Auranofin/Vitamin C: A Novel Drug Combination Targeting Triple-Negative Breast Cancer. J. Natl. Cancer Inst. 2018, djy149. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs R D 2015, 15, 13–20. [Google Scholar] [CrossRef]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S.D.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef]
- Hou, G.X.; Liu, P.P.; Zhang, S.; Yang, M.; Liao, J.; Yang, J.; Hu, Y.; Jiang, W.Q.; Wen, S.; Huang, P. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis. Cell Death Dis. 2018, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Saba, N.; Ghias, M.; Manepalli, R.; Schorno, K.; Weir, S.; Austin, C.; Maddocks, K.; Byrd, J.C.; Kambhampati, S.; Bhalla, K. Auranofin Induces a Reversible In-Vivo Stress Response That Correlates with a Transient Clinical Effect in Patients with Chronic Lymphocytic Leukemia. Blood 2013, 122, 3819. [Google Scholar]
- Leary, M.; Heerboth, S.; Lapinska, K.; Sarkar, S. Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy. Cancers 2018, 10, 483. [Google Scholar] [CrossRef] [PubMed]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-rasG12V transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2011, 22, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, H.; Abe, M.; Yang, Y.; Cui, D.; Seki, T.; Nakamura, M.; Hosaka, K.; Lim, S.; Wu, J.; He, X.; et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab. 2018, 28, 104–117. [Google Scholar] [CrossRef]
- Nulton-Persson, A.C.; Szweda, L.I. Modulation of mitochondrial function by hydrogen peroxide. J. Biol. Chem. 2001, 276, 23357–23361. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug. Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Mai, C.; Wei, Y.L.; Zhao, J.J.; Hu, Y.M.; Zeng, Z.L.; Yang, J.; Lu, W.H.; Xu, R.H.; Huang, P. Decreased expression of the mitochondrial metabolic enzyme aconitase (ACO2) is associated with poor prognosis in gastric cancer. Med. Oncol. 2013, 30, 552. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Fang, H.; Liu, J.; Vartak, R.; Deng, J.; Bai, Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum. Mol. Genet. 2011, 20, 4605–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelicano, H.; Xu, R.H.; Du, M.; Feng, L.; Sasaki, R.; Carew, J.S.; Hu, Y.; Ramdas, L.; Hu, L.; Keating, M.J.; et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 2006, 175, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Bertagnolli, M.M.; Eagle, C.J.; Zauber, A.G.; Mark, R.; Solomon, S.D.; Kyungmann, K.; Jie, T.; Rosenstein, R.B.; Janet, W.; Donald, C. Celecoxib for the prevention of sporadic colorectal adenomas. N. Engl. J. Med. 2006, 355, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Gridley, G.; Mclaughlin, J.K.; Ekbom, A.; Klareskog, L.; Adami, H.O.; Hacker, D.G.; Hoover, R.; Fraumeni, J.F., Jr. Incidence of cancer among patients with rheumatoid arthritis. J. Natl. Cancer Inst. 1993, 85, 307–311. [Google Scholar] [CrossRef]
- Damdimopoulos, A.E.; Miranda-Vizuete, A.; Pelto-Huikko, M.; Gustafsson, J.A.; Spyrou, G. Human mitochondrial thioredoxin. Involvement in mitochondrial membrane potential and cell death. J. Biol. Chem. 2002, 277, 33249–33257. [Google Scholar] [CrossRef]
- Veltman, J.D.; Lambers, M.E.; van Nimwegen, M.; Hendriks, R.W.; Hoogsteden, H.C.; Aerts, J.G.; Hegmans, J.P. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 2010, 10, 464. [Google Scholar] [CrossRef]
- Lampiasi, N.; Azzolina, A.; Umezawa, K.; Montalto, G.; McCubrey, J.A.; Cervello, M. The novel NF-kappaB inhibitor DHMEQ synergizes with celecoxib to exert antitumor effects on human liver cancer cells by a ROS-dependent mechanism. Cancer Lett. 2012, 322, 35–44. [Google Scholar] [CrossRef]
- Liu, X.; Yue, P.; Zhou, Z.; Khuri, F.R.; Sun, S.Y. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J. Natl. Cancer Inst. 2004, 96, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Rudner, J.; Elsaesser, S.J.; Muller, A.C.; Belka, C.; Jendrossek, V. Differential effects of anti-apoptotic Bcl-2 family members Mcl-1, Bcl-2, and Bcl-xL on celecoxib-induced apoptosis. Biochem. Pharmacol. 2010, 79, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Leng, J.; Han, C.; Demetris, A.J. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol. Cancer Ther. 2004, 3, 299–307. [Google Scholar] [PubMed]
- Pritchard, R.; Rodríguez-Enríquez, S.; Pacheco-Velà zquez, S.C.; Bortnik, V.; Moreno-Sã, N.R.; Ralph, S. Celecoxib inhibits mitochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochem. Pharmacol. 2018, 154, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.P.; Liao, J.; Tang, Z.J.; Wu, W.J.; Yang, J.; Zeng, Z.L.; Hu, Y.; Wang, P.; Ju, H.Q.; Xu, R.H.; et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014, 21, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Van Riel, P.L.; Gribnau, F.W.; Van de Putte, L.B.; Arts, C.W.; Van Aernsbergen, A. Serum gold concentrations during treatment with auranofin. Clin. Rheumatol. 1987, 6, 50–54. [Google Scholar] [CrossRef]
- Rapaport, M.H.; Delrahim, K.K.; Bresee, C.J.; Maddux, R.E.; Ahmadpour, O.; Dolnak, D. Celecoxib augmentation of continuously ill patients with schizophrenia. Biol. Psychiatry 2005, 57, 1594–1596. [Google Scholar] [CrossRef]
- Soni, P.; Shell, B.; Cawkwell, G.; Li, C.; Ma, H. The hepatic safety and tolerability of the cyclooxygenase-2 selective NSAID celecoxib: Pooled analysis of 41 randomized controlled trials. Curr. Med. Res. Opin. 2009, 25, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, P.; Wang, K.; Glorieux, C.; Hu, Y.; Wen, S.; Jiang, W.; Huang, P. Chemotherapy induces tumor immune evasion by upregulation of programmed cell death ligand 1 expression in bone marrow stromal cells. Mol. Oncol. 2017, 11, 358–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, Y.M.; Jones, D.P. Thioredoxin redox western analysis. Curr. Protoc. Toxicol. 2009, 41, 17.12.1–17.12.12. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, W.; Lu, W.; Song, M.; Liu, K.; Chen, P.; Chang, A.; Ling, J.; Chiao, P.J.; Hu, Y.; et al. Identification of cisplatin sensitizers through high-throughput combinatorial screening. Int. J. Oncol. 2018, 53, 1237–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Chen, P.; Zhang, Y.; Lu, W.; Ding, W.; Luo, Y.; Wen, S.; Xu, R.; Liu, P.; Huang, P. Synergy between Auranofin and Celecoxib against Colon Cancer In Vitro and In Vivo through a Novel Redox-Mediated Mechanism. Cancers 2019, 11, 931. https://doi.org/10.3390/cancers11070931
Han Y, Chen P, Zhang Y, Lu W, Ding W, Luo Y, Wen S, Xu R, Liu P, Huang P. Synergy between Auranofin and Celecoxib against Colon Cancer In Vitro and In Vivo through a Novel Redox-Mediated Mechanism. Cancers. 2019; 11(7):931. https://doi.org/10.3390/cancers11070931
Chicago/Turabian StyleHan, Yi, Ping Chen, Yanyu Zhang, Wenhua Lu, Wenwen Ding, Yao Luo, Shijun Wen, Ruihua Xu, Panpan Liu, and Peng Huang. 2019. "Synergy between Auranofin and Celecoxib against Colon Cancer In Vitro and In Vivo through a Novel Redox-Mediated Mechanism" Cancers 11, no. 7: 931. https://doi.org/10.3390/cancers11070931