Characterization of Driver Mutations in Anaplastic Thyroid Carcinoma Identifies RAS and PIK3CA Mutations as Negative Survival Predictors



Abstract
:1. Introduction
2. Results
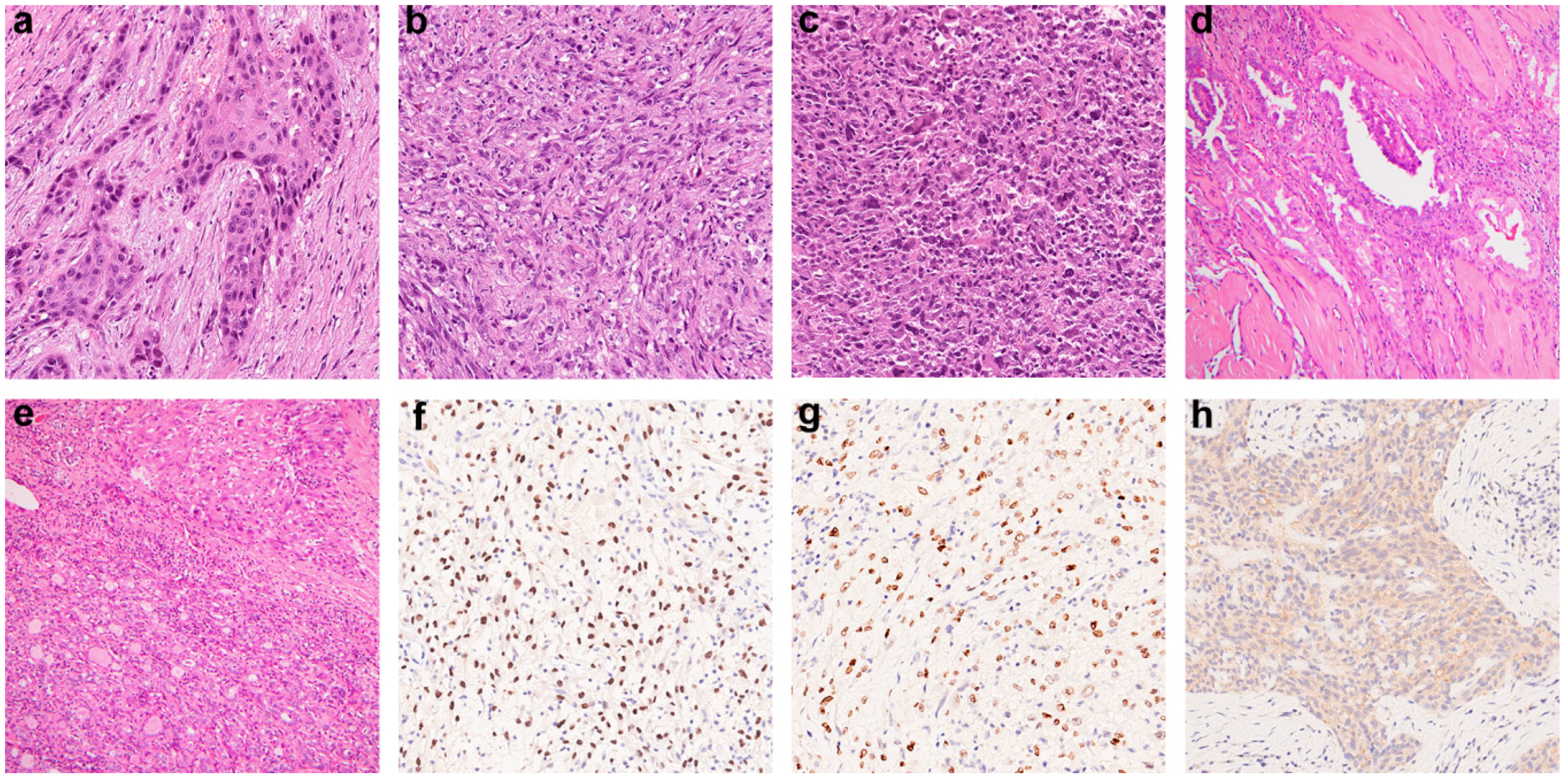
2.1. Clinicopathological Features of ATC
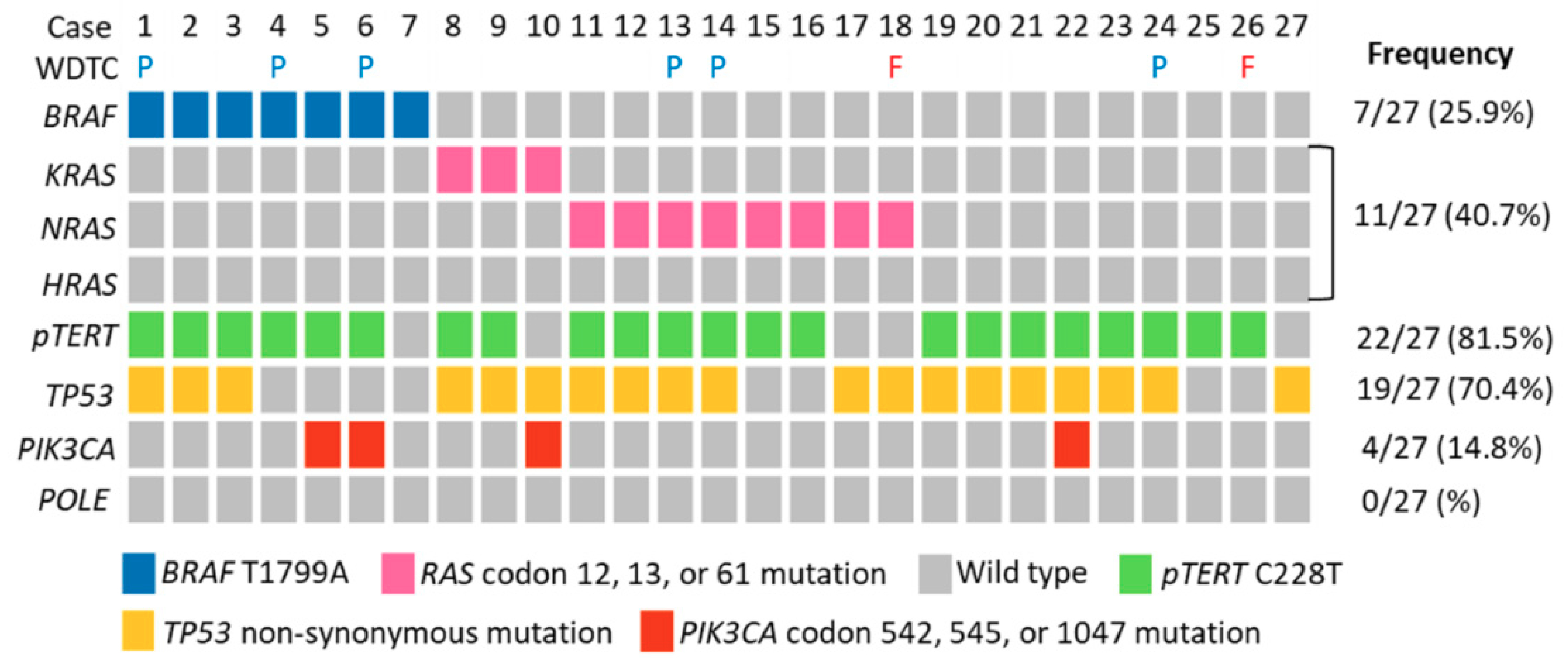
2.2. Molecular Profiles of ATC
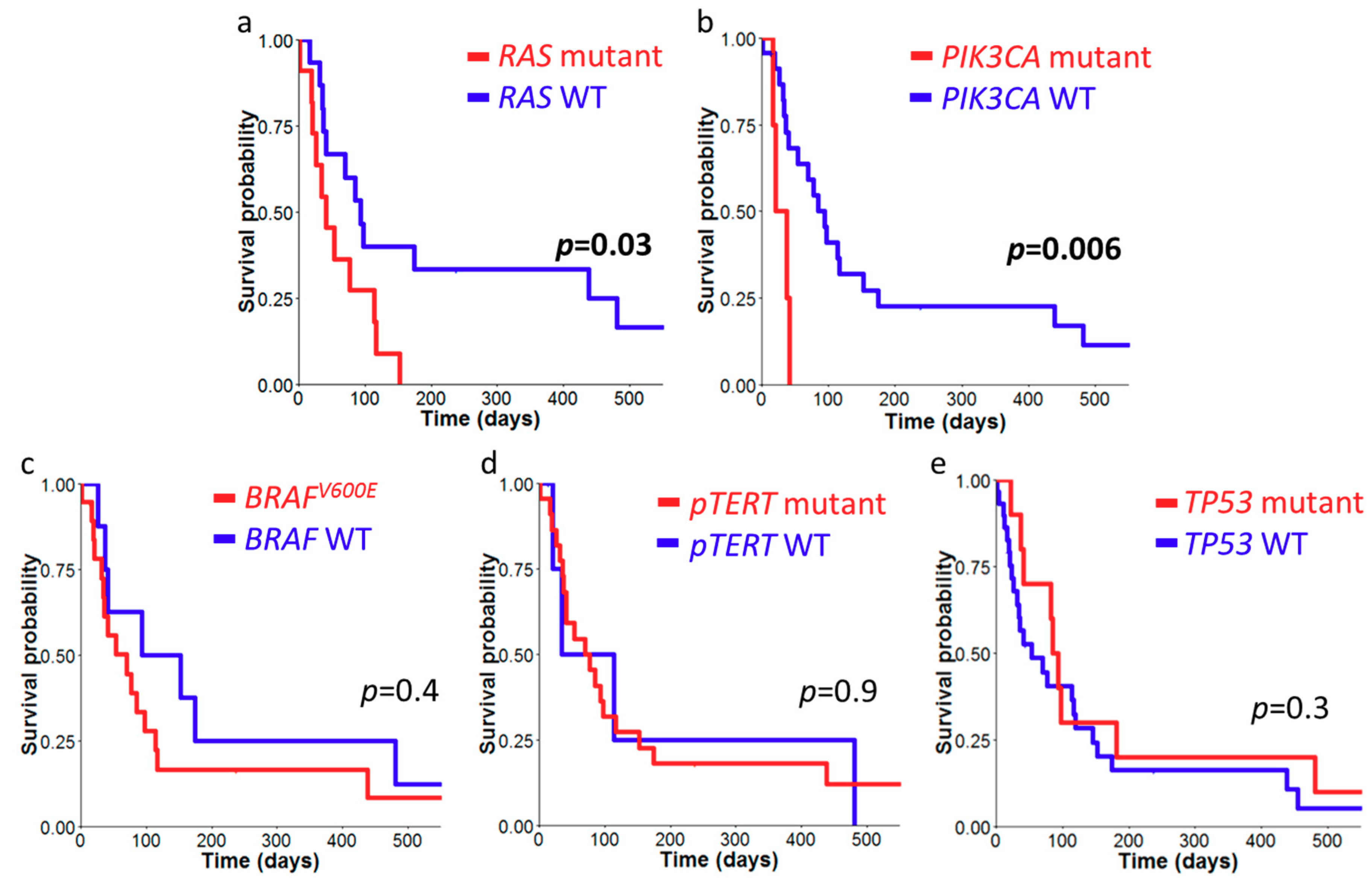
2.3. Survival Analysis of ATC Cohort
3. Discussion

{kind=link}
{kind=link}
{kind=link}
| References | Case No. | BRAFV600E | RAS | TP53 | TERT | PIK3CA | POLE | MMR Alternation b |
|---|---|---|---|---|---|---|---|---|
| Current Study | 27 | 25.9% | 40.7% | 70.4% | 81.5% | 14.8% | 0% | 0% |
| Xu 2020 [8] a | 102 | 43.1% | 22% | 63% | 75% | 18% | 4% | 8% |
| Khan 2019 [16] | 90 | 32% | 26% | 65.6% | 32% | 12.2% | na | na |
| Duan 2019 [10] | 25 | 56% | 24% | 60% | 56% | 44% | na | na |
| Yoo 2019 [9] | 27 | 40.1% | 44.4% | 48.1% | 55.6% | 11.1% | na | na |
| Ravi 2019 [33] | 11 | 18% | 18% | 55% | 36% | 18% | 9% | 9% |
| Chen 2018 [17] | 12 | 25% | 33% | 25% | na | 0% | na | na |
| Bonhomme 2017 [13] | 94 | 12.8% | 42.6% | 54.4% | 54% | 6.4% | na | na |
| Kunstman 2015 [15] | 22 | 27.3% | 27.3% | 27.3% | na | 9.1% | na | 13.6% |
4. Materials and Methods
4.1. Case Selection
4.2. Construction of Tissue Microarray and Immunohistochemistry
4.3. DNA Extraction
4.4. Targeted Next-Generation Sequencing (NGS)
4.5. Mass Spectrometry for PI3KCA Mutations
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, F.-C.; Lin, H.-T.; Lin, S.-F.; Kuo, C.-F.; Chung, T.-T.; Yu, H.-P. Nationwide cohort study on the epidemiology and survival outcomes of thyroid cancer. Oncotarget 2017, 8, 78429–78451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic thyroid carcinoma: From clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 2017, 13, 644–660. [Google Scholar] [CrossRef]
- Brignardello, E.; Gallo, M.; Baldi, I.; Palestini, N.; Piovesan, A.; Grossi, E.; Ciccone, G.; Boccuzzi, G. Anaplastic thyroid carcinoma: Clinical outcome of 30 consecutive patients referred to a single institution in the past 5 years. Eur. J. Endocrinol. 2007, 156, 425–430. [Google Scholar] [CrossRef]
- Kebebew, E.; Greenspan, F.S.; Clark, O.H.; Woeber, K.A.; McMillan, A. Anaplastic thyroid carcinoma. Treatment outcome and prognostic factors. Cancer 2005, 103, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-K.; Lee, S.; Kim, S.-j.; Jee, H.-G.; Kim, B.-A.; Cho, H.; Song, Y.S.; Cho, S.W.; Won, J.-K.; Shin, J.-Y.; et al. Comprehensive analysis of the transcriptional and mutational landscape of follicular and papillary thyroid cancers. PLoS Genet. 2016, 12, e1006239. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa Sylvia, L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Xu, B.; Fuchs, T.L.; Dogan, S.; Landa, I.; Katabi, N.; Fagin, J.A.; Tuttle, R.M.T.; Sherman, E.J.; Gill, A.; Ghossein, R. Dissecting anaplastic thyroid carcinoma (ATC): A comprehensive clinical, histologic, immunophenotypic, and molecular study of 360 cases. Thyroid 2020, in press. [Google Scholar] [CrossRef]
- Yoo, S.-K.; Song, Y.S.; Lee, E.K.; Hwang, J.; Kim, H.H.; Jung, G.; Kim, Y.A.; Kim, S.-j.; Cho, S.W.; Won, J.-K.; et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat. Commun. 2019, 10, 2764–2775. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.; Li, Y.; Hu, P.; Gao, J.; Ying, J.; Xu, W.; Zhao, D.; Wang, Z.; Ye, J.; Lizaso, A.; et al. Mutational profiling of poorly differentiated and anaplastic thyroid carcinoma by the use of targeted next-generation sequencing. Histopathology 2019, 75, 890–899. [Google Scholar] [CrossRef]
- Deeken-Draisey, A.; Yang, G.-Y.; Gao, J.; Alexiev, B.A. Anaplastic thyroid carcinoma: An epidemiologic, histologic, immunohistochemical, and molecular single-institution study. Hum. Pathol. 2018, 82, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Oishi, N.; Kondo, T.; Ebina, A.; Sato, Y.; Akaishi, J.; Hino, R.; Yamamoto, N.; Mochizuki, K.; Nakazawa, T.; Yokomichi, H.; et al. Molecular alterations of coexisting thyroid papillary carcinoma and anaplastic carcinoma: Identification of TERT mutation as an independent risk factor for transformation. Mod. Pathol. 2017, 30, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, B.; Godbert, Y.; Perot, G.; Al Ghuzlan, A.; Bardet, S.; Belleannée, G.; Crinière, L.; Do Cao, C.; Fouilloux, G.; Guyetant, S.; et al. Molecular pathology of anaplastic thyroid carcinomas: A retrospective study of 144 cases. Thyroid 2017, 27, 682–692. [Google Scholar] [CrossRef]
- Jeon, M.J.; Chun, S.-M.; Kim, D.; Kwon, H.; Jang, E.K.; Kim, T.Y.; Kim, W.B.; Shong, Y.K.; Jang, S.J.; Song, D.E.; et al. Genomic alterations of anaplastic thyroid carcinoma detected by targeted massive parallel sequencing in a BRAF V600E mutation-prevalent area. Thyroid 2016, 26, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Kunstman, J.W.; Juhlin, C.C.; Goh, G.; Brown, T.C.; Stenman, A.; Healy, J.M.; Rubinstein, J.C.; Choi, M.; Kiss, N.; Nelson-Williams, C.; et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum. Mol. Genet. 2015, 24, 2318–2329. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Ci, B.; Xie, Y.; Gerber, D.E.; Beg, M.S.; Sherman, S.I.; Cabanillas, M.E.; Busaidy, N.L.; Burtness, B.A.; Heilmann, A.M.; et al. Unique mutation patterns in anaplastic thyroid cancer identified by comprehensive genomic profiling. Head Neck 2019, 41, 1928–1934. [Google Scholar] [CrossRef]
- Chen, H.; Luthra, R.; Routbort, M.J.; Patel, K.P.; Cabanillas, M.E.; Broaddus, R.R.; Williams, M.D. Molecular profile of advanced thyroid carcinomas by next-generation sequencing: Characterizing tumors beyond diagnosis for targeted therapy. Mol. Cancer Ther. 2018, 17, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Ghossein, R. Genomic landscape of poorly differentiated and anaplastic thyroid carcinoma. Endocr. Pathol. 2016, 27, 205–212. [Google Scholar] [CrossRef]
- Haroon Al Rasheed, M.R.; Xu, B. Molecular alterations in thyroid carcinoma. Surg. Pathol. Clin. 2019, 12, 921–930. [Google Scholar] [CrossRef]
- Acquaviva, G.; Visani, M.; Repaci, A.; Rhoden, K.J.; de Biase, D.; Pession, A.; Giovanni, T. Molecular pathology of thyroid tumours of follicular cells: A review of genetic alterations and their clinicopathological relevance. Histopathology 2018, 72, 6–31. [Google Scholar] [CrossRef]
- Wong, K.S.; Lorch, J.H.; Alexander, E.K.; Nehs, M.A.; Nowak, J.A.; Hornick, J.L.; Barletta, J.A. Clinicopathologic features of mismatch repair-deficient anaplastic thyroid carcinomas. Thyroid 2019, 29, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Laird, P.W.; Park, P.J. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 2013, 155, 858–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600–mutant anaplastic thyroid cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.R.; Zafereo, M.E.; Dadu, R.; Ferrarotto, R.; Busaidy, N.L.; Lu, C.; Ahmed, S.; Gule-Monroe, M.K.; Williams, M.D.; Sturgis, E.M.; et al. Complete surgical resection following neoadjuvant dabrafenib plus trametinib in BRAFV600E-mutated anaplastic thyroid carcinoma. Thyroid 2019, 29, 1036–1043. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the “undruggable”. Biochim. Biophys. Acta 2020, 1867, 118570. [Google Scholar] [CrossRef] [PubMed]
- Yakushina, V.D.; Lerner, L.V.; Lavrov, A.V. Gene fusions in thyroid cancer. Thyroid 2017, 28, 158–167. [Google Scholar] [CrossRef]
- ElMokh, O.; Ruffieux-Daidié, D.; Roelli, M.A.; Stooss, A.; Phillips, W.A.; Gertsch, J.; Dettmer, M.S.; Charles, R.-P. Combined MEK and PI3-kinase inhibition reveals synergy in targeting thyroid cancer in vitro and in vivo. Oncotarget 2017, 8, 24604–24620. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; de la Chapelle, A.; Pritchard, C.C. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 2014, 147, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Wang, C.; Lee, P.P.; Chu, P.; Fakih, M. Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J. Natl. Compr. Cancer Netw. 2017, 15, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinopoulos, P.A.; Luo, W.; Liu, J.F.; Gulhan, D.C.; Krasner, C.; Ishizuka, J.J.; Gockley, A.A.; Buss, M.; Growdon, W.B.; Crowe, H.; et al. Phase II study of avelumab in patients with mismatch repair deficient and mismatch repair proficient recurrent/persistent endometrial cancer. J. Clin. Oncol. 2019, 37, 2786–2794. [Google Scholar] [CrossRef] [PubMed]
- Ravi, N.; Yang, M.; Gretarsson, S.; Jansson, C.; Mylona, N.; Sydow, S.R.; Woodward, E.L.; Ekblad, L.; Wennerberg, J.; Paulsson, K. Identification of targetable lesions in anaplastic thyroid cancer by genome profiling. Cancers 2019, 11, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.-C.; et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, R.V.; Osamura, R.Y.; Klöppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Santarpia, L.; El-Naggar, A.K.; Cote, G.J.; Myers, J.N.; Sherman, S.I. Phosphatidylinositol 3-kinase/Akt and Ras/Raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J. Clin. Endocr. Metab. 2008, 93, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Church, D.N.; Briggs, S.E.W.; Palles, C.; Domingo, E.; Kearsey, S.J.; Grimes, J.M.; Gorman, M.; Martin, L.; Howarth, K.M.; Hodgson, S.V.; et al. DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013, 22, 2820–2828. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- García-Rostán, G.; Costa, A.M.; Pereira-Castro, I.; Salvatore, G.; Hernandez, R.; Hermsem, M.J.A.; Herrero, A.; Fusco, A.; Cameselle-Teijeiro, J.; Santoro, M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005, 65, 10199–10207. [Google Scholar] [CrossRef] [Green Version]



| Age, n (%) | ||
| ≤55 | 4 (10.3) | |
| >55 | 35 (89.7) | |
| Gender, n (%) | ||
| Female | 20 (51.3) | |
| Male | 19 (48.7) | |
| Stage, n (%) a | ||
| IVA | 5 (13.9) | |
| IVB | 11 (30.6) | |
| IVC | 20 (55.6) | |
| Surgery, n (%) | ||
| Subtotal thyroidectomy | 9 (23.1) | |
| Total thyroidectomy | 24 (61.5) | |
| Biopsy only | 6 (15.4) | |
| Other Therapies b, n (%) | ||
| Chemotherapy | 7 (24.1) | |
| Radioiodine | 6 (21.4) | |
| External radiation | 7 (25) | |
| Targeted therapy | 5 (15.1) | |
| Tumor size c (cm) | 7.14 | |
| Histologic Pattern, n (%) | ||
| Epithelial | + | 28 (71.8%) |
| - | 11 (28.2%) | |
| Giant cell | + | 5 (12.8%) |
| - | 34 (87.2%) | |
| Sarcomatoid | + | 17 (43.6%) |
| - | 22 (56.4%) | |
| Coexisting WDTC, n (%) | ||
| PTC | + | 10 (25.6) |
| - | 29 (74.4) | |
| FTC | + | 3 (7.7) |
| - | 36 (92.3) | |
| Case Number | n (%) | ||||
|---|---|---|---|---|---|
| PAX8 | TTF-1 | BRAF VE1 | Retained MMR Proteins | ||
| All | 39 | 22 (56.4) | 2 (5.1) | 10 (25.6) | 39 (100) |
| Coexisting WDTC | |||||
| None | 26 | 15 (5.8) | 2 (7.7) | 4 (15.4) | 26 (100) |
| PTC | 10 | 7 (70) | 0 | 6 (60) | 10 (100) |
| FTC | 3 | 0 | 0 | 0 | 3 (100) |
| BRAF | RAS | Non-BRAF/RAS | p Value | ||
|---|---|---|---|---|---|
| Case Number | 7 | 11 | 9 | ||
| Age, n (%) | |||||
| ≤55 | 1 (14.3) | 1 (9.1) | 2 (22.2) | 0.803 | |
| >55 | 6 (85.7) | 10 (90.9) | 7 (77.8) | ||
| Gender, n (%) | |||||
| Female | 2 (28.6) | 10 (90.9) | 4 (44.4) | 0.012 * | |
| Male | 5 (71.4) | 1 (9.1) | 5 (55.6) | ||
| Stage, n (%) a | |||||
| IVA | 1 (14.3) | 0 (0) | 2 (25) | 0.445 | |
| IVB | 3 (42.9) | 4 (44.4) | 1 (12.5) | ||
| IVC | 3 (42.9) | 5 (55.6) | 5 (50) | ||
| Surgery, n (%) | |||||
| Subtotal Tx | 1 (14.3) | 2 (18.2) | 4 (44.4) | 0.488 | |
| Total Tx | 6 (85.7) | 8 (72.7) | 4 (44.4) | ||
| Biopsy only | 0 | 1 (9.1) | 1 (11.1) | ||
| Other Therapies b, n (%) | |||||
| Chemotherapy | 1 (16.7) | 3 (33.3) | 2 (33.3) | 0.851 | |
| Radioiodine | 1 (16.7) | 2 (22.2) | 0 (0) | 0.763 | |
| External radiation | 1 (16.7) | 4 (44.4) | 1 (20) | 0.568 | |
| Targeted therapy | 2 (33.3) | 1 (10) | 0 (0) | 0.314 | |
| Tumor size, cm | 5.1 | 7.8 | 7.5 | 0.253 | |
| Histologic Pattern, n (%) | |||||
| Epithelial | + | 6 (85.7) | 8 (72.7) | 6 (66.7) | 0.860 |
| - | 1(14.3) | 3 (27.3) | 3 (33.3) | ||
| Giant cell | + | 1 (14.3) | 1 (9.1) | 1 (11.1) | 1 |
| - | 6 (85.7) | 10 (90.9) | 8 (88.9) | ||
| Sarcomatoid | + | 1 (14.3) | 5 (45.5) | 7 (77.8) | 0.045 * |
| - | 6 (85.7) | 6 (54.5) | 2 (22.2) | ||
| Coexisting WDTC, n (%) | |||||
| PTC | + | 3 (42.9) | 2 (18.2) | 1 (11.1) | 0.369 |
| - | 4 (57.1) | 9 (81.8) | 8 (88.9) | ||
| FTC | + | 0 (0) | 1 (9.1) | 1 (11.1) | 1.000 |
| - | 7 (100) | 10 (90.9) | 8 (88.9) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, W.-A.; Liu, C.-Y.; Lin, S.-Y.; Chen, C.-C.; Hang, J.-F. Characterization of Driver Mutations in Anaplastic Thyroid Carcinoma Identifies RAS and PIK3CA Mutations as Negative Survival Predictors. Cancers 2020, 12, 1973. https://doi.org/10.3390/cancers12071973
Lai W-A, Liu C-Y, Lin S-Y, Chen C-C, Hang J-F. Characterization of Driver Mutations in Anaplastic Thyroid Carcinoma Identifies RAS and PIK3CA Mutations as Negative Survival Predictors. Cancers. 2020; 12(7):1973. https://doi.org/10.3390/cancers12071973
Chicago/Turabian StyleLai, Wei-An, Chih-Yi Liu, Shih-Yao Lin, Chien-Chin Chen, and Jen-Fan Hang. 2020. "Characterization of Driver Mutations in Anaplastic Thyroid Carcinoma Identifies RAS and PIK3CA Mutations as Negative Survival Predictors" Cancers 12, no. 7: 1973. https://doi.org/10.3390/cancers12071973