Novel Targeted Therapies for Metastatic Thyroid Cancer—A Comprehensive Review


Thyroid Tumors and Functional Thyroid Disorders Section, Metabolic Disease Branch, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20814, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cancers 2020, 12(8), 2104; https://doi.org/10.3390/cancers12082104
Submission received: 14 July 2020
/
Revised: 22 July 2020
/
Accepted: 25 July 2020
/
Published: 29 July 2020
(This article belongs to the Special Issue Thyroid Cancer Metastases (Biological and Clinical Aspects))
Abstract
:The knowledge on thyroid cancer biology has grown over the past decade. Thus, diagnostic and therapeutic strategies to manage thyroid cancer are rapidly evolving. With new insights into tumor biology and cancer genetics, several novel therapies have been approved for the treatment of thyroid cancer. Tyrosine kinase inhibitors (TKIs), such as lenvatinib and sorafenib, have been successfully utilized for the treatment of radioactive iodine (RAI)-refractory metastatic differentiated thyroid cancer (DTC). In addition, pretreatment with mitogen-activated protein kinase (MAPK) inhibitors (trametinib and selumetinib) has been shown to restore RAI avidity in previously RAI-refractory DTCs. Local therapies, such as external beam radiation and radiofrequency/ethanol ablation, have also been employed for treatment of DTC. Vandetanib and cabozantinib are the two TKIs currently approved by the Food and Drug Administration (FDA) for the treatment of medullary thyroid cancer (MTC). Other novel therapies, such as peptide receptor radionuclide therapy and carcinoembryonic antigen (CEA) vaccine, have also been utilized in treating MTC. Ongoing trials on selective rearranged-during-transfection (RET) protooncogene inhibitors, such as LOXO-292 and BLU-667, have demonstrated promising results in the treatment of metastatic MTC resistant to non-selective TKIs. The FDA-approved BRAF/MEK inhibitor combination of dabrafenib and trametinib has revolutionized treatment of BRAFV600E mutation positive anaplastic thyroid cancer. Several other emerging classes of medications, such as gene fusion inhibitors and immune checkpoint inhibitors, are being actively investigated in several clinical trials. In this review, we describe the molecular landscape of thyroid cancer and novel targeted therapies and treatment combinations available for the treatment of metastatic thyroid cancer.
1. Introduction
Thyroid cancer is predicted to affect 52,890 new patients in the USA in 2020, with the incidence being three times higher in women as compared with men [1]. Until recently, thyroid cancer was the most rapidly increasing cancer in the USA, largely, but not solely, due to increasing use of sensitive diagnostic procedures. However, the increase of about 7% per year during the 2000s has slowed to 2% per year in men, and rates have stabilized in women from the period of 2012 to 2016, as a result of the more conservative diagnostic approach that has been recently implemented [1]. Differentiated thyroid cancer (DTC) that arises from follicular cells is further subclassified into papillary thyroid cancer (PTC), which is the most common histological type, follicular thyroid cancer (FTC), and Hürthle cell cancer (HTC). De-differentiated thyroid cancer is classified as poorly differentiated thyroid cancer (PDTC) and anaplastic thyroid cancer (ATC). Medullary thyroid cancer (MTC) arises from neuroendocrine C cells that are derived from the neural crest [2].
The standard of care for DTC consists of surgery (lobectomy or total/near-total thyroidectomy, with or without lymph node dissection), dependent on the tumor size, extrathyroidal extension, and metastatic potential. Very low-risk PTCs can be serially followed with active surveillance, using neck ultrasound (US) without surgical intervention, if the size remains stable over time [3].
Following surgery, all patients classified as high-risk for persistent/recurrent disease with evidence of radioactive iodine (RAI)-avid metastases, as well as patients classified as intermediate risk for persistence or recurrence, receive RAI treatment, as it has shown to decrease mortality and/or morbidity, respectively [3,4]. Metastatic RAI-non-avid disease carries a worse prognosis, as the five-year survival is estimated to be as low as 10% [3].
In contrast to DTC, all patients with MTC undergo total thyroidectomy with or without lymph node dissection. Patients with MTC do not benefit from RAI therapy, as the neuroendocrine tumors lack the sodium-iodide (NIS) symporter necessary to incorporate RAI within the cell [5]. External beam radiation to the neck may be used for persistent or residual local disease [5]. The overall 10-year survival for MTC confined to the thyroid gland is 95.6% but is as low as 40% for patients with distant metastatic disease at the time of diagnosis [6].
Since standard RAI therapy for DTC is not effective in 5–22% of patients, and surgical treatment is not curative in patients with MTC presenting with metastatic disease, small molecules have been developed that target aberrant signaling pathways specifically found in thyroid cancer [3]. Therefore, understanding of the molecular landscape of thyroid cancer is crucial to provide individualized targeted therapies.
2. Molecular Landscape of Thyroid Cancer
Dysregulation of multiple signaling pathways, such as mitogen-activated protein kinase (MAPK), phosphoinositide-3-kinase (PI3K), receptor tyrosine kinase (RTK), and Wingless/Integrated (WNT), has been reported to contribute toward the pathogenesis of thyroid cancer and is generally associated with the genetic alterations of genes involved in these pathways [7]. The most common driver genetic alterations observed in DTC include mutations in murine sarcoma viral oncogene homolog B (BRAF), rat sarcoma (RAS), and RET (rearranged-during-transfection)/PTC rearrangements; in PDTC and ATC, TP53 (tumor protein p53) mutations are often observed; and in MTC, point mutations in RET oncogene and pathogenic RAS variants are common. These genetic alterations are affecting MAPK and PI3K signaling [8]. In this section, we discuss the molecular alterations that have been identified in different types of thyroid cancers, including DTC (PTC, FTC, and HTC), ATC, PDTC, and MTC.
2.1. Molecular Alterations in MAPK and PI3K Signaling Pathway
The most common genetic alterations associated with MAPK and PI3K signaling pathways in thyroid cancer include mutations in BRAF, RAS, PTEN (Phosphatase and tensin homolog), AKT (Protein Kinase B), and PI3KCA (Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha) [7] (Figure 1 and Table 1). BRAFV600E is the predominant mutation in PTC (18–87%) but has also been detected in PDTC, ATC, and HTC [9,10]. Other rarely detected BRAF variants include BRAFK601E mutation, large insertion/deletions, fusions, and small deletions [11]. RAS mutations also have been detected with a prevalence rate of 30–50% in FTC, 30–45% in follicular-variant of PTC (FVPTC), 15% in HTC, 20–40% in PDTC and ATC, and 10–30% in MTC and rarely in classical PTC patients [7,10,12,13,14,15]. The most common RAS mutations occur at codons 12, 13, and 61, which are considered mutational hot-spot regions and may affect any of the three RAS genes—HRAS, KRAS, and NRAS [16]. The most predominant RAS mutation to be detected in thyroid neoplasms is NRAS61 [17].
BRAF and RAS mutations are mutually exclusive genetic alterations, suggesting that the presence of either of these genetic variants is sufficient for thyroid tumorigenesis [40] (Figure 1). Based on the differences of the gene profile of the BRAFV600E and RAS mutant tumors, a BRAFV600E-RAS score (BRS) has been developed that can categorize PTCs into either BRAFV600E-like PTCs or RAS-like PTCs [11].
Members of the PI3K pathway that are mutated in thyroid cancer include PTEN, AKT, and PIK3CA. However, the frequency of their presence is low [41] (Figure 1 and Table 1). PTEN is a tumor suppressor gene, and mutations, deletions, and epigenetic changes leading to its loss of expression have been observed in DTC, PDTC, ATC, and HTC [7,10,11,42]. Somatic single nucleotide variants (SSNVs) of AKT1, AKT2, and AKT3 have been detected in thyroid cancer at a very low frequency [11,43]. An AKT1 (G49A) mutation has been seen in thyroid cancer metastasis, suggesting that this mutation arises late during progression [44]. Copy number gain of AKT1 has also been reported in ATC and FTC [21]. The genetic alterations reported in the PIK3CA gene in thyroid cancer include gene amplification and, less commonly, mutations [45]. As in AKT1, genetic alterations in PIK3CA appear late during malignant progression of thyroid cancer and are more common in ATC than DTC [44,45] (Figure 1 and Table 1).
In MTCs, the most common genetic alteration related to MAPK/PI3K signaling pathways is RAS mutation. Mutations in KRAS and HRAS genes have been identified in RET wild-type sporadic MTCs and are mutually exclusive events in MTCs [46].
2.2. Molecular Alterations in RTK Signaling Pathway
RTKs play a crucial role in the regulation of processes such as proliferation, differentiation, and metabolism, and genetic alterations in RTKs have been reported frequently in cancer [33]. In thyroid cancer, the RET proto-oncogene is one of the most common RTKs to be altered. The most prevalent genetic alterations of RET in PTC are RET–PTC fusions. The RET/PTC rearrangements are the predominant genetic alterations in sporadic childhood DTCs and in radiation-related DTCs [47]. The RET/PTC1 rearrangement was a common oncogenic alteration to be observed in childhood thyroid cancer cases after the Chernobyl nuclear power plant accident, which resulted in radiation exposures [47]. Moreover, RET fusions have been observed in almost 50% of atomic bomb survivors with PTCs who had high radiation exposure [48].
Various RET fusions have been reported with genes such as CCCD6 (Coiled Coil Domain Containing 6), NCOA4 (Nuclear Receptor Coactivator 4), ERC1 (ELKS/RAB6-Interacting/CAST Family Member 1), AKAP13 (A-Kinase Anchoring Protein 13), FKBP15 (FKBP Prolyl Isomerase 15), SPECC1L (Sperm Antigen With Calponin Homology And Coiled-Coil Domains 1 Like), TBL1XR1 (Transducin Beta Like 1 X-Linked Receptor 1), and TRIM27 (Tripartite Motif Containing 27) [7,11]. The genetic alterations in RET can lead to constitutive RET activation and stimulation of the MAPK and PI3K signaling pathways [7].
Neurotrophic receptor tyrosine kinase (NTRK) rearrangements have been reported in PTCs. NTRKs are receptor tyrosine kinases encoded by the NTRK1, NTRK2, and NTRK3 genes [11,49,50]. The NTRK fusion proteins are phosphorylated at tyrosine residues, leading to the constitutive tyrosine-kinase activity and stimulation of MAPK, PI3K, and phospholipase C-signaling pathway [7,49,50] (Figure 1 and Table 1).
Mutations and gene rearrangements have also been reported in anaplastic lymphoma kinase (ALK) gene in thyroid cancer. The ALK gene fusions are mainly observed in ATC and PDTC, but are also present in PTC, at low frequencies [7,51]. Two ALK mutations (C3592T and G3602A) have been identified in ATC. These mutations lead to amino acid changes (L1198F and G1201E) within the ALK tyrosine kinase domain, resulting in increased tyrosine kinase activities [31]. The ALK-associated genetic alterations activate the downstream MAPK and PI3K signaling pathways [7,31] (Figure 1 and Table 1).
Other RTK genetic alterations include FGFR2 (Fibroblast Growth Factor Receptor 2) gene fusions with OFD1 (Oral-Facial-Digital Syndrome 1) and VCL (Vinculin) in PTC [11]; missense mutations in EGFR (Epidermal Growth Factor Receptor), FGFR2, and FLT3 (Fms-Relat.ed Receptor Tyrosine Kinase 3) in PDTC [32]; and copy number gains in EGFR, PDGFRα (Platelet-Derived Growth Factor Receptor Alpha), PDGFRβ (Platelet-Derived Growth Factor Receptor Beta), VEGFR1 (Vascular endothelial growth factor receptor 1), VEGFR2 (Vascular endothelial growth factor receptor 2), KIT (KIT Proto-Oncogene), and MET (MET Proto-Oncogene) in ATC and FTC [21]. In HTC, mutations of various RTK genes (EGFR, ERBB2-Erb-B2 Receptor Tyrosine Kinase 2, PDGFR, MET, and RET) were observed in 20% of the tumors [10].
In MTCs, the most common RTK alteration is the RET gain of function point mutations in the RET oncogene. MTC can be sporadic (70–80% cases) or hereditary (20–30% cases), and about 40–70% of the sporadic and 95% of the hereditary cases harbor mutations in the RET oncogene [28,29]. The RET mutations associated with MTC occur at the cysteine-rich or tyrosine kinase domains located within the seven exons (8, 10, 11, 13, 14, 15, and 16) [52]. These mutations can lead to one of the three clinically distinctive subtypes of hereditary MTC—multiple endocrine neoplasia (MEN) type 2A (MEN2A), and MEN2B, and familial MTC [53]. The most frequent RET point mutation to be detected in 85–90% of MEN2A patients and a small subset of sporadic MTC patients occurs at codon 634 [28,52]. On the contrary, the most commonly observed RET mutation in 95% of the MEN2B and 75–95% of the sporadic cases is M918T [54]. To this date, several RET mutations have been identified in sporadic and hereditary MTC. It has been shown that different RET mutations can produce distinct phenotypes that differ in terms of the aggressiveness of MTC, age of onset of MTC, and the presence/absence of other endocrine malignancies [52]. Based on the genotype–phenotype associations, the RET mutations have been stratified into three risk groups (highest risk, high risk, and moderate risk) that reflects the aggressiveness of MTC [55]. The highest risk RET mutation is M918T mutation, and the patients with this mutation develop the aggressive form of MTC and metastasis at a very young age [55]. The RET mutations that fall in the high-risk group include mutations in codons 634 and 883, and the patients with these mutations are at risk of developing an aggressive form of MTC at an early age [55]. The moderate risk RET mutations occur at codons 533, 609, 611, 618, 620, 790, 804, and 891, and the patients with these mutations typically show a less aggressive form of MTC in comparison with the high-risk and highest-risk groups and later onset [55]. Moreover, RET and ALK gene fusions have also been observed in MTC at low frequencies [51,56].
Receptor tyrosine kinases (RTKs) are upstream of the MAPK and PI3K pathways. The mutations in the RTKs and RTK-gene fusions can alter the downstream RAS/MAPK and PI3K/AKT pathways, leading to oncogenic transformation/progression. Similarly, the mutations in the components of the MAPK and PI3K pathways can promote tumorigenesis. Genes that have shown to be mutated in thyroid cancer are shown in red with an asterisk.
2.3. Other Molecular Alterations
PAX8-PPARγ (Paired Box 8-Peroxisome Proliferator Activated Receptor Gamma) rearrangement is present in 30–35% of FTC and 2–13% of follicular adenomas (FA) [36,57]. Besides this, PAX8-PPARγ gene fusion also appears with variable frequency in the FVPTC [36,57]. This gene fusion can act as a dominant-negative activator of PPAR-γ and can act as a transcriptional activator of a subset of PPARγ-inducible genes leading to its oncogenic effects [36] (Figure 2).
TP53 mutations leading to the inactivation of this tumor suppressor gene are common in ATC (50–80%) and PDTC (10–35%), but have been detected with variable frequency in FTC and PTC as well [7,35] (Figure 2, Table 1). These mutations usually occur between exons 5–8 and promote tumor development as well as progression [35]. Telomerase reverse transcriptase (TERT) promoter mutations are also detected in ATC (40–70%), PDTC (40%), an aggressive form of HTC (32%), PTC (10%) and FTC (20%) [7,10,37], (Figure 2, Table 1). The other common mutations in ATC and PDTC include mutations of the components of the Wnt pathway [34]. This includes the gain of function mutations of CTNNB1 (β-catenin gene) and loss of function mutations of the Axin1 gene (Figure 2 and Table 1). The mutations in β-catenin promote its nuclear localization and may increase transcription [34]. The other genetic alterations to be predominantly observed in less differentiated thyroid cancer include mutations in the members of the DNA Mismatch Repair pathway (MSH2—MutS Homolog 2, MSH6—MutS Homolog 6, and MLH1—MutL Homolog 1); members of the SWI-SNF chromatin remodeling complex; eukaryotic translation initiation factor 1A (EIF1AX); histone methyl-transferases (HMTs); and isocitrate dehydrogenase 1 (IDH1) [7] (Figure 2 and Table 1).
A comprehensive analysis of the genomic landscape of HTC was recently performed which revealed unique genetic alterations in addition to the common genetic alterations that are seen in other types of thyroid cancer [10]. The common genetic alterations in HTC include mutations in mitochondrial genes, epigenetic modifiers, and components of DNA damage and repair pathway; chromosome 5 and 7 duplications; loss of heterozygosity; and in-frame gene fusions [10].
The molecular landscape of thyroid cancer forms a basis for the implementation of targeted therapies.
2.4. Mechanisms of RAI-Refractoriness in DTC
RAI-refractoriness is the loss of ability of the DTC cells to take up and concentrate radioactive 131I, which is necessary to eliminate foci of persistent/recurrent disease [58]. Approximately 5–15% of DTC patients become refractory to RAI by losing the expression of sodium iodide symporter (NIS), which occurs as a result of the loss of thyroid differentiation features [58,59]. There are multiple mechanisms responsible for the loss of NIS expression on the cell membrane of the thyroid cancer cells. In the thyroid, TSH regulates NIS expression through stimulation of TSHR, which activates adenylyl cyclase and results in the accumulation of cyclic AMP (cAMP) within thyroid cells (Figure 3) [60]. This increase in cAMP induces NIS transcription by stimulating thyroid-specific transcriptional factors (TTFs), like paired box 8 (PAX8) [59,60]. Studies have suggested that aberrant activation of MAPK/ERK and PI3K/AKT pathways are responsible for the TTF repression and loss of NIS expression (Figure 3) [59]. The presence of BRAFV600E mutation has been strongly correlated to the loss of NIS expression and RAI refractoriness in PTC patients [61]. BRAF activation has been shown to repress PAX8 binding to the NIS promoter by activating TGFβ (transforming growth factor β)/Smad3 signaling [62,63]. In addition, activated BRAF can also promote NIS silencing by driving histone deacetylation of the NIS promoter [63,64]. Targeted treatment with BRAF and MAPK inhibitors have been shown to re-sensitize thyroid cancer cells/tumors to RAI therapy by improving NIS expression (Figure 3) [65,66,67].
Activation of the PI3K–AKT pathway has also been correlated to loss of NIS expression. Various growth factors, such as insulin, insulin-like growth factor 1 (IGF-1), hepatocyte growth factor (HGF), or epidermal growth factor (EGF), leading to PI3K activation and AKT phosphorylation, downregulated NIS expression in thyrocytes [68]. Conversely, inhibition of the PI3K pathway promoted NIS expression in rat thyroid cells and human PTC cells [69]. Activation of mTOR—a downstream target of PI3K/AKT pathway—also resulted in decreased NIS expression in thyrocytes, thus suggesting the involvement of the PI3K–AKT pathway in controlling NIS expression in thyroid cells (Figure 3) [68]. Similarly, RET/PTC rearrangement has also been reported to reduce NIS expression in thyroid cells [69]. In addition to this, the presence of TERT promoter mutations has been correlated to radioiodine refractoriness in distant metastatic DTC patients [70].
2.5. Progression from DTC to Advanced Thyroid Carcinomas
The integrative analysis of the genomic landscape of well-differentiated thyroid tumors revealed the presence of a low frequency of somatic alterations, with the majority of those tumors harboring mutually exclusive mutations in BRAF and RAS genes, as well as gene fusions that primarily involve RTKs [38]. The progression of DTC to advanced thyroid carcinomas involves the acquisition of mutational hits in either oncogenes or tumor suppressor genes. The molecular profiling of advanced thyroid carcinomas showed that the presence of mutations is a much more common event in comparison to well-differentiated thyroid tumors [71]. The predominant mutations in the advanced thyroid tumors were in the components of the MAPK and PI3K/AKT pathway [71]. BRAFV600E mutation was prevalent in advanced PTC, PDTC, and ATC, whereas NRAS mutations were common in advanced FTC and PDTC. The predominant tumor suppressor mutation occurred in TP53 and was prevalent in HTC, ATC, and PDTC [71]. The presence of co-mutations is also a frequent event in advanced thyroid cancers. Concomitant mutations in RET/PTC, RAS, and BRAF have been observed frequently in advanced PTC and were associated with poor prognosis [72]. In another study, the integrative analysis of aggressive thyroid carcinomas, including ATC and advanced DTC, revealed the frequent presence of TERT, AKT1, PIK3CA, and EIF1AX mutations in combination with BRAFV600E and RAS mutations in these advanced forms of thyroid cancers [73]. The most common oncogenic duet that has been investigated for its association with aggressive thyroid cancer phenotype is BRAFV600E and mutated TERT promoter. The presence of TERT promoter mutations in synergy with BRAF mutations has been associated with aggressive thyroid tumor characteristics, which include lymph node metastasis, distant metastasis, tumor recurrence, multifocality, extrathyroidal extension, and patient mortality [74,75,76,77]. The two parts of this oncogenic duet (BRAFV600E and mutated TERT promoter) have been shown to work cooperatively together to promote oncogenesis, leading to aggressive thyroid cancer phenotype [78]. The underlying mechanism involves increased expression of an E26 transformation-specific (ETS) transcription factor, GA-binding protein subunit beta (GABPβ) through BRAFV600E driven MAPK pathway leading to the formation of GABPα–GABPβ complex, which binds to the mutant TERT promoter and increases TERT expression [78]. Besides BRAFV600E, TERT promoter mutations have also been observed to coexist with RAS mutations in aggressive thyroid tumors; however, the clinical significance of this oncogenic duet is still not fully understood [37]. It is likely that RAS mutations work cooperatively together with TERT promoter mutations through the activated PI3K pathway, to promote aggressive tumor characteristics [37], but this needs to be investigated further.
3. Targeted Therapies for Thyroid Cancer
3.1. Tyrosine Kinase Inhibitors
3.1.1. DTC
In November 2013, sorafenib was the first tyrosine kinase inhibitor (TKI) to be approved by the Food and Drug Administration (FDA), for the treatment of progressive metastatic DTC refractory to RAI treatment. Sorafenib targets VEGFR 1-3, PDGFR, RET, FLT, and c-kit. The approval was based on the results of the DECISION trial, which was a phase III, multicenter, double-blind, placebo-controlled trial conducted in 417 patients with progressive DTC which failed standard treatment. Sorafenib was associated with a significantly longer median progression-free survival (PFS) (10.8 months) when compared to the placebo (5.8 months). The most common side effects encountered in patients treated with sorafenib were hand–foot skin reaction (76.3%), diarrhea (68.6%), alopecia (67.1%), and rash or desquamation (50.2%) [79,80].
In February 2015, a second TKI lenvatinib, which targets VEGFR2, VEGFR3, EGFR, PDGFR, KIT, and RET, was approved for the treatment of progressive DTC refractory to RAI, based on the SELECT trial—A phase III, randomized, double-blind, multicenter study involving 261 patients with progressive DTC. Lenvatinib was associated with a longer median PFS of 18.3 months vs. 3.6 months in the placebo group. Of the 20 deaths that occurred on lenvatinib, six were attributed to the treatment itself, as TKIs lead to QT interval prolongation and fatal tachyarrhythmias [81]. Adverse reactions led to dose reductions in 68% of patients receiving lenvatinib and discontinuation in 18% of patients [82]. Given lack of permanent complete remissions after therapy with the FDA-approved agents, the use of alternative TKIs, as well as combination therapies involving TKIs and mTOR inhibitors or immune checkpoint inhibitors, is being evaluated in several ongoing clinical trials [83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102] (Table 2).
Since BRAFV600E is one of the most common mutations in PTC, BRAF inhibitors such as vemurafenib or dabrafenib have been implemented in the management of thyroid cancer. Interestingly, a basket trial of vemurafenib in BRAFV600E mutation-positive tumors showed significant responses in patients with ATC [105]. However, thyroid cancers exhibit primary resistance to RAF kinase inhibition due to reduced negative feedback, and hence a combination of RAF and MEK kinase inhibitors is necessary to effect MAPK pathway inhibition [115] (Figure 1). An open-label phase II study of 16 patients with BRAFV600E-mutant ATC treated with dabrafenib in combination with the MEK inhibitor trametinib showed a remarkable overall response rate of 69% in patients with ATC, with one patient achieving complete response [106]. The 12-month duration of response, PFS, and overall survival were estimated at 90%, 79%, and 80%, respectively, a phenomenon not previously seen in ATC, which is usually characterized by overall survival of 3–6 months post-diagnosis [116]. This observation has revolutionized the management of this very aggressive cancer and led to the FDA approval of combination therapy with dabrafenib and trametinib in BRAFV600E-mutant ATC in May 2018 [106].
3.1.2. MTC
Two additional TKIs—vandetanib, which targets VEGFR2, VEGFR3, EGFR, KIT, and RET, and cabozantinib, which targets VEGFR2, RET, MET, FLT3, and AXL (AXL Receptor Tyrosine Kinase)—have been FDA-approved for the treatment of metastatic progressive MTC. The treatment with the first one was associated with a longer median PFS of 22.6 compared with 16.4 months in individuals exposed to placebo, while the latter led to a PFS of 11.2 months in the treated group vs. four months in the placebo arm in multicenter, randomized, double-blind phase III clinical trials [103,104,117,118]. Again, the side effect profile was similar to other TKIs and included QT prolongation and arrhythmias, severe hypertension leading to reversible posterior leukoencephalopathy syndrome, headaches, hand–foot syndrome, fistula formation, profound fatigue, decreased appetite, nausea, diarrhea, and abdominal pain [104]. In addition, interstitial lung disease and Stevens Johnson syndrome have been associated with use of vandetanib [104].
The large number of severe side effects of TKIs result from its ability to target multiple kinases (Figure 4) which play a role not only in cancer progression but also in many important physiological processes. Therefore, more targeted therapeutic strategies have been recently developed and include inhibition of RET-only in RET-mutated MTCs and PTCs and inhibition of BRAF-only in BRAFV600E-mutated tumors [106,115].
There are two RET inhibitors currently implemented in the management of RET mutation-positive tumors in the clinical trial setting—BLU-667 and selpercatinib (LOXO-292) (Table 2) [119,120,121,122]. BLU-667 inhibits the protein product of RETM918T, as well as RETV804L/M gatekeeper mutations conferring resistance to TKIs, while LOXO-292 is a highly selective RET kinase inhibitor with nanomolar potency against the canonical RET MTC drivers, RET gatekeeper mutations, and RET fusions [115]. Preliminary data from the ongoing clinical trials documented minimal side effects, excellent tolerability, and high efficacy, with objective response rates (complete and partial responses) ranging from 47% to 62% (Table 2) [115,119,120,121,122].
3.2. Therapies Targeting Gene Fusions
An additional development in the management of thyroid cancer which has been proven highly effective in preliminary clinical trials is the use of therapy aimed at targeting NTRK and ALK gene fusions (Table 2 and Figure 4). Larotrectinib is a selective inhibitor of tropomyosin receptor kinase (TRKA), TRKB, and TRKC which has been approved by the FDA for treatment of solid tumors with NTRK fusion [107,123]. The objective response rate in phase I and II clinical trials was remarkably high (80%) with 63% of patients with NTRK-positive tumors experiencing partial response and 13% complete response [107,123]. Most of the adverse events reported in the primary analysis were grade 1 and grade 2, with the most common being elevated ALT and AST (42%), fatigue (36%), vomiting (33%), dizziness (31%), nausea (31%), diarrhea (29%), and anemia (29%). (Table 2 and Figure 4).
Entrectinib is another selective inhibitor of TRKA, TRKB, and TRKC that also inhibits ALK and ROS1 tyrosine kinases. In August 2019, it received FDA approval for treatment of TRK-positive solid tumors, based on results of phase 1 and 2 clinical trials documenting an objective response rate of 57%, including a partial response in 50% and complete response in 7% [108] (Table 2 and Figure 4). The most common adverse events included dysgeusia (43%), dizziness (33%), constipation (33%), diarrhea (28%), and weight increase (26%) [108].
Unfortunately, at the moment, there are no clinical trials targeting THADA or other described above fusions in thyroid cancer, such as PAX8/PPARγ, apart from a one case report describing application of pioglitazone in PPARγ fusion protein positive metastatic HTC and resulting in the reduction of tumor size and improved pain control [124].
3.3. Restoration of RAI Uptake via MEK and BRAF Inhibition
Another important strategy implemented in the management of de-differentiated DTC and PDTC relies on upregulation of sodium iodide symporter NIS via inhibition of its negative regulators MEK and RAF signaling. This strategy re-enables incorporation of iodine within the cancer cell and thus consists of the pretreatment with MEK and BRAF inhibitors, followed by RAI therapy [125,126].
The landmark study by Ho et al. has proven the principle that the inhibition of MEK1 and MEK2 by selumetinib induces RAI uptake in RAI-non-avid DTC (Figure 3) [109]. Furthermore, an individualized approach consisting of evaluation of the amount of restored RAI uptake, as measured by 124I positron emission tomography/computed tomography (PET/CT)-based tumor dosimetry, has identified patients meeting the tumor accumulation threshold warranting RAI therapy. Among eight patients subsequently treated with RAI, five had a partial response and three had stable disease [109]. Interestingly, these preliminary data suggest a particularly high efficacy of this therapeutic approach in NRAS-mutated tumors. Given these promising results, there are several ongoing clinical trials utilizing selumetinib as an adjunct to RAI therapy in larger patients populations (Table 2) [127]. The second-generation MEK inhibitor trametinib is being evaluated in an ongoing phase II trial implementing pretreatment with trametinib, followed by 124I PET/CT-based lesional dosimetry and RAI therapy in patients with sufficiently restored RAI uptake (clinicaltrials.gov identifier NCT02152995) (Table 2).
Additional clinical trials utilize individualized approaches to patients with RAS-mutated and BRAF-mutated tumors (Table 2). Patients with tumors harboring an NRAS mutations are treated with the MEK inhibitor trametinib, while those with tumors characterized by BRAFV600E mutation are treated with combination therapies, including BRAF inhibitors such as dabrafenib and vemurafenib and MEK inhibitors (Figure 3). Preliminary data suggest high efficacy of such an approach with partial response to RAI therapy at three months’ follow-up observed in all patients who achieved sufficient restoration of RAI uptake [110]. A similar concept is being utilized in another ongoing multicentric prospective non-randomized phase II trial with two independent arms studying the use of trametinib for NRAS mutation-harboring tumors and dabrafenib for BRAFV600E-harboring tumors (clinicaltrials.gov identifier NCT03244956) (Table 2).
3.4. Peptide Receptor Radionuclide Therapy in Thyroid Cancer
Another therapeutic concept utilizing radiolabeled agents for treatment of metastatic thyroid cancer had been based on a subset of thyroid cancer expresses somatostatin receptors (SSTR) that could be targeted with peptide receptor radionuclide therapy (PRRT) [128,129].
Radiolabeled somatostatin receptor (SSTR) analogs, such as 68Ga-DOTATATE, which recognize predominantly SSTR type 2, are utilized for positron emission tomography/computed tomography (PET/CT) imaging. Of note, 68Ga-DOTATATE was FDA approved in June 2016 for clinical use for imaging of patients with neuroendocrine tumors, including MTC. Identification of patients with RAI-non-avid DTC and metastatic MTC characterized by positive 68Ga-DOTATATE uptake enables identification of individuals who may benefit from PRRT. There were small pilot studies performed in Europe that documented the utility of treatment of progressive RAI-non-avid metastatic DTC and MTC with SSTR agonists radiolabeled with 177Lutetium or 86Ytrium. The overall response rate was similar to current standard-of-care therapy with TKIs, while the quality of life was better and complication rate was lower after the therapy with PRRT (Table 2) [111,112,113,130,131,132].
3.5. Immunotherapy in Thyroid Cancer
3.5.1. DTC
The tumor microenvironment has been acknowledged as the major player in cancer progression and response to therapy. Therefore, the immune landscape of the thyroid cancer has emerged as a potential therapeutic target. Immune checkpoints, such as programmed cell death protein 1 (PD1) and its ligand-PDL-1, as well as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) inhibitors, exhibit antitumor effects by altering the interaction between the immune system cells and tumor cells [133]. The expression of PD1/PDL1 in thyroid cancer has been extensively studied for both diagnostic and prognostic purposes [134]. In one meta-analysis by Aghajani et al. the expression of PDL1 in thyroid cancer was associated with tumor recurrence and poor survival [135]. Based upon analysis of the TCGA database, a higher level of PD-L1 mRNA was associated with lymph node metastasis, extrathyroidal invasion, and shorter disease-specific survival [136]. The latter was further supported by two independent cohort studies [89,137]. Nevertheless, DTC is thought to be poorly immunogenic due to a relatively low mutation burden. Consistently, published studies have shown that thyroid cancer has relatively poor response to immunotherapy with checkpoint inhibitors [114,138] (Table 2). In order to enhance the efficacy of immunotherapy, combination treatments are being tested in several clinical trials in solid tumors, including thyroid cancer (Table 2). The efficacy of these therapies might be better particularly in de-differentiated tumors, such as widely invasive HTC and ATC, as they are characterized by high mutation burden and higher likelihood of introducing immunogenicity [10,139].
3.5.2. MTC
Another interesting concept utilizing the ability of MTC cells to produce carcinoembryonic antigen (CEA) consisted of the introduction of dendritic cell vaccination with anti-CEA vaccine [140,141,142]. Yeast-CEA (GI-6207) is a therapeutic cancer vaccine genetically modified to express recombinant carcinoembryonic antigen (CEA) protein. In a phase 1 trial involving 25 patients with CEA expressing cancers, the treatment with Yeast-CEA showed stabilization of disease and its biochemical biomarkers. It was also well tolerated, with the most common adverse effect being grade 1/2 injection-site reaction [142]. A phase II trial was designed at the National Cancer Institute to evaluate the use of GI-6207 in recurrent medullary thyroid cancer and documented biochemical response without a significant tumor response [143].
4. Limitations of Targeted Therapies
The major limitation in applying targeted therapies is its side-effects profile, as well as transient efficacy due to the development of escape mechanisms by the tumors. The first limitation could be overcome by dose de-escalation and adjunct therapy with agents targeting adverse symptoms. The latter requires a thorough investigation of the mechanisms of tumor resistance and analysis of acquired second hit mutations, in order to tailor therapy against changing over time the molecular landscape of the tumor.
4.1. Management of Side Effects of Different Targeted Therapies
4.1.1. Tyrosine Kinase Inhibitors
Systemic treatment with TKIs often leads to side effects affecting different organs and systems. Fatigue is a common side effects that is reported with different agents (Table 2). It can be severe enough (grade 3 and grade 4) to significantly affect the quality of life, leading to interruption or discontinuation of treatment [88,103]. Another common side effect is diarrhea, which can be mild, moderate, or severe. It is thought that it results from the effect of the agent on VEGF and EGF receptors in the bowel [144]. When diarrhea is of grade 1 or grade 2, the treatment is conservative, with antimotility agents such as loperamide and/or diphenoxylate/atropine, as well as cholestyramine and tincture of opium [144,145,146].
The most common cardiovascular adverse event associated with TKIs is elevated blood pressure, which is mediated by the anti-angiogenic effect on VEGF receptors, leading to reduction in endothelial nitric oxide, causing increased vascular tone [147,148]. Given the associated morbidity and mortality from hypertension in these patients, blood pressure should be frequently monitored with a target of <140/90 [148]. If blood pressure is above the target, a treatment with any of the first line anti-hypertensive agent should be initiated [148]. If hypertension is of grade 3 (SBP > 160 and/or DBP > 100) despite optimal treatment with anti-hypertensive agents, then treatment with TKI should be transiently discontinued till blood pressure is at target [148]. Grade 4 hypertension is an indication to stop treatment and consider other treatment options [148]. Other encountered cardiovascular adverse event of TKIs, particularly vandetanib, include QT prolongation, which can cause torsades de pointes ventricular tachyarrhythmia and subsequent cardiac arrest [104]. Therefore, ECG should be routinely done, to monitor QT interval, and QT interval prolongation to >500 milliseconds is an indication to stop treatment [149].
Another organ that can be affected by TKIs is the skin, as muco-cutaneous adverse reactions such as dryness, rash, hyperkeratosis, hand–foot syndrome, alopecia and mucositis occur frequently [150]. Hand–foot syndrome can manifest as hyperkeratosis, skin ulceration, or vesicles and present with pain, tingling, or paresthesia in affected skin areas which, if severe enough, may lead to interruption discontinuation of treatment [151,152]. The management of mucocutaneous side effects relies on preventive measures such as good skin hydration, urea creams, symptomatic treatment such as targeting neuropathic pain with gabapentin, dose reduction, or discontinuation of treatment with TKI [145,150,151].
Some adverse effects are more specific to certain TKIs; for example, pazopanib causes skin and hair hypopigmentation, while sorafenib causes skin hyperpigmentation [79,90]. Treatment with BRAF inhibitors dabrafenib and vemurafenib can be associated with the abnormal skin growth, forming papilloma, keratoacanthoma, nevi, or even skin cancers such as squamous cell carcinoma and melanoma [99,153].
Other encountered adverse effects of antiangiogenic targeted therapies are impaired wound healing, hemorrhage (within the tumor), fistula formation, or viscus perforation (in GI or respiratory tract) [144,154]. Therefore, it is important not to start these agents directly after the surgery for thyroid cancer.
4.1.2. Therapies Targeting Gene Fusion—Tropomyosin Receptor Kinase Inhibitor
The most common side effect of larotrectinib is abnormalities in liver function test [107]. However, transaminitis was rarely severe (grade > 3 or more) and required dose de-escalation only in four patients out 55 involved in the clinical trial targeting NTRK-positive tumors [107]. Other side effects that were less frequent and led to dose de-escalation were neutrophilia and dizziness [107]. Of note, none of the patients who received larotrectinib discontinued treatment due to adverse events [107].
The most frequent adverse events seen with entrectinib were dysgeusia, which was mild and reversible [108]. Other side effects, such as central nervous system related events, weight gain, congestive heart failure anemia, elevated creatinine level, and fatigue, were more severe, requiring dose reduction, treatment interruption, or even discontinuation [108].
4.1.3. Peptide Receptor Radionuclide Treatment
The treatment with (90)Yttrium and (177)Lutetium labeled somatostatin analogs in one study was associated with minor hematological abnormalities, such as leukopenia, erthryocytopenia, and/or decreased hemoglobin level [111]. Most of the hematological abnormalities seen with PRRT are self-limiting, but some patients may develop chronic complications such as myelodysplastic syndrome or leukemia [155]. Elevated liver enzymes, including ALT, AST, GGT, and ALP abnormalities, were also observed [111]. PRRT-related renal toxicity can lead to chronic renal disease/failure especially in patients with comorbidities that affect kidney function, such as hypertension and diabetes [156,157]. This complication is more likely to be observed with 90Ytrium-labeled somatostatin analogs rather than 177 Lutetium radio-labeled. In addition, amino acids infusion before the therapy has reno-protective properties. Unfortunately, amino acids infusion is often associated with nausea and vomiting, which are usually transient [112].
4.1.4. mTOR Inhibitors
A very common side effect of mTOR inhibitor everolimus is stomatitis, which presents as mouth pain, difficulty swallowing, or loss of taste [158]. When severe enough, stomatitis may require stopping oral intake and initiation of parenteral nutrition [158]. Patients receiving stomatitis should be counseled regarding this side effect and encouraged to keep good oral hygiene, to prevent stomatitis [159]. Treatment options include use of mouthwash, topical analgesics, and/or topical corticosteroids [158]. When symptoms of stomatitis are severe and affect the patient, oral-intake treatment should be interrupted till symptoms resolve [158]. In life-threatening situations, everolimus should be permanently discontinued [158].
Another common side effect of everolimus is skin rash, which usually presents as macules and/or papules. This is often associated with pruritis [160]. The mainstay for prevention of skin rash is the use of moisturizers and avoidance of sun exposure [161]. For acne-like rash topical treatment is the first option, but there may be a need to oral antibiotics in severe cases [161].
Other side effects seen with mTOR inhibitors include metabolic derangements such as hyperglycemia and hyperlipidemia, which mandate close monitoring of blood glucose and lipid profile routinely while on everolimus and utilization of anti-hyperglycemic agents and statins for the biochemical control [162].
4.1.5. Immunotherapy
Immune checkpoint inhibitors are associated with immune related adverse effects involving different organs and systems in the human body. Skin is commonly affected by immune checkpoint inhibitors, and the presentation can vary from mild dry skin to life-threatening Steven Johnson Syndrome (SJS) or Toxic Epidermal Necrolysis (TEN) [163,164]. Management of skin manifestation depends on severity; for mild-skin-lesion treatment with antipruritic agents or topical steroids is indicated. Severe manifestations such as SJS require inpatient or even intensive critical care unit admission and treatment with systemic steroids. Grade 4 reactions mandate permanent discontinuation of treatment [164,165]. Another common side effect of immune checkpoint inhibitors is colitis, which often presents as diarrhea. Once other causes of diarrhea are ruled out and the diagnosis is confirmed, treatment can be initiated with oral steroids (budesonide) and antimotility agents [166]. In more severe/refractory cases, treatment interruption or discontinuation and the use systemic steroids or infliximab is indicated [167,168]. Immune checkpoint inhibitors can have different effects on the endocrine system. The effect of immune checkpoint inhibitors on the thyroid gland can lead to hypothyroidism and, less frequently, hyperthyroidism, which is usually not an important consideration in thyroidectomized patients with thyroid cancer [169]. Other less common endocrinological adverse manifestations of immune checkpoint inhibitors are hypophysitis, adrenalitis, and development of type 1 diabetes mellitus [169]. Inflammation of other organs has also been described and includes myocarditis, pneumonitis, hepatitis, nephritis, pancreatitis, and others [170,171,172,173].
5. Mechanisms of Tumor Escape from Targeted Therapies
The targeted therapies have shown promising results in the treatment of thyroid cancer, but their biggest limitation is that the tumor cells develop resistance against those therapies over a period of time. The tumor cells acquire resistance to the treatment by developing an escape mechanism against the targeting drugs [174]. Some tumor types have intrinsic resistance to certain drugs; for example, MTCs with V804M-RET and V804L-RET gatekeeper mutations do not respond to vandetanib treatment. These mutations can also be acquired, over time, upon exposure to vandetanib if they were not already present before treatment [175]. Most of the tumors develop resistance against targeted drugs by acquiring new mutations that result in overactivation of pathways or through induction of alternate pathways to bypass the action of the drug [174].
MAPK and PI3K pathways are the overlapping pathways, and the upregulation of either of these pathways can lead to the same outcome. One possible mechanism by which thyroid tumors overcome the action of multiple kinase inhibitors is through induction of alternate signaling pathways or by upregulating the expression of tyrosine kinase receptors on the cell surface [176]. For example, targeting RAS and RET/PTC oncogenes in DTC can lead to the induction of PI3K pathway and activation of downstream targets, such as mTOR, forkhead family of transcription factor (FoxO), and others, thus helping tumors escape the drug action [176].
Thyroid tumors acquire RAI resistance by multiple mechanisms, and not all of them are well understood. One of the mechanisms by which thyroid tumors acquire RAI resistance is through the upregulation of the human epidermal receptor (HER) family of receptor tyrosine kinases [176]. HER2 and HER3 are the members of the EGFR family and are upstream of both the MAPK and PI3K pathways [176]. Preclinical data have shown that BRAF-mutant cells escape from the vemurafenib (BRAF inhibitor) effects by overexpressing HER2 and HER3 receptors, leading to the activation of mTOR and MAPK signaling pathways [63,176]. The tumor escape can be overcome by the combination treatment of vemurafenib and HER2 inhibitor trastuzumab, to improve RAI sensitivity (Figure 3) [63].
ALK fusion proteins also help thyroid tumors to escape from the effects of TKIs. ALK fusions are primarily seen in aggressive thyroid-cancer types, and they are known to activate MAPK and PI3K signaling pathways [177]. EML4-ALK fusion and various other ALK translocations have been identified in RAI resistant DTC [63]. STRN-ALK fusions have been observed in up to 4% ATC and 9% PDTC cases [176]. Combining TKIs with ALK specific inhibitors such as crizotinib will be beneficial to patients with ALK-positive tumors [63].
Thyroid tumors can escape targeted therapies by multiple mechanisms, and to this date, we have very little understanding of those pathways. The biggest challenge in overcoming the tumor escape mechanisms is to identify the mutations or alterations that play a role in the development of resistance against targeting drugs. Identification of the tumor escape mechanism will be helpful in the development of alternative therapies that can be combined with standard therapies to overcome the development of resistance against targeted therapies.
6. Conclusions
Several novel targeted therapies have recently been approved by the FDA for use in advanced thyroid cancer. There are many ongoing national and multi-national studies implementing targeted therapies in thyroid cancer. There is a shift in the management paradigm, with the molecular landscape rather than histology/morphology driving an individualized treatment approach. Even amongst the same histological groups of thyroid cancer, the response to therapy is different based on genetic and immune signature of the tumor, further confirming that “one size does not fit all”. The use of the therapeutic agents should be individualized and based on shared decision-making after informing the patient about the possible benefits and disadvantages of the therapy. Encouraged by the results of individualized therapies documenting even complete responses when targeting driver mutations, the aim of future research should focus on finding efficacious treatments that will have a long-lasting curative effect.
Author Contributions
Conceptualization, J.K.-G.; writing—original draft preparation, M.A.-J., S.T., S.G. and J.K.-G.; writing—review and editing, M.A.-J., S.T., S.G. and J.K.-G.; supervision, J.K.-G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank Lee S. Weinstein for the critical review of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2020. [Google Scholar]
- Fagin, J.A.; Wells, S.A., Jr. Biologic and clinical perspectives on thyroid cancer. N. Engl. J. Med. 2016, 375, 1054–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid Off. J. Am. Thyroid Assoc. 2016, 26, 1–133. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, R.M.; Ahuja, S.; Avram, A.M.; Bernet, V.J.; Bourguet, P.; Daniels, G.H.; Dillehay, G.; Draganescu, C.; Flux, G.; Führer, D. Controversies, Consensus, and Collaboration in the Use of 131I Therapy in Differentiated Thyroid Cancer: A Joint Statement from the American Thyroid Association, the European Association of Nuclear Medicine, the Society of Nuclear Medicine and Molecular Imaging, and the European Thyroid Association. Thyroid 2019, 29, 461–470. [Google Scholar] [PubMed] [Green Version]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma: The American Thyroid Association Guidelines Task Force on medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Roman, S.; Lin, R.; Sosa, J.A. Prognosis of medullary thyroid carcinoma: Demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2006, 107, 2134–2142. [Google Scholar] [CrossRef]
- Tirro, E.; Martorana, F.; Romano, C.; Vitale, S.R.; Motta, G.; Di Gregorio, S.; Massimino, M.; Pennisi, M.S.; Stella, S.; Puma, A.; et al. Molecular Alterations in Thyroid Cancer: From Bench to Clinical Practice. Genes 2019, 10, 709. [Google Scholar] [CrossRef] [Green Version]
- Khatami, F.; Tavangar, S.M. A Review of Driver Genetic Alterations in Thyroid Cancers. Iran. J. Pathol. 2018, 13, 125–135. [Google Scholar] [CrossRef]
- Kebebew, E.; Weng, J.; Bauer, J.; Ranvier, G.; Clark, O.H.; Duh, Q.Y.; Shibru, D.; Bastian, B.; Griffin, A. The prevalence and prognostic value of BRAF mutation in thyroid cancer. Ann. Surg. 2007, 246, 466–470. [Google Scholar] [CrossRef]
- Ganly, I.; Makarov, V.; Deraje, S.; Dong, Y.; Reznik, E.; Seshan, V.; Nanjangud, G.; Eng, S.; Bose, P.; Kuo, F.; et al. Integrated Genomic Analysis of Hurthle Cell Cancer Reveals Oncogenic Drivers, Recurrent Mitochondrial Mutations, and Unique Chromosomal Landscapes. Cancer Cell 2018, 34, 256–270.e5. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Censi, S.; Cavedon, E.; Bertazza, L.; Galuppini, F.; Watutantrige-Fernando, S.; De Lazzari, P.; Nacamulli, D.; Pennelli, G.; Fassina, A.; Iacobone, M.; et al. Frequency and Significance of Ras, Tert Promoter, and Braf Mutations in Cytologically Indeterminate Thyroid Nodules: A Monocentric Case Series at a Tertiary-Level Endocrinology Unit. Front. Endocrinol. (Lausanne) 2017, 8, 273. [Google Scholar] [CrossRef] [PubMed]
- Ciampi, R.; Mian, C.; Fugazzola, L.; Cosci, B.; Romei, C.; Barollo, S.; Cirello, V.; Bottici, V.; Marconcini, G.; Rosa, P.M.; et al. Evidence of a low prevalence of RAS mutations in a large medullary thyroid cancer series. Thyroid Off. J. Am. Thyroid Assoc. 2013, 23, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Ciampi, R.; Romei, C.; Ramone, T.; Prete, A.; Tacito, A.; Cappagli, V.; Bottici, V.; Viola, D.; Torregrossa, L.; Ugolini, C.; et al. Genetic Landscape of Somatic Mutations in a Large Cohort of Sporadic Medullary Thyroid Carcinomas Studied by Next-Generation Targeted Sequencing. iScience 2019, 20, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, M. Clinical utility of RAS mutations in thyroid cancer: A blurred picture now emerging clearer. BMC Med. 2016, 14, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulten, H.J.; Salama, S.; Al-Ahmadi, A.; Al-Mansouri, Z.; Mirza, Z.; Al-Ghamdi, K.; Al-Hamour, O.A.; Huwait, E.; Gari, M.; Al-Qahtani, M.H.; et al. Comprehensive survey of HRAS, KRAS, and NRAS mutations in proliferative thyroid lesions from an ethnically diverse population. Anticancer Res. 2013, 33, 4779–4784. [Google Scholar] [PubMed]
- Howell, G.M.; Hodak, S.P.; Yip, L. RAS mutations in thyroid cancer. Oncologist 2013, 18, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.; Westra, W.H.; Tufano, R.P.; Cohen, Y.; Rosenbaum, E.; Rhoden, K.J.; Carson, K.A.; Vasko, V.; Larin, A.; Tallini, G.; et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2005, 90, 6373–6379. [Google Scholar] [CrossRef] [Green Version]
- Paragliola, R.M.; Corsello, A.; Del Gatto, V.; Papi, G.; Pontecorvi, A.; Corsello, S.M. Lenvatinib for thyroid cancer treatment: Discovery, pre-clinical development and clinical application. Expert Opin. Drug Discov. 2020, 15, 11–26. [Google Scholar] [CrossRef]
- Bond, J.A.; Wyllie, F.S.; Rowson, J.; Radulescu, A.; Wynford-Thomas, D. In vitro reconstruction of tumour initiation in a human epithelium. Oncogene 1994, 9, 281–290. [Google Scholar]
- Liu, Z.; Hou, P.; Ji, M.; Guan, H.; Studeman, K.; Jensen, K.; Vasko, V.; El-Naggar, A.K.; Xing, M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 2008, 93, 3106–3116. [Google Scholar] [CrossRef] [Green Version]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.C.; et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [Green Version]
- Guigon, C.J.; Zhao, L.; Willingham, M.C.; Cheng, S.Y. PTEN deficiency accelerates tumour progression in a mouse model of thyroid cancer. Oncogene 2009, 28, 509–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, M.; Chung, Y.J.; Saji, M.; Ringel, M.D. AKT in thyroid tumorigenesis and progression. Endocrinology 2007, 148, 942–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringel, M.D.; Hayre, N.; Saito, J.; Saunier, B.; Schuppert, F.; Burch, H.; Bernet, V.; Burman, K.D.; Kohn, L.D.; Saji, M. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001, 61, 6105–6111. [Google Scholar] [PubMed]
- Garcia-Rostan, G.; Costa, A.M.; Pereira-Castro, I.; Salvatore, G.; Hernandez, R.; Hermsem, M.J.; Herrero, A.; Fusco, A.; Cameselle-Teijeiro, J.; Santoro, M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005, 65, 10199–10207. [Google Scholar] [CrossRef] [Green Version]
- Jhiang, S.M.; Sagartz, J.E.; Tong, Q.; Parker-Thornburg, J.; Capen, C.C.; Cho, J.Y.; Xing, S.; Ledent, C. Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology 1996, 137, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Putzer, B.M.; Drosten, M. The RET proto-oncogene: A potential target for molecular cancer therapy. Trends Mol. Med. 2004, 10, 351–357. [Google Scholar] [CrossRef]
- Taccaliti, A.; Silvetti, F.; Palmonella, G.; Boscaro, M. Genetic alterations in medullary thyroid cancer: Diagnostic and prognostic markers. Curr. Genom. 2011, 12, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.W.; Feng, F.Y. A Tumor-Agnostic NTRK (TRK) Inhibitor. Cell 2019, 177, 8. [Google Scholar] [CrossRef]
- Murugan, A.K.; Xing, M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011, 71, 4403–4411. [Google Scholar] [CrossRef] [Green Version]
- Gerber, T.S.; Schad, A.; Hartmann, N.; Springer, E.; Zechner, U.; Musholt, T.J. Targeted next-generation sequencing of cancer genes in poorly differentiated thyroid cancer. Endocr. Connect. 2018, 7, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Perona, A.; Santisteban, P. Role of the wnt pathway in thyroid cancer. Front. Endocrinol. (Lausanne) 2012, 3, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzella, L.; Stella, S.; Pennisi, M.S.; Tirro, E.; Massimino, M.; Romano, C.; Puma, A.; Tavarelli, M.; Vigneri, P. New Insights in Thyroid Cancer and p53 Family Proteins. Int. J. Mol. Sci. 2017, 18, 1325. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.; Koenig, R.J. Pax-8-PPAR-gamma fusion protein in thyroid carcinoma. Nat. Rev. Endocrinol. 2014, 10, 616–623. [Google Scholar] [CrossRef]
- Liu, R.; Xing, M. TERT promoter mutations in thyroid cancer. Endocr. Relat. Cancer 2016, 23, R143–R155. [Google Scholar] [CrossRef] [Green Version]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [Green Version]
- Murugan, A.K.; Bojdani, E.; Xing, M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem. Biophys. Res. Commun. 2010, 393, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Xing, M. BRAF mutation in thyroid cancer. Endocr. Relat. Cancer 2005, 12, 245–262. [Google Scholar] [CrossRef] [Green Version]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Hou, P.; Ji, M.; Xing, M. Association of PTEN gene methylation with genetic alterations in the phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid tumors. Cancer 2008, 113, 2440–2447. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Wang, R.; Zou, K.; Zou, L. Genetic alterations in anaplastic thyroid carcinoma and targeted therapies. Exp. Med. 2019, 18, 2369–2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar] [CrossRef] [Green Version]
- Paes, J.E.; Ringel, M.D. Dysregulation of the phosphatidylinositol 3-kinase pathway in thyroid neoplasia. Endocrinol. Metab. Clin. N. Am. 2008, 37, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, N.; Jiao, Y.; Sausen, M.; Leary, R.; Bettegowda, C.; Roberts, N.J.; Bhan, S.; Ho, A.S.; Khan, Z.; Bishop, J.; et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J. Clin. Endocrinol. Metab. 2013, 98, E364–E369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Saenko, V.; Yamashita, S.; Mitsutake, N. Radiation-Induced Thyroid Cancers: Overview of Molecular Signatures. Cancers 2019, 11, 1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, M.; Moccia, M.; Federico, G.; Carlomagno, F. RET Gene Fusions in Malignancies of the Thyroid and Other Tissues. Genes 2020, 11, 424. [Google Scholar] [CrossRef] [Green Version]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef]
- Greco, A.; Miranda, C.; Pierotti, M.A. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol. Cell Endocrinol. 2010, 321, 44–49. [Google Scholar] [CrossRef]
- Yakushina, V.D.; Lerner, L.V.; Lavrov, A.V. Gene Fusions in Thyroid Cancer. Thyroid 2018, 28, 158–167. [Google Scholar] [CrossRef]
- Krampitz, G.W.; Norton, J.A. RET gene mutations (genotype and phenotype) of multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma. Cancer 2014, 120, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Kouvaraki, M.A.; Shapiro, S.E.; Perrier, N.D.; Cote, G.J.; Gagel, R.F.; Hoff, A.O.; Sherman, S.I.; Lee, J.E.; Evans, D.B. RET proto-oncogene: A review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 2005, 15, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernock, R.D.; Hagemann, I.S. Molecular pathology of hereditary and sporadic medullary thyroid carcinomas. Am. J. Clin. Pathol. 2015, 143, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Frank-Raue, K.; Raue, F. Hereditary Medullary Thyroid Cancer Genotype-Phenotype Correlation. Recent Results Cancer Res. 2015, 204, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Hillier, K.; Hughes, A.; Shamberger, R.C.; Shusterman, S.; Perez-Atayde, A.R.; Wassner, A.J.; Iafrate, A.J.; Dubuc, A.; Janeway, K.A.; Rothenberg, S.M.; et al. A Novel ALK Fusion in Pediatric Medullary Thyroid Carcinoma. Thyroid 2019, 29, 1704–1707. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Yang, H.; Yip, L.; Ohori, N.P.; McCoy, K.L.; Stang, M.T.; Hodak, S.P.; Nikiforova, M.N.; Carty, S.E.; Nikiforov, Y.E. PAX8/PPARgamma rearrangement in thyroid nodules predicts follicular-pattern carcinomas, in particular the encapsulated follicular variant of papillary carcinoma. Thyroid 2014, 24, 1369–1374. [Google Scholar] [CrossRef] [Green Version]
- Worden, F. Treatment strategies for radioactive iodine-refractory differentiated thyroid cancer. Adv. Med. Oncol. 2014, 6, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Paladino, S.; Melillo, R.M. Editorial: Novel Mechanism of Radioactive Iodine Refractivity in Thyroid Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Kogai, T.; Brent, G.A. The sodium iodide symporter (NIS): Regulation and approaches to targeting for cancer therapeutics. Pharmacol. Ther. 2012, 135, 355–370. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, Y.; Lin, Y.; Liang, J. Radioactive Iodine-Refractory Differentiated Thyroid Cancer and Redifferentiation Therapy. Endocrinol. Metab. (Seoul) 2019, 34, 215–225. [Google Scholar] [CrossRef]
- Costamagna, E.; Garcia, B.; Santisteban, P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. J. Biol. Chem. 2004, 279, 3439–3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aashiq, M.; Silverman, D.A.; Na’ara, S.; Takahashi, H.; Amit, M. Radioiodine-Refractory Thyroid Cancer: Molecular Basis of Redifferentiation Therapies, Management, and Novel Therapies. Cancers 2019, 11, 1382. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, D.; Murugan, A.K.; Liu, Z.; Xing, M. Histone deacetylation of NIS promoter underlies BRAFV600E-promoted NIS silencing in thyroid cancer. Endocr. Relat. Cancer 2014, 21, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Jaber, T.; Waguespack, S.G.; Cabanillas, M.E.; Elbanan, M.; Vu, T.; Dadu, R.; Sherman, S.I.; Amit, M.; Santos, E.B.; Zafereo, M.; et al. Targeted Therapy in Advanced Thyroid Cancer to Resensitize Tumors to Radioactive Iodine. J. Clin. Endocrinol. Metab. 2018, 103, 3698–3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Chen, D. Synergistic inhibition of MEK/ERK and BRAFV600E with PD98059 and PLX4032 induces sodium/iodide symporter (NIS) expression and radioiodine uptake in BRAF mutated papillary thyroid cancer cells. Thyroid Res. 2018, 11, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.M.; Kalimuthu, S.; Gangadaran, P.; Baek, S.H.; Zhu, L.; Lee, H.W.; Rajendran, R.L.; Hong, C.M.; Jeong, S.Y.; Lee, S.W.; et al. Reverting iodine avidity of radioactive-iodine refractory thyroid cancer with a new tyrosine kinase inhibitor (K905-0266) excavated by high-throughput NIS (sodium iodide symporter) enhancer screening platform using dual reporter gene system. Oncotarget 2018, 9, 7075–7087. [Google Scholar] [CrossRef] [Green Version]
- Vaisman, F.; Carvalho, D.P.; Vaisman, M. A new appraisal of iodine refractory thyroid cancer. Endocr. Relat. Cancer 2015, 22, R301–R310. [Google Scholar] [CrossRef]
- Kogai, T.; Sajid-Crockett, S.; Newmarch, L.S.; Liu, Y.Y.; Brent, G.A. Phosphoinositide-3-kinase inhibition induces sodium/iodide symporter expression in rat thyroid cells and human papillary thyroid cancer cells. J. Endocrinol. 2008, 199, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, J.; Li, X.; Liang, Z.; Gao, W.; Liang, J.; Cheng, S.; Lin, Y. TERT Promoter Mutation Predicts Radioiodine-Refractory Character in Distant Metastatic Differentiated Thyroid Cancer. J. Nucl. Med. 2017, 58, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Luthra, R.; Routbort, M.J.; Patel, K.P.; Cabanillas, M.E.; Broaddus, R.R.; Williams, M.D. Molecular Profile of Advanced Thyroid Carcinomas by Next-Generation Sequencing: Characterizing Tumors Beyond Diagnosis for Targeted Therapy. Mol. Cancer 2018, 17, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.; Baitei, E.Y.; Alzahrani, A.S.; BinHumaid, F.S.; Alkhafaji, D.; Al-Rijjal, R.A.; Meyer, B.F.; Shi, Y. Concomitant RAS, RET/PTC, or BRAF mutations in advanced stage of papillary thyroid carcinoma. Thyroid 2014, 24, 1256–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.K.; Song, Y.S.; Lee, E.K.; Hwang, J.; Kim, H.H.; Jung, G.; Kim, Y.A.; Kim, S.J.; Cho, S.W.; Won, J.K.; et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat. Commun. 2019, 10, 2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Yan, C.; Xiao, J.; Wang, T.; Ling, R. Relevance and clinicopathologic relationship of BRAFV600E, TERT and NRAS mutations for papillary thyroid carcinoma patients in Northwest China. Diagn. Pathol. 2019, 14, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.E.; Hwang, T.S.; Choi, Y.L.; Han, H.S.; Kim, W.S.; Jang, M.H.; Kim, S.K.; Yang, J.H. Prognostic Significance of TERT Promoter Mutations in Papillary Thyroid Carcinomas in a BRAF(V600E) Mutation-Prevalent Population. Thyroid 2016, 26, 901–910. [Google Scholar] [CrossRef]
- Liu, X.; Qu, S.; Liu, R.; Sheng, C.; Shi, X.; Zhu, G.; Murugan, A.K.; Guan, H.; Yu, H.; Wang, Y.; et al. TERT promoter mutations and their association with BRAFV600E mutation and aggressive clinicopathological characteristics of thyroid cancer. J. Clin. Endocrinol. Metab. 2014, 99, E1130–E1136. [Google Scholar] [CrossRef] [Green Version]
- Vuong, H.G.; Altibi, A.M.A.; Duong, U.N.P.; Hassell, L. Prognostic implication of BRAF and TERT promoter mutation combination in papillary thyroid carcinoma-A meta-analysis. Clin. Endocrinol. (Oxf.) 2017, 87, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Zhang, T.; Zhu, G.; Xing, M. Regulation of mutant TERT by BRAFV600E/MAP kinase pathway through FOS/GABP in human cancer. Nat. Commun. 2018, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; De La Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Food and Drug Administration. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021923s008s009lbl.pdf (accessed on 5 June 2020).
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Food and Drug Administration. Available online: https://www.accessd.ata.fda.gov/drugsatfda_docs/label/2018/206947s007lbl.pdf (accessed on 5 June 2020).
- Sun, Y.; Du, F.; Gao, M.; Ji, Q.; Li, Z.; Zhang, Y.; Guo, Z.; Wang, J.; Chen, X.; Wang, J. Anlotinib for the treatment of patients with locally advanced or metastatic medullary thyroid cancer. Thyroid 2018, 28, 1455–1461. [Google Scholar] [CrossRef]
- Li, D.; Tang, P.Z.; Chen, X.; Ge, M.; Zhang, Y.; Guo, Z.; Wang, J.; Shi, F.; Zhang, J.; Cheng, Y. Anlotinib Treatment in Locally Advanced or Metastatic Medullary Thyroid Carcinoma: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Phase IIB Trial. J. Clin. Oncol. 2019, 37, 6019. [Google Scholar] [CrossRef]
- Cohen, E.E.; Rosen, L.S.; Vokes, E.E.; Kies, M.S.; Forastiere, A.A.; Worden, F.P.; Kane, M.A.; Sherman, E.; Kim, S.; Bycott, P. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: Results from a phase II study. J. Clin. Oncol. 2008, 26, 4708. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, J.; Trigo, J.M.; Aller, J.; Manzano, J.L.; Adrián, S.G.; Llopis, C.Z.; Reig, Ò.; Bohn, U.; y Cajal, T.R.; Duran-Poveda, M. Axitinib treatment in advanced RAI-resistant differentiated thyroid cancer (DTC) and refractory medullary thyroid cancer (MTC). Eur. J. Endocrinol. 2017, 177, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Chung, W.Y.; Nam, K.-H.; Kang, S.-W.; Lim, J.Y.; Kim, H.-G.; Shin, S.H.; Sun, J.-M.; Kim, S.-G.; Kim, J.-H. An open label, multicenter, phase II study of dovitinib in advanced thyroid cancer. Eur. J. Cancer 2015, 51, 1588–1595. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Jarzab, B.; Cabanillas, M.E.; Robinson, B.; Pacini, F.; Ball, D.W.; McCaffrey, J.; Newbold, K.; Allison, R.; Martins, R.G. A phase II trial of the multitargeted tyrosine kinase inhibitor lenvatinib (E7080) in advanced medullary thyroid cancer. Clin. Cancer Res. 2016, 22, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Tahara, M.; Kiyota, N.; Yamazaki, T.; Chayahara, N.; Nakano, K.; Inagaki, L.; Toda, K.; Enokida, T.; Minami, H.; Imamura, Y. Lenvatinib for anaplastic thyroid cancer. Front. Oncol. 2017, 7, 25. [Google Scholar] [CrossRef]
- Bible, K.C.; Suman, V.J.; Molina, J.R.; Smallridge, R.C.; Maples, W.J.; Menefee, M.E.; Rubin, J.; Sideras, K.; Morris III, J.C.; McIver, B. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: Results of a phase 2 consortium study. Lancet Oncol. 2010, 11, 962–972. [Google Scholar] [CrossRef] [Green Version]
- Bible, K.C.; Suman, V.J.; Molina, J.R.; Smallridge, R.C.; Maples, W.J.; Menefee, M.E.; Rubin, J.; Karlin, N.; Sideras, K.; Morris III, J.C. A multicenter phase 2 trial of pazopanib in metastatic and progressive medullary thyroid carcinoma: MC057H. J. Clin. Endocrinol. Metab. 2014, 99, 1687–1693. [Google Scholar] [CrossRef]
- Bible, K.C.; Suman, V.J.; Menefee, M.E.; Smallridge, R.C.; Molina, J.R.; Maples, W.J.; Karlin, N.J.; Traynor, A.M.; Kumar, P.; Goh, B.C. A multiinstitutional phase 2 trial of pazopanib monotherapy in advanced anaplastic thyroid cancer. J. Clin. Endocrinol. Metab. 2012, 97, 3179–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, E.T.; Ringel, M.D.; Kloos, R.T.; Prior, T.W.; Knopp, M.V.; Liang, J.; Sammet, S.; Hall, N.C.; Wakely, P.E., Jr.; Vasko, V.V. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J. Clin. Oncol. 2010, 28, 2323. [Google Scholar] [CrossRef] [Green Version]
- Capdevila, J.; Iglesias, L.; Halperin, I.; Segura, A.; Martínez-Trufero, J.; Vaz, M.Á.; Corral, J.; Obiols, G.; Grande, E.; Grau, J.J. Sorafenib in metastatic thyroid cancer. Endocr. Relat. Cancer 2012, 19, e209. [Google Scholar] [CrossRef] [Green Version]
- Bikas, A.; Kundra, P.; Desale, S.; Mete, M.; O’Keefe, K.; Clark, B.G.; Wray, L.; Gandhi, R.; Barett, C.; Jelinek, J.S. Phase 2 clinical trial of sunitinib as adjunctive treatment in patients with advanced differentiated thyroid cancer. Eur. J. Endocrinol. 2016, 174, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, L.L.; Mankoff, D.A.; Goulart, B.H.; Eaton, K.D.; Capell, P.T.; Kell, E.M.; Bauman, J.E.; Martins, R.G. Phase II study of daily sunitinib in FDG-PET–positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin. Cancer Res. 2010, 16, 5260–5268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravaud, A.; de la Fouchardière, C.; Caron, P.; Doussau, A.; Do Cao, C.; Asselineau, J.; Rodien, P.; Pouessel, D.; Nicolli-Sire, P.; Klein, M. A multicenter phase II study of sunitinib in patients with locally advanced or metastatic differentiated, anaplastic or medullary thyroid carcinomas: Mature data from the THYSU study. Eur. J. Cancer 2017, 76, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Leboulleux, S.; Bastholt, L.; Krause, T.; de la Fouchardiere, C.; Tennvall, J.; Awada, A.; Gómez, J.M.; Bonichon, F.; Leenhardt, L.; Soufflet, C. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 2 trial. Lancet Oncol. 2012, 13, 897–905. [Google Scholar] [CrossRef]
- Falchook, G.S.; Millward, M.; Hong, D.; Naing, A.; Piha-Paul, S.; Waguespack, S.G.; Cabanillas, M.E.; Sherman, S.I.; Ma, B.; Curtis, M. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid 2015, 25, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in patients with BRAFV600E-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Chang, H.; Yoon, M.; Hong, Y.; Kim, H.; Chung, W.; Park, C.; Nam, K.; Kang, S.; Kim, M. A multicenter, phase II trial of everolimus in locally advanced or metastatic thyroid cancer of all histologic subtypes. Ann. Oncol. 2013, 24, 3089–3094. [Google Scholar] [CrossRef]
- Hanna, G.J.; Busaidy, N.L.; Chau, N.G.; Wirth, L.J.; Barletta, J.A.; Calles, A.; Haddad, R.I.; Kraft, S.; Cabanillas, M.E.; Rabinowits, G. Genomic correlates of response to everolimus in aggressive radioiodine-refractory thyroid cancer: A phase II study. Clin. Cancer Res. 2018, 24, 1546–1553. [Google Scholar] [CrossRef] [Green Version]
- Elisei, R.; Schlumberger, M.J.; Müller, S.P.; Schöffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639. [Google Scholar] [CrossRef] [Green Version]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Iravani, A.; Solomon, B.; Pattison, D.A.; Jackson, P.; Ravi Kumar, A.; Kong, G.; Hofman, M.S.; Akhurst, T.; Hicks, R.J. Mitogen-Activated Protein Kinase Pathway Inhibition for Redifferentiation of Radioiodine Refractory Differentiated Thyroid Cancer: An Evolving Protocol. Thyroid 2019, 29, 1634–1645. [Google Scholar] [CrossRef]
- Budiawan, H.; Salavati, A.; Kulkarni, H.R.; Baum, R.P. Peptide receptor radionuclide therapy of treatment-refractory metastatic thyroid cancer using (90)Yttrium and (177)Lutetium labeled somatostatin analogs: Toxicity, response and survival analysis. Am. J. Nucl. Med. Mol. Imaging 2013, 4, 39–52. [Google Scholar]
- Versari, A.; Sollini, M.; Frasoldati, A.; Fraternali, A.; Filice, A.; Froio, A.; Asti, M.; Fioroni, F.; Cremonini, N.; Putzer, D.; et al. Differentiated thyroid cancer: A new perspective with radiolabeled somatostatin analogues for imaging and treatment of patients. Thyroid 2014, 24, 715–726. [Google Scholar] [CrossRef]
- Lapa, C.; Werner, R.A.; Schmid, J.S.; Papp, L.; Zsótér, N.; Biko, J.; Reiners, C.; Herrmann, K.; Buck, A.K.; Bundschuh, R.A. Prognostic value of positron emission tomography-assessed tumor heterogeneity in patients with thyroid cancer undergoing treatment with radiopeptide therapy. Nucl. Med. Biol. 2015, 42, 349–354. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Varga, A.; Brose, M.S.; Aggarwal, R.R.; Lin, C.-C.; Prawira, A.; De Braud, F.; Tamura, K.; Doi, T.; Piha-Paul, S.A. Safety and antitumor activity of the anti–PD-1 antibody pembrolizumab in patients with advanced, PD-L1–positive papillary or follicular thyroid cancer. BMC Cancer 2019, 19, 196. [Google Scholar] [CrossRef] [PubMed]
- Tiedje, V.; Fagin, J.A. Therapeutic breakthroughs for metastatic thyroid cancer. Nat. Rev. Endocrinol. 2020, 16, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Smallridge, R.C.; Ain, K.B.; Asa, S.L.; Bible, K.C.; Brierley, J.D.; Burman, K.D.; Kebebew, E.; Lee, N.Y.; Nikiforov, Y.E.; Rosenthal, M.S. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid 2012, 22, 1104–1139. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/022405s007lbl.pdf (accessed on 23 March 2020).
- Food and Drug Administration. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/208692s003lbl.pdf (accessed on 23 March 2020).
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, V.; Taylor, M.; Lin, J.; Hu, M.; Ou, S.; Brose, M.; Garralda, E.; Clifford, C.; Palmer, M.; Tugnait, M. Highly potent and selective RET inhibitor, BLU-667, achieves proof of concept in ARROW, a phase 1 study of advanced, RET-altered solid tumors. In Proceedings of the American Association for Cancer Research Annual Meeting, Chicago, IL, USA, 14–18 April 2018; pp. 14–18. [Google Scholar]
- Subbiah, V.; Velcheti, V.; Tuch, B.; Ebata, K.; Busaidy, N.L.; Cabanillas, M.E.; Wirth, L.; Stock, S.; Smith, S.; Lauriault, V. Selective RET kinase inhibition for patients with RET-altered cancers. Ann. Oncol. 2018, 29, 1869–1876. [Google Scholar] [CrossRef]
- Taylor, M.H.; Gainor, J.F.; Hu, M.I.-N.; Zhu, V.W.; Lopes, G.; Leboulleux, S.; Brose, M.S.; Schuler, M.H.; Bowles, D.W.; Kim, D.-W.; et al. Activity and tolerability of BLU-667, a highly potent and selective RET inhibitor, in patients with advanced RET-altered thyroid cancers. J. Clin. Oncol. 2019, 37, 6018. [Google Scholar] [CrossRef]
- Lassen, U.; Albert, C.; Kummar, S.; van Tilburg, C.; Dubois, S.; Geoerger, B.; Mascarenhas, L.; Federman, N.; Schilder, R.; Doz, F. 409O Larotrectinib efficacy and safety in TRK fusion cancer: An expanded clinical dataset showing consistency in an age and tumor agnostic approach. Ann. Oncol. 2018, 29, mdy279.397. [Google Scholar] [CrossRef]
- Giordano, T.J.; Haugen, B.R.; Sherman, S.I.; Shah, M.H.; Caoili, E.M.; Koenig, R.J. Pioglitazone Therapy of PAX8-PPAR γ Fusion Protein Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2018, 103, 1277–1281. [Google Scholar] [CrossRef]
- Haddad, R.I.; Nasr, C.; Bischoff, L.; Busaidy, N.L.; Byrd, D.; Callender, G.; Dickson, P.; Duh, Q.Y.; Ehya, H.; Goldner, W.; et al. NCCN Guidelines Insights: Thyroid Carcinoma, Version 2.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 1429–1440. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Efficacy of MEK (Trametinib) and BRAFV600E (Dabrafenib) Inhibitors with Radioactive Iodine (RAI) for the Treatment of Refractory Metastatic Differentiated Thyroid Cancer (MERAIODE). Available online: https://clinicaltrials.gov/ct2/show/NCT03244956?cond=MEK+and+BRAF%2C+RAI&draw=2&rank=1 (accessed on 23 March 2020).
- Brown, S.R.; Hall, A.; Buckley, H.L.; Flanagan, L.; Gonzalez de Castro, D.; Farnell, K.; Moss, L.; Gregory, R.; Newbold, K.; Du, Y.; et al. Investigating the potential clinical benefit of Selumetinib in resensitising advanced iodine refractory differentiated thyroid cancer to radioiodine therapy (SEL-I-METRY): Protocol for a multicentre UK single arm phase II trial. BMC Cancer 2019, 19, 582. [Google Scholar] [CrossRef]
- Papotti, M.; Croce, S.; Bellò, M.; Bongiovanni, M.; Allìa, E.; Schindler, M.; Bussolati, G. Expression of somatostatin receptor types 2, 3 and 5 in biopsies and surgical specimens of human lung tumours. Virchows Arch. 2001, 439, 787–797. [Google Scholar] [CrossRef]
- Zatelli, M.C.; Tagliati, F.; Taylor, J.E.; Rossi, R.; Culler, M.D.; degli Uberti, E.C. Somatostatin receptor subtypes 2 and 5 differentially affect proliferation in vitro of the human medullary thyroid carcinoma cell line tt. J. Clin. Endocrinol. Metab. 2001, 86, 2161–2169. [Google Scholar] [CrossRef]
- Vaisman, F.; de Castro, P.H.R.; Lopes, F.P.P.L.; Kendler, D.B.; Pessoa, C.H.; Bulzico, D.A.; de Carvalho Leal, D.; Vilhena, B.; Vaisman, M.; Carneiro, M. Is there a role for peptide receptor radionuclide therapy in medullary thyroid cancer? Clin. Nucl. Med. 2015, 40, 123–127. [Google Scholar] [CrossRef]
- Iten, F.; Müller, B.; Schindler, C.; Rochlitz, C.; Oertli, D.; Mäcke, H.R.; Müller-Brand, J.; Walter, M.A. Response to [90Yttrium-DOTA]-TOC treatment is associated with long-term survival benefit in metastasized medullary thyroid cancer: A phase II clinical trial. Clin. Cancer Res. 2007, 13, 6696–6702. [Google Scholar] [CrossRef] [Green Version]
- Beukhof, C.M.; Brabander, T.; van Nederveen, F.H.; van Velthuysen, M.-L.F.; de Rijke, Y.B.; Hofland, L.J.; Franssen, G.J.; Fröberg, L.A.; Kam, B.L.; Visser, W.E. Peptide receptor radionuclide therapy in patients with medullary thyroid carcinoma: Predictors and pitfalls. BMC Cancer 2019, 19, 325. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Fu, G.; Polyakova, O.; MacMillan, C.; Ralhan, R.; Walfish, P.G. Programmed death-ligand 1 expression distinguishes invasive encapsulated follicular variant of papillary thyroid carcinoma from noninvasive follicular thyroid neoplasm with papillary-like nuclear features. EBioMedicine 2017, 18, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Aghajani, M.; Graham, S.; McCafferty, C.; Shaheed, C.A.; Roberts, T.; DeSouza, P.; Yang, T.; Niles, N. Clinicopathologic and prognostic significance of programmed cell death ligand 1 expression in patients with non-medullary thyroid cancer: A systematic review and meta-analysis. Thyroid 2018, 28, 349–361. [Google Scholar] [CrossRef]
- Ulisse, S.; Tuccilli, C.; Sorrenti, S.; Antonelli, A.; Fallahi, P.; D’Armiento, E.; Catania, A.; Tartaglia, F.; Amabile, M.I.; Giacomelli, L. PD-1 Ligand Expression in Epithelial Thyroid Cancers: Potential Clinical Implications. Int. J. Mol. Sci. 2019, 20, 1405. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.; Veyhl, J.; Jessa, F.; Polyakova, O.; Alenzi, A.; MacMillan, C.; Ralhan, R.; Walfish, P.G. Programmed death-ligand 1 overexpression is a prognostic marker for aggressive papillary thyroid cancer and its variants. Oncotarget 2016, 7, 32318. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Gopal, R.K.; Kubler, K.; Calvo, S.E.; Polak, P.; Livitz, D.; Rosebrock, D.; Sadow, P.M.; Campbell, B.; Donovan, S.E.; Amin, S.; et al. Widespread Chromosomal Losses and Mitochondrial DNA Alterations as Genetic Drivers in Hurthle Cell Carcinoma. Cancer Cell 2018, 34, 242–255.e5. [Google Scholar] [CrossRef] [Green Version]
- Schott, M.; Seissler, J.; Lettmann, M.; Fouxon, V.; Scherbaum, W.A.; Feldkamp, J. Immunotherapy for medullary thyroid carcinoma by dendritic cell vaccination. J. Clin. Endocrinol. Metab. 2001, 86, 4965–4969. [Google Scholar] [CrossRef]
- Bachleitner-Hofmann, T.; Friedl, J.; Hassler, M.; Hayden, H.; Dubsky, P.; Sachet, M.; Rieder, E.; Pfragner, R.; Brostjan, C.; Riss, S. Pilot trial of autologous dendritic cells loaded with tumor lysate (s) from allogeneic tumor cell lines in patients with metastatic medullary thyroid carcinoma. Oncol. Rep. 2009, 21, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Bilusic, M.; Heery, C.R.; Arlen, P.M.; Rauckhorst, M.; Apelian, D.; Tsang, K.Y.; Tucker, J.A.; Jochems, C.; Schlom, J.; Gulley, J.L.; et al. Phase I trial of a recombinant yeast-CEA vaccine (GI-6207) in adults with metastatic CEA-expressing carcinoma. Cancer Immunol. Immunother. 2014, 63, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Madan, R.A.; Singh, N.K.; Gramza, A.W.; Fojo, A.T.; Heery, C.R.; Kim, J.W.; McMahon, S.; Rauckhorst, M.; King, T.H.; Apelian, D.; et al. A phase II study of a yeast-based therapeutic cancer vaccine, GI-6207, targeting CEA in patients with minimally symptomatic, metastatic medullary thyroid cancer. J. Clin. Oncol. 2013, 31, TPS3127. [Google Scholar] [CrossRef]
- Dy, G.K.; Adjei, A.A. Understanding, recognizing, and managing toxicities of targeted anticancer therapies. CA Cancer J. Clin. 2013, 63, 249–279. [Google Scholar] [CrossRef]
- Brose, M.S.; Bible, K.C.; Chow, L.Q.; Gilbert, J.; Grande, C.; Worden, F.; Haddad, R. Management of treatment-related toxicities in advanced medullary thyroid cancer. Cancer Treat. Rev. 2018, 66, 64–73. [Google Scholar] [CrossRef]
- Jimenez, C.; Mimi, I.; Hu, N.; Gagel, R.F. Management of medullary thyroid carcinoma. Endocrinol. Metab. Clin. N. Am. 2008, 37, 481–496. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Martin-Manso, G.; Maxhimer, J.B.; Roberts, D.D. Regulation of nitric oxide signalling by thrombospondin 1: Implications for anti-angiogenic therapies. Nat. Rev. Cancer 2009, 9, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Kappers, M.H.; van Esch, J.H.; Sluiter, W.; Sleijfer, S.; Danser, A.J.; van den Meiracker, A.H. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension 2010, 56, 675–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, J. Tyrosine kinase inhibitors and vascular toxicity: Impetus for a classification system? Curr. Oncol. Rep. 2016, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.B.; Macdonald, B.; Golitz, L.E.; LoRusso, P.; Sekulic, A. Cutaneous adverse effects of targeted therapies: Part II: Inhibitors of intracellular molecular signaling pathways. J. Am. Acad. Dermatol. 2015, 72, 221–236. [Google Scholar] [CrossRef]
- Stanculeanu, D.L.; Daniela, Z.; Oana Catalina, T.; Georgescu, B.; Papagheorghe, L.; Mihaila, R.I. Cutaneous toxicities of molecular targeted therapies. Maedica 2017, 12, 48. [Google Scholar]
- Sibaud, V.; Dalenc, F.; Chevreau, C.; Roché, H.; Delord, J.-P.; Mourey, L.; Lacaze, J.-L.; Rahhali, N.; Taïeb, C. HFS-14, a specific quality of life scale developed for patients suffering from hand–foot syndrome. Oncologist 2011, 16, 1469. [Google Scholar] [CrossRef] [Green Version]
- Cebollero, A.; Puértolas, T.; Pajares, I.; Calera, L.; Antón, A. Comparative safety of BRAF and MEK inhibitors (vemurafenib, dabrafenib and trametinib) in first-line therapy for BRAF-mutated metastatic melanoma. Mol. Clin. Oncol. 2016, 5, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Blevins, D.P.; Dadu, R.; Hu, M.; Baik, C.; Balachandran, D.; Ross, W.; Gunn, B.; Cabanillas, M.E. Aerodigestive fistula formation as a rare side effect of antiangiogenic tyrosine kinase inhibitor therapy for thyroid cancer. Thyroid 2014, 24, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Kwekkeboom, D.J.; Krenning, E.P. Peptide Receptor Radionuclide Therapy in the Treatment of Neuroendocrine Tumors. Hematol. Oncol. Clin. N. Am. 2016, 30, 179–191. [Google Scholar] [CrossRef]
- Rolleman, E.J.; Valkema, R.; de Jong, M.; Kooij, P.P.; Krenning, E.P. Safe and effective inhibition of renal uptake of radiolabelled octreotide by a combination of lysine and arginine. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 9–15. [Google Scholar] [CrossRef]
- Bodei, L.; Cremonesi, M.; Ferrari, M.; Pacifici, M.; Grana, C.M.; Bartolomei, M.; Baio, S.M.; Sansovini, M.; Paganelli, G. Long-term evaluation of renal toxicity after peptide receptor radionuclide therapy with 90 Y-DOTATOC and 177 Lu-DOTATATE: The role of associated risk factors. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1847–1856. [Google Scholar] [CrossRef]
- De Oliveira, M.A.; e Martins, F.M.; Wang, Q.; Sonis, S.; Demetri, G.; George, S.; Butrynski, J.; Treister, N.S. Clinical presentation and management of mTOR inhibitor-associated stomatitis. Oral Oncol. 2011, 47, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.E. Management of adverse events in patients with hormone receptor-positive breast cancer treated with everolimus: Observations from a phase III clinical trial. Supportive Care Cancer 2013, 21, 2341–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Fort, M.K.; Case, E.C.; Rosen, A.C.; Cerci, F.B.; Wu, S.; Lacouture, M.E. Rash to the mTOR inhibitor everolimus: Systematic review and meta-analysis. Am. J. Clin. Oncol. 2014, 37, 266–271. [Google Scholar] [CrossRef]
- Campistol, J.M.; de Fijter, J.W.; Flechner, S.M.; Langone, A.; Morelon, E.; Stockfleth, E. mTOR inhibitor-associated dermatologic and mucosal problems. Clin. Transpl. 2010, 24, 149–156. [Google Scholar] [CrossRef]
- Hutson, T.E.; Figlin, R.A.; Kuhn, J.G.; Motzer, R.J. Targeted therapies for metastatic renal cell carcinoma: An overview of toxicity and dosing strategies. Oncologist 2008, 13, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Shi, V.J.; Rodic, N.; Gettinger, S.; Leventhal, J.S.; Neckman, J.P.; Girardi, M.; Bosenberg, M.; Choi, J.N. Clinical and histologic features of lichenoid mucocutaneous eruptions due to anti–programmed cell death 1 and anti–programmed cell death ligand 1 immunotherapy. JAMA Derm. 2016, 152, 1128–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saw, S.; Lee, H.Y.; Ng, Q.S. Pembrolizumab-induced Stevens–Johnson syndrome in non-melanoma patients. Eur. J. Cancer 2017, 81, 237–239. [Google Scholar] [CrossRef]
- Scarpati, G.D.V.; Fusciello, C.; Perri, F.; Sabbatino, F.; Ferrone, S.; Carlomagno, C.; Pepe, S. Ipilimumab in the treatment of metastatic melanoma: Management of adverse events. Oncotargets Ther. 2014, 7, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; De Felice, K.; Loftus, E.V., Jr.; Khanna, S. Systematic review: Colitis associated with anti-CTLA-4 therapy. Aliment. Pharmacol. Ther. 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714. [Google Scholar] [CrossRef]
- Yanai, S.; Nakamura, S.; Matsumoto, T. Nivolumab-induced colitis treated by infliximab. Clin. Gastroenterol. Hepatol. 2017, 15, e80–e81. [Google Scholar] [CrossRef] [Green Version]
- Barroso-Sousa, R.; Barry, W.T.; Garrido-Castro, A.C.; Hodi, F.S.; Min, L.; Krop, I.E.; Tolaney, S.M. Incidence of endocrine dysfunction following the use of different immune checkpoint inhibitor regimens: A systematic review and meta-analysis. JAMA Oncol. 2018, 4, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Wang, X.; Woo, K.M.; Iyriboz, T.; Halpenny, D.; Cunningham, J.; Chaft, J.E.; Segal, N.H.; Callahan, M.K.; Lesokhin, A.M. Pneumonitis in patients treated with anti–programmed death-1/programmed death ligand 1 therapy. J. Clin. Oncol. 2017, 35, 709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.W.; Ramaiya, N.H.; Krajewski, K.M.; Jagannathan, J.P.; Tirumani, S.H.; Srivastava, A.; Ibrahim, N. Ipilimumab associated hepatitis: Imaging and clinicopathologic findings. Investig. New Drugs 2013, 31, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L. Fulminant myocarditis with combination immune checkpoint blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef]
- Cortazar, F.B.; Marrone, K.A.; Troxell, M.L.; Ralto, K.M.; Hoenig, M.P.; Brahmer, J.R.; Le, D.T.; Lipson, E.J.; Glezerman, I.G.; Wolchok, J. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. 2016, 90, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Viola, D.; Valerio, L.; Molinaro, E.; Agate, L.; Bottici, V.; Biagini, A.; Lorusso, L.; Cappagli, V.; Pieruzzi, L.; Giani, C.; et al. Treatment of advanced thyroid cancer with targeted therapies: Ten years of experience. Endocr. Relat. Cancer 2016, 23, R185–R205. [Google Scholar] [CrossRef] [Green Version]
- Chau, N.G.; Haddad, R.I. Vandetanib for the treatment of medullary thyroid cancer. Clin. Cancer Res. 2013, 19, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Naoum, G.E.; Morkos, M.; Kim, B.; Arafat, W. Novel targeted therapies and immunotherapy for advanced thyroid cancers. Mol. Cancer 2018, 17, 51. [Google Scholar] [CrossRef]
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic lymphoma kinase: Signalling in development and disease. Biochem. J. 2009, 420, 345–361. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Genetic alterations affecting MAPK and PI3K signaling pathways in thyroid cancer.

Figure 2.
Genetic alterations affecting nuclear processes in thyroid cancer. The figure shows mutations/gene fusions affecting nuclear processes such as transcription, DNA repair, and epigenetic regulation of genes to promote oncogenesis. The genes that have been observed to be mutated in thyroid cancer are shown in red with an asterisk.
Figure 2.
Genetic alterations affecting nuclear processes in thyroid cancer. The figure shows mutations/gene fusions affecting nuclear processes such as transcription, DNA repair, and epigenetic regulation of genes to promote oncogenesis. The genes that have been observed to be mutated in thyroid cancer are shown in red with an asterisk.

Figure 3.
Mechanism of RAI resistance and its reversal with targeted therapies. (A) Under normal conditions, expression of sodium-iodine symporter (NIS) is regulated through TSHR, resulting in stimulation of thyroid-specific transcriptional factors such as PAX8, which promotes NIS transcription and its expression on the cell surface. (B) In thyroid tumor cells with hyperactive MAPK and PI3K/AKT signaling, the NIS transcription is repressed, resulting in loss of its cell surface expression and RAI resistance. (C) Targeted treatment with inhibitors that block the MAPK/PI3K signaling pathways improves NIS expression and RAI avidity. * Trastuzumab has been tested in breast cancer model to target HER2 signaling but not in thyroid cancer model.
Figure 3.
Mechanism of RAI resistance and its reversal with targeted therapies. (A) Under normal conditions, expression of sodium-iodine symporter (NIS) is regulated through TSHR, resulting in stimulation of thyroid-specific transcriptional factors such as PAX8, which promotes NIS transcription and its expression on the cell surface. (B) In thyroid tumor cells with hyperactive MAPK and PI3K/AKT signaling, the NIS transcription is repressed, resulting in loss of its cell surface expression and RAI resistance. (C) Targeted treatment with inhibitors that block the MAPK/PI3K signaling pathways improves NIS expression and RAI avidity. * Trastuzumab has been tested in breast cancer model to target HER2 signaling but not in thyroid cancer model.

Figure 4.
Targeted therapies for the treatment of thyroid cancer. The figure shows inhibitor drug molecules targeting various RTKs, components of MAPK and PI3K signaling pathways, and gene fusions in thyroid cancer. The immunotherapeutic agents, PRRT molecules, and a vaccine with the potential for the treatment of thyroid cancer are also included in the figure.
Figure 4.
Targeted therapies for the treatment of thyroid cancer. The figure shows inhibitor drug molecules targeting various RTKs, components of MAPK and PI3K signaling pathways, and gene fusions in thyroid cancer. The immunotherapeutic agents, PRRT molecules, and a vaccine with the potential for the treatment of thyroid cancer are also included in the figure.


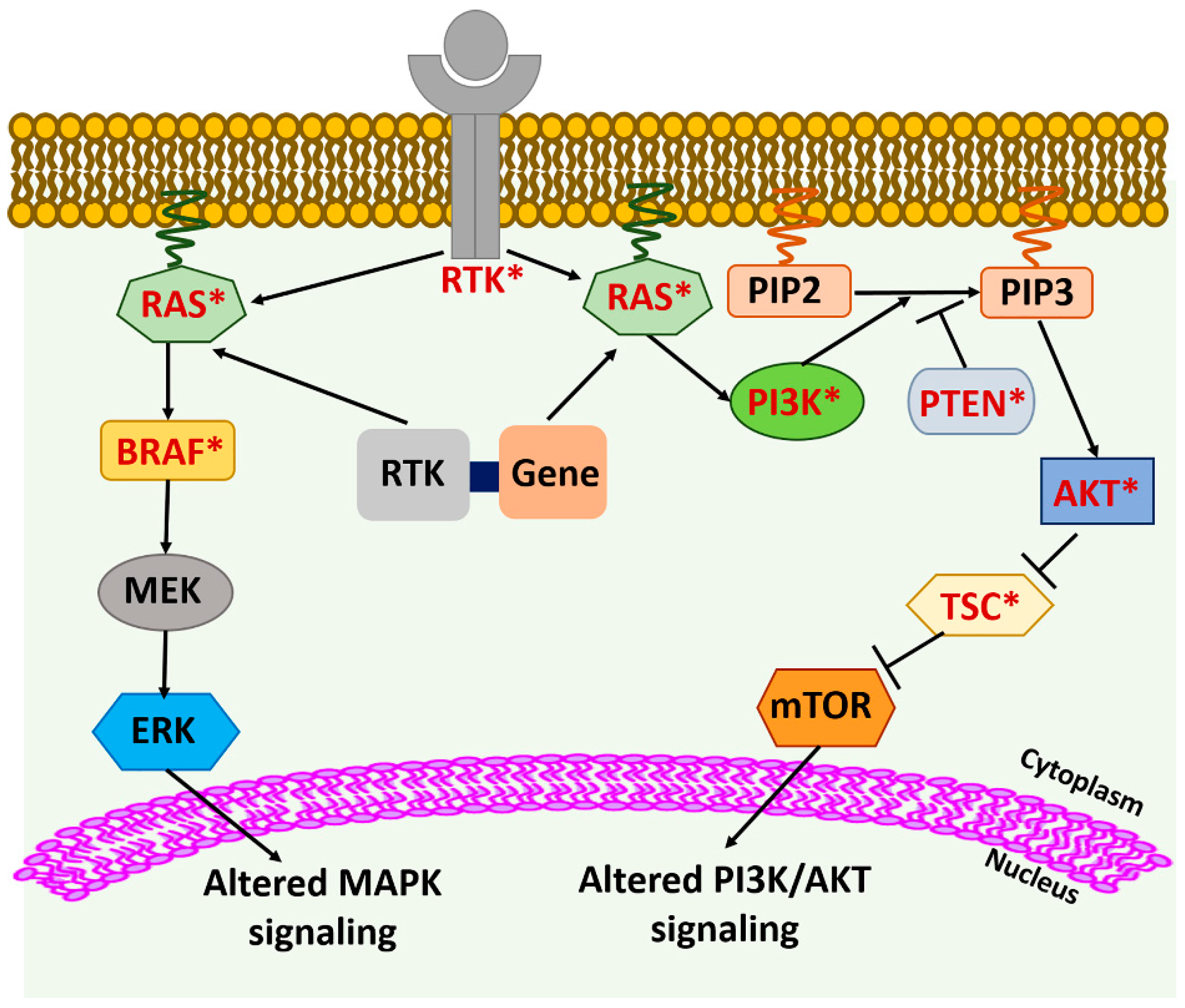{kind=link}
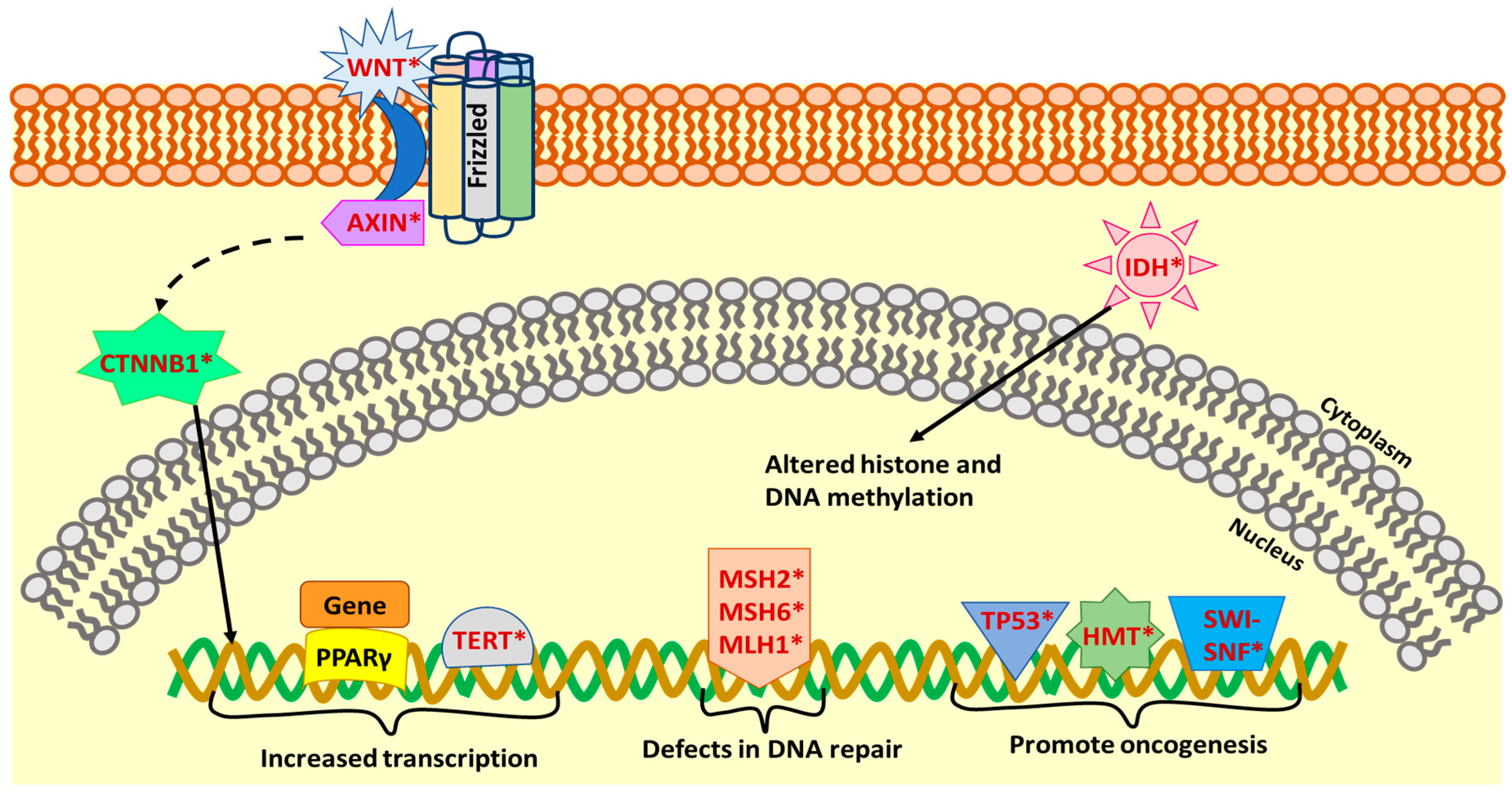{kind=link}
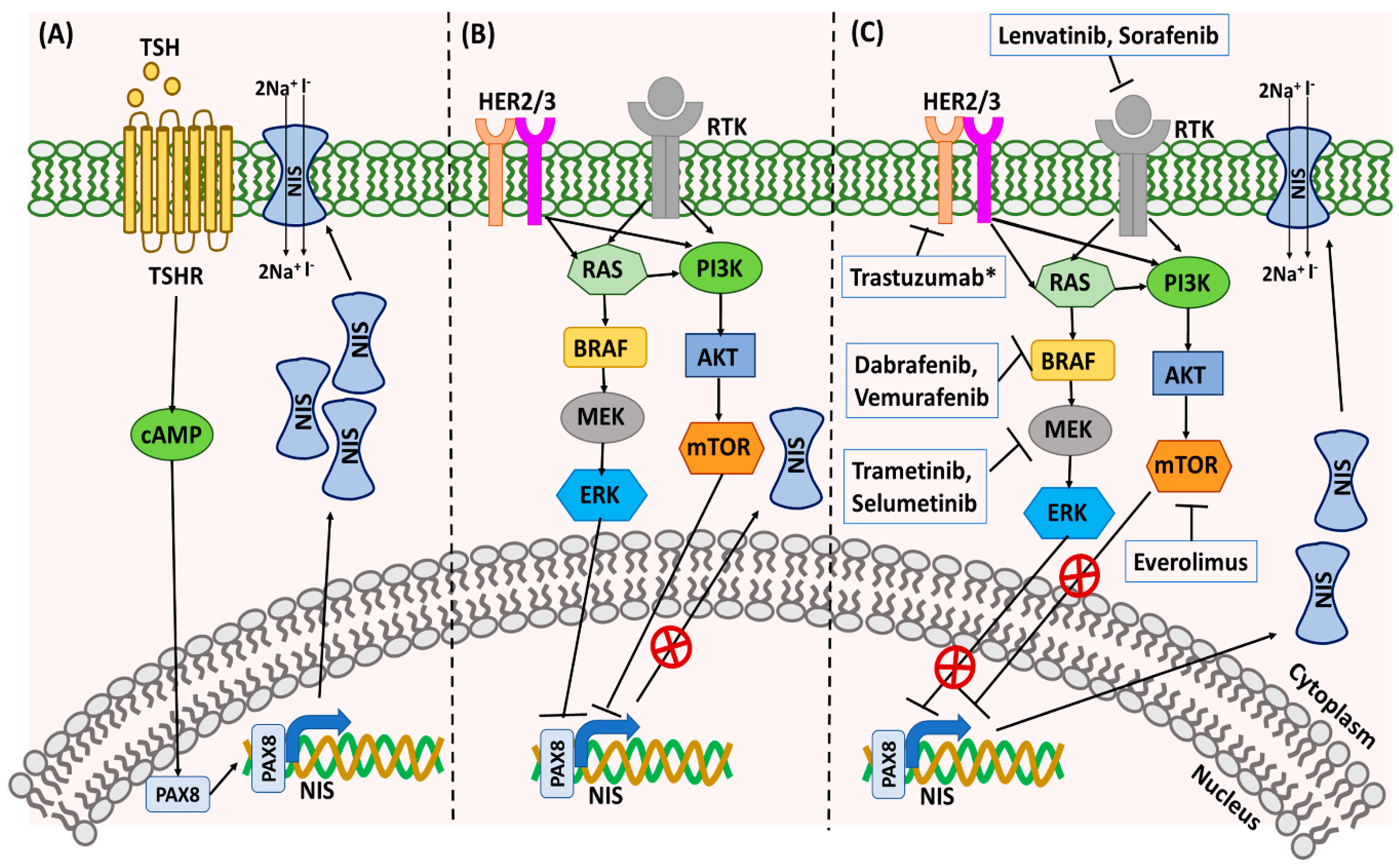{kind=link}
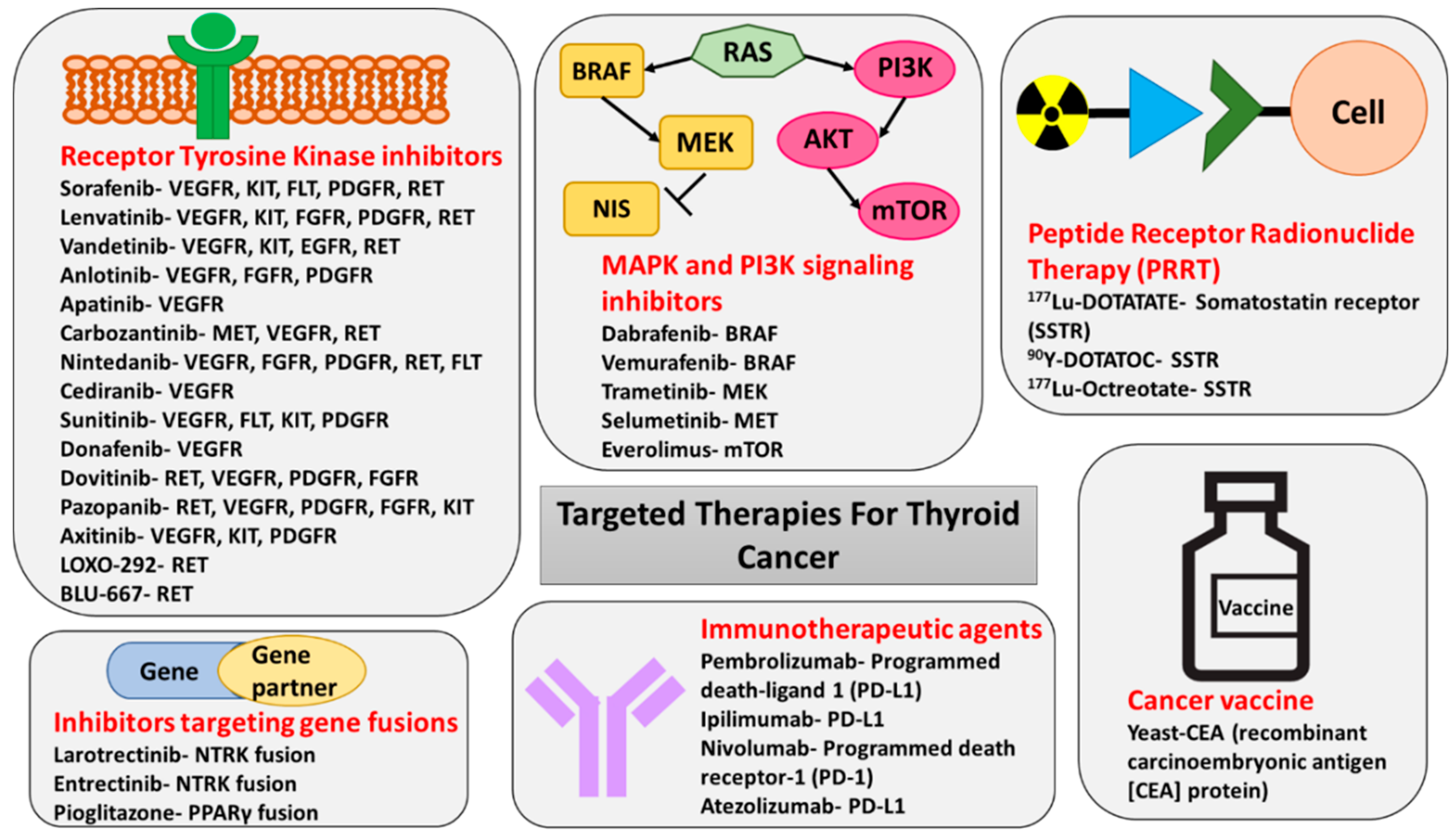{kind=link}
Table 1.
Molecular alterations in thyroid cancer.
| Pathways/Cell Components | Genes | Type of Molecular Alterations | Type of Thyroid Cancers (Frequency) | Role in Thyroid Cancer |
|---|---|---|---|---|
| MAPK and PI3K/AKT signaling pathway | BRAF | Activating mutations | PTC (40–80%), | Promotes aggressive tumor phenotype and is associated with poor clinical prognosis and higher risk of recurrence [9,18]. |
| PDTCs (5–35%), | ||||
| ATCs (10–50%) [7] | ||||
| RAS | Activating mutation | FTC (30–50%), | Promotes early tumorigenesis and may predispose DTC to de-differentiate into aggressive forms of thyroid cancer [17,19,20]. | |
| FVPTC (30–45%), | ||||
| HTC (15%), | ||||
| PDTC (20–40%), | ||||
| ATC (10–20%), | ||||
| MTC (10–30%), | ||||
| classical PTC (rarely) [7,10,12,13,14,15] | ||||
| PTEN | Inactivating mutation, inactivating deletion, epigenetic changes leading to loss of expression | PTC (2%) | PTEN loss promotes thyroid cancer progression and invasion [23]. | |
| FTC (2–14%), | ||||
| HTC (4–17%), | ||||
| ATC (11–15%), | ||||
| PDTC (4%) [7,10,21,22] | ||||
| AKT1 | Copy number gain and activating mutations | ATC (19%), | Promotes thyroid tumorigenesis and progression [24,25]. | |
| FTC (8%), | ||||
| PDTC (19%) [7,21] | ||||
| PIK3CA | Gene amplification and activating mutations | ATC (18%), FTC (1%), | Promotes oncogenic transformation [26]. | |
| PDTC (2%) [7] | ||||
| RTK signaling pathway | RET-PTC | Activating gene fusions | PTC (5–25%) [7] | Promotes oncogenic transformation [19,27] |
| RET | Activating point mutations | Hereditary MTC (95%), | Promotes oncogenic transformation [28]. | |
| Sporadic MTC (40–70%) [28,29] | ||||
| NTRK | Gene fusions | PTC (5–25%) [30] | Biological consequence of NTRK fusions in thyroid cancer remains to be explored. | |
| ALK | Activating gene fusions | ATC, DTC [7] | Promotes thyroid cancer progression and aggressiveness [7]. | |
| ALK | Activating mutations | ATC (11%) [31] | Promotes thyroid tumorigenesis via anchorage-independent growth, and cell invasion in in vitro model [31]. | |
| Other RTKs | Activating gene mutations | PDTC, HTC [10,32] | Activate transcriptional pathways involved in regulation of various cellular processes [33]. | |
| Copy number gains | ATC, FTC [21] | |||
| Wnt pathway | CTNNB1 | Gain of function mutations | ATC (60–65%), | Involved in thyroid cancer progression [34]. |
| Axin1 | Loss of function mutations | PDTC (25%) [7] | ||
| Tumor suppressor | TP53 | Inactivating mutations | ATC (50–80%), | Promotes tumor progression [7]. |
| PDTC (10–35%), | ||||
| DTC (variable frequency) [7,35] | ||||
| Transcription factors | PAX8-PPARγ | Gene fusions | FTC (30–35%), | Promotes oncogenesis by transcriptional activation of downstream effector genes [36]. |
| FA (2–13%) [7] | ||||
| Enzyme | TERT | Activating promoter mutations | ATC (40–70%), | Associated with aggressive thyroid cancer phenotype and poor prognosis [37]. |
| PDTC (40%), | ||||
| HTC (32%), | ||||
| PTC (10%), | ||||
| FTC (20%) [7,10,37] | ||||
| DNA Mismatch Repair pathway | MSH2, MSH6, and MLH1 | Loss of function mutations | PDTC (2%), | Defective mismatch repair pathway promotes tumorigenesis [38] |
| ATC (12%) [38] | ||||
| Other pathways/cell components | SWI-SNF chromatin remodeling complex | Nature of mutations remain to be explored | PDTC (6%), | Biological consequence of these mutations in thyroid cancer remain to be explored. |
| ATC (36%) [38] | ||||
| EIF1AX | Loss of function mutations | PDTC (11%), | Leads to defects in the protein translation [7]. | |
| ATC (9%), PTC (1–2%) [7] | ||||
| Histone methyltransferases (HMTs) | Nature of mutations remain to be explored | PDTC (7%), | Biological consequence of these mutations in thyroid cancer remain to be explored. | |
| ATC (24%) [38] | ||||
| IDH1 | Probably inactivating mutations | ATC (11%), FTCs (5%) [7] | Associated with loss of IDH1 enzymatic activity, but the tumorigenic role of these mutations in thyroid cancer remain to be explored [7,39]. | |
| Mutations in mitochondrial genes, epigenetic modifiers, and components of DNA damage and repair pathway; chromosome 5 and 7 duplications; loss of heterozygosity; and in-frame gene fusions | Nature of mutations remain to be explored | HTC [10] | Biological consequence of these mutations in thyroid cancer remain to be explored. |
Table 2.
Novel targeted therapies for thyroid cancer treatment.
| Drug/ ClinicalTrials.gov ID/Reference | Mechanism of Action | Enrolled Patients * | Primary Outcome | Study Design | Results | Reported Adverse Events |
|---|---|---|---|---|---|---|
| Tyrosine Kinase Inhibitors | ||||||
| Anlotinib Sun et al. [83] | VEGFR, PDGFR, FGFR1 | MTC: 58 (locally advanced or metastatic) | PFS | Phase II, single arm, open label | PR: 56.9% PFS at 48 weeks: 85.5% Drop of calcitonin and CEA > 50% in 78% of patients | Hand–foot syndrome, lipid profile abnormalities, fatigue, diarrhea, proteinuria |
| Anlotinib Li et al. [84] | MTC: 91 (no previous exposure to antiangiogenic agents) | PFS | Phase II/III, two arms, randomized, double blinded, placebo controlled (ALTER0103) | Median PFS: 20.67 months in Anlotinib arm vs. 11.07 months in placebo arm | ||
| Anlotinib NCT04309136 | DTC/MTC (locally advanced thyroid cancer with/without distant metastasis) | ORR | Phase II, single arm, open label, recruiting | N/A Estimated End Date 6/2022 (clinicaltrials.gov) | ||
| Anlotinib NCT02586337 | DTC (RAI refractory disease) | PFS | Phase II/III, two arms, randomized, double blinded, placebo controlled, active, not recruiting (ALTER01032) | N/A Estimated End Date 12/2019 (clinicaltrials.gov) | ||
| Apatinib NCT03048877 | VEGFR | DTC (locally advanced or metastatic disease) | PFS | Phase III, two arms, Randomized, double blinded, placebo controlled, active, not recruiting | N/A Estimated End Date 6/2021 (clinicaltrials.gov) | Hypertension, hand–foot syndrome, proteinuria, fatigue |
| Apatinib NCT03167385 | DTC (locally advanced or metastatic disease) | Disease control rate | Phase II, single arm, open label, recruiting | N/A Estimated End Date 12/2020 (clinicaltrials.gov) | ||
| Axitinib Cohen et al. [85] | VEGFR, PDGFR, KIT | DTC: 45 (resistant to or not appropriate for RAI) MTC: 11 ATC 11 | ORR | Phase II, single arm, open label | ORR of 30% SD for ≥ 16 weeks: 38% PFS: 18.1 months | Fatigue, diarrhea, nausea, anorexia, hypertension, stomatitis |
| Axitinib Capdevila et al. [86] | DTC: 34 (RAI refractory) MTC: 13 | ORR | Retrospective study, compassionate use program (CUP) in Spain | ORR for PTC: 29.4% PFS for PTC: 7.4 months ORR for MTC: 23.1% PFS for MTC: 9.4 months | ||
| Cabozantinib Elisei et al. [103] | VEGFR, RET, MET, FLT3, AXL | MTC: 219 (locally advanced, metastatic with radiographic progression in the past 14 months) | PFS | Phase III, randomized, double blinded, placebo controlled | PFS: 11.2 vs. 4.0 months for placebo (regardless of RET mutation status) PR: 28% (regardless of RET mutation status) | Diarrhea, hand–foot syndrome, weight loss, decreased appetite, nausea, fatigue |
| Cabozantinib NCT03690388 | DTC (RAI disease that progressed after use of VEGFR–TKI therapy) | PFS ORR | Phase III, two arms, randomized, double blinded, placebo controlled, recruiting | N/A Estimated End Date 12/2022 (clinicaltrials.gov) | ||
| Cabozantinib NCT02041260 | DTC (RAI refractory disease with radiographic progression in the past 14 months) | Number of AEs | Phase II, single arm, open label, active, not recruiting | N/A Estimated End Date 3/2021 (clinicaltrials.gov) | ||
| Donafenib NCT03602495 | VEGFR | DTC (RAI refractory/resistant disease) | PFS | Phase III, two arms, randomized, double blinded, placebo controlled, recruiting | N/A Estimated End Date 3/2021 (clinicaltrials.gov) | Hand–foot syndrome, diarrhea, rash, hair loss, hypertension, tachycardia |
| Dovitinib Lim et al. [87] | DTC: 28 (RAI refractory or not appropriate) MTC: 12 | ORR | Phase II, single arm, open label | ORR: 20.5% Median PFS: 5.4 months | Diarrhea, anorexia, nausea, vomiting, fatigue | |
| Lenvatinib Schlumberger et al. [81] | DTC: 261 (RAI refractory progressive disease) | PFS | Phase III, two arms, randomized, double blinded, placebo controlled | Median PFS: 18.3 vs. 3.6 for placebo ORR: 64.8% (CR 4/261 + PR 165/261) | Hypertension, diarrhea, fatigue, anorexia, weight loss, nausea | |
| Lenvatinib Schlumberger et al. [88] | VEGFR, PDGFR, EGFR, RET, KIT | MTC: 59 (unresectable progressive disease) | ORR | Phase II, single arm, open label | PR 36% | |
| Lenvatinib Tahara et al. [89] | Enrolled all types of thyroid cancer, but results reported one cohort for 17 patients with ATC | Serious/non-serious AE | Phase II, single arm, open label | Most frequent AE (Decreased appetite, 82%; HTN, 82%; Fatigue, 59%; Nausea, 59%; Proteinuria, 59%) Secondary Endpoints: ORR: 24% Median PFS: 7.4 months Median OS | ||
| Lenvatinib NCT03573960 | DTC (locally recurrent or metastatic progressive RAI refractory disease) | -Grade 3 and higher TEAEs -Number of dose reductions -Time to 1st dose reduction | Phase IV, single arm, open label, recruiting | N/A Estimated End Date 12/2020 (clinicaltrials.gov) | ||
| Lenvatinib NCT03506048 | DTC (progressive despite RAI in the past 12 months) | Time to progression | Phase II, single arm, open label, recruiting | N/A Estimated End Date 1/2021 (clinicaltrials.gov) | ||
| Lenvatinib NCT02702388 | DTC (RAI refractory disease) | -PFS -TEAEs | Phase II, two arms, randomized, double blinded, evaluating the starting dose 18 mg vs. 24 mg, active, not recruiting | N/A Estimated End Date 9/2020 (clinicaltrials.gov) | ||
| Lenvatinib NCT02966093 | DTC (RAI refractory disease in China) | PFS | Phase III, two arms, randomized, double blinded, placebo controlled, active, not recruiting | N/A Estimated End Date 4/2021 (clinicaltrials.gov) | ||
| Nintedanib NCT01788982 | VEGFR, PDGFR, FGFR, RET, FLT, | DTC MTC (as second-line therapy if progressive disease after first-line therapy) | PFS | Phase II, two arms, randomized, double blinded, active not recruiting | N/A Estimated End Date 9/2019 (clinicaltrials.gov) | Diarrhea, nausea, vomiting, abdominal pain |
| Pazopanib Bible et al. [90] | VEGFR, FGFR, PDGFR, RET, KIT | DTC: 37 (progressive RAI refractory disease) | Tumor response rate | Phase II, two arms, open label | PR: 49% | Fatigue, skin and hair hypopigmentation, diarrhea, nausea |
| Pazopanib Bible et al. [91] | MTC: 35 (advanced or metastatic disease) | Tumor response rate | Phase II, two arms, open label | PR: 14.3% PFS: 9.4 months OS: 19.9 months | ||
| Pazopanib Bible et al. [92] | ATC: 15 (advanced or metastatic disease) | Tumor response rate | Phase II, two arms, open label | No response | ||
| Sorafenib Brose et al. [79] | VEGFR, PDGFR, RET, KIT, FLT | DTC: 209 (locally advanced or metastatic RAI refractory disease) | PFS | Phase III, two arms, randomized, double blinded, placebo controlled (decision trial) | PFS: 10.8 months vs. 5.8 for placebo (regardless of mutation status) PR: 12% | Hand–foot skin reaction, diarrhea, alopecia, skin rash or desquamation |
| Sorafenib Lam et al. [93] | MTC: 16 (locally advanced or metastatic, arm A—Hereditary MTC, arm B—Sporadic MTC) | ORR | Phase II, single arm, open label | PR: 6.3% SD: 87.5% | ||
| Sorafenib Capdevila et al. [94] | DTC: 16 MTC: 15 ATC: 3 (metastatic progressive unsuitable for surgery, RAI, or radiotherapy) | ORR | Retrospective, Spanish off-label-sorafenib-use program | DTC PR: 19% MTC PR: 47% ATC PR: 33% | ||
| Sunitinib Bikas et al. [95] | VEGFR, PDGFR, RET, KIT, FLT | DTC: 23 (metastatic, residual, recurrent, or progressive disease) | ORR | Phase II, single arm, open label, as adjunctive treatment | PR: 26% SD: 57% | Cytopenia, diarrhea, fatigue, hand–foot skin reaction, nausea, musculoskeletal pain, hypertension |
| Sunitinib Carr et al. [96] | DTC: 28 (RAI refractory disease, FDG–PET avid disease) MTC: 7 (FDG–PET avid disease) | ORR | Phase II, Single Arm, Open Label | DTC ORR: 28% MTC ORR: 50% | ||
| Sunitinib Ravaud et al. [97] | DTC: 41 (RAI resistant) MTC: 26 ATC: 4 (sunitinib as a first-line anti-angiogenic therapy) | ORR | Phase II, single arm, open label | DTC PR: 22% MTC PR: 38.5% ATC: no response | ||
| Vandetanib Wells et al. [104] | VEGFR, EGFR, RET, KIT | MTC: 231 (unresectable locally advanced or metastatic disease) | PFS | Phase III, two arms, randomized, double blinded, placebo controlled | PFS HR 0.46 compared to placebo (predicted median PFS 30.5 vs. 19.3 months for placebo) PR 45% | Diarrhea, rash, nausea, hypertension, headache |
| Vandetanib Leboulleux et al. [98] | DTC: 72 (locally advanced or metastatic disease) | PFS | Phase II, randomized, double blind, placebo controlled | PFS 11.1 vs. 5.9 months for placebo | ||
| Vandetanib NCT01876784 | DTC: 119 (locally advanced or metastatic RAI refractory or unsuitable disease) | PFS | Phase III, two arms, randomized, double blind, placebo controlled | PFS (no statistically significant difference—10.0 months vs. 5.7 months with p value 0.080) | ||
| Selective Ret Inhibitors | ||||||
| LOXO-292 (Selpercatinib) NCT03157128 | RET | MTC (among multiple RET-fusion and RET-activation-positive solid tumors) | Phase I: maximum tolerated dose (MTD), recommended phase II dose Phase II: ORR | Phase I/II, single arm, open label (LIBRETTO-001 trial), recruiting | N/A Estimated End Date 3/2022 (clinicaltrials.gov) | Fatigue, dyspnea, joint pain, insomnia, abnormal liver enzymes (from proof-of-concept study) |
| LOXO-292 (Selpercatinib) NCT03899792 | MTC PTC (among multiple RET-altered solid and CNS tumors in pediatrics) | Phase I: safety Phase II: ORR | Phase I/II, single arm, open label (LIBRETTO-121), recruiting | N/A Estimated End Date 10/2022 (clinicaltrials.gov) | ||
| BLU-667 (Pralsetinib) NCT03037385 | RET | MTC: 49 RET mutant PTC: 5 RET mutant (among multiple RET-altered solid tumors) | Phase I: maximum tolerated dose, number of patients with TEAEs Phase II: ORR, number of patients with TEAEs | Phase I/II, seven groups in Phase II, open label (ARROW TRIAL), recruiting | MTC ORR: 47% MTC PR: 21/49 MTC CR: 2/49 MTC SD: 25/49 Rapid reduction of calcitonin and CEA levels PTC PR 2 of 4 evaluable Estimated End Date 2/2024 (clinicaltrials.gov) | Constipation, elevated liver enzymes, diarrhea, fatigue, elevated serum creatinine, WBC count decrease, hypertension |
| BRAF Inhibitors | ||||||
| Dabrafenib Falchook et al. [99] | BRAFV600E | DTC: 13 (BRAFV600E mutant disease) | ORR | Subset of phase I study | PR: 29% | Skin papilloma hyperkeratosis, alopecia, fatigue, fever, diarrhea |
| Vemurafenib Brose et al. [100] | BRAFV600E | DTC: 51 (unresectable and metastatic RAI refractory BRAFV600E mutant disease) | ORR | Phase II, parallel assignment, open label | PR 38.5% (VEGFR multikinase inhibitor naïve cohort) PR 27% (prior treatment with VEGFR multikinase inhibitors) | Rash, fatigue, arthralgia |
| Vemurafenib Hytman et al. [105] | ATC: 7 (multiple BRAFV600E mutant tumors) | ORR | Phase II, basket trial | PR: 14% CR: 14% | ||
| BRAF/MEK Inhibitor Combination | ||||||
| Dabrafenib and Trametinib Subbiah et al. [106] | Dabrafenib: BRAFV600E Trametinib: MEK1, MEK2 | ATC: 16 locally advanced or metastatic BRAFV600E mutant disease | ORR | Phase II, single arm, open label | PR: 63% CR: 6% | See Dabrafenib See Trametinib |
| Tropomyosin Receptor Kinase Inhibitor | ||||||
| Larotrectinib Drilon et al. [107] | TRKI: TRKA, TRKB, TRKC | NTRK harboring solid tumors in pediatrics and adults (thyroid cancer: 5 patients) | ORR | NCT02122913 (Phase I adults, open label) NCT02637687 (Phase I/II, pediatrics, open label) NCT02576431 (Phase II pediatrics and adults, basket study) | PR: 100% (5/5) | Elevated ALT and AST, fatigue, nausea, vomiting, dizziness |
| Entrectinib Doebele et al. [108] | TRKI: TRKA, TRKB, TRKC ALK, ROS1 | Locally advanced and metastatic NTRK-fusion solid tumors (thyroid cancer: 4 patients) | ORR Median duration of response | (STARTRK-1): NCT02097810 Phase I (STARTRK-2): NCT02568267 Phase II (ALKA-372-001): EudraCT 2012-000148-88 Phase I | Thyroid cancer PR 50% (2/4) | Dysgeusia, dizziness, constipation, diarrhea, weight gain |
| Radioactive Iodine Restoration Treatments | ||||||
| Selumetinib Ho et al. [109] | MEK1, MEK2 | DTC: 20 (RAI refractory disease—9 BRAF mutation, 5 NRAS mutation) | The percentage of patients with selumetinib-induced increases in iodine uptake in the index tumor | Open label, single arm, treatment with selumetinib, then evaluate by RAI uptake study | Increased I-124 uptake in 12/20 (4/9 with BRAF mutations) (5 of 5 with NRAS mutations) (8/12 reached dosimetry threshold for RAI treatment) | Fatigue, maculopapular rash, elevated liver enzymes, acneiform rash |
| Selumetinib ISRCTN17468602 (UK) | Locally advanced and metastatic RAI refractory DTC | PFS | Phase II, single arm, treatment with selumetinib, then evaluate by RAI uptake study (SEL-I-METRY Trial) | N/A | ||
| Trametinib with RAI NCT02152995 | MEK1, MEK2 | DTC: RAS mutant or RAS/RAF wild-type RAI-refractory recurrent and/or metastatic disease | PFS | Phase II, single arm, open label, recruiting | N/A Estimated End Date 12/2020 (clinicaltrials.gov) | Acneiform rash |
| Trametinib OR Dabrafenib NCT03244956 | DTC RAI refractory with RAS (trametinib) or BRAFV600E (dabrafenib) mutation | ORR | Phase II, two arms, open label, recruiting | N/A Estimated End Date 12/2022 | See Trametinib See Dabrafenib | |
| Trametinib/ combination Dabrafenib and Trametinib or Vemurafenib and Cobimetinib Irvani et al. [110] | Trametinib (see above), Dabrafenib (see above), Vemurafenib (see above), Cobimetinib: MEK1, MEK2 | DTC: 6 (3 BRAFV600E positive treated with combination, 3 NRAS-positive treated with trametinib) | Restoration of RAI uptake | Retrospective, cohort study | RAI uptake restoration: BRAFV600E (3/3), NRAS (1/3) with median follow-up 16.6 months | See Trametinib See Dabrafenib See Vemurafenib Cobimetinib: diarrhea, pyrexia, photosensitivity reaction, abnormal LFT, hyponatremia |
| Peptide Receptor Radionuclide Therapy | ||||||
| PRRT 90Y-DOTATATE or 177Lu-DOTATATE Budiawan et al. [111] | DTC: 7 MTC: 8 Mixed DTC and MTC: 1 (non-RAI avid and RAI refractory thyroid cancer) | Treatment response Treatment related toxicity | Phase II trial, single arm, open label | PR 18.2% SD 36.4% Median PFS 25 months Mean survival 4.2 years | Mild hematological toxicity, abnormal liver enzymes, mild nephrotoxicity | |
| PRRT (90)Y-DOTATOC Versari et al. [112] | DTC: 41 (RAI negative enrolled in the study 11/41 patients were treated with PRRT) | Treatment response | Phase II trial, single arm, open label | ORR 63% (PR 2/11, SD 4/11) | ||
| PRRT 177Lu-DOTATATE or 90Y-DOTATOC Lapa et al. [113] | DTC: 8 (progressive RAI refractory) MTC: 4 | Assess tumor heterogenicity in predicting PFS and OS | Phase II trial, single arm, open label | Mean PFS 221 days Mean OS 450 days | ||
| mTOR Inhibitors | ||||||
| Everolimus Lim et al. [101] | mTOR | Thyroid cancer (all subtypes): 38 | Disease control rate (PR + SD > 12 weeks) | Phase II, single arm, open label | PR: 5% (2/38, one PTC patient and one FTC) SD: 76% | Mucositis, anorexia, abnormal liver enzymes, acneiform rash |
| Everolimus Hanna et al. [102] | DTC: 33 MTC: 10 ATC: 7 | PFS | Phase II, single arm, open label | DTC: Median PFS 12.9 months, PR 1/38 MTC: Median PFS 13.1 months, PR 1/10 ATC: Median PFS 2.2 months, PR 1/7 | ||
| Immunotherapy | ||||||
| Pembrolizumab Mehnert et al. [114] | PD-1 receptor | DTC: 22 (Refractory to standard therapy, with PD-L1 expression) | ORR | Phase 1b, single arm, open label (KEYNOTE-28 Trial) | ORR: 9% PR: 2/22 (9%) SD: 13/22 (59%) Median PFS (PR+SD): 7 months | Diarrhea, fatigue, pruritis, rash, colitis (grade 3 in one patient) |
| Combination Therapies Under Investigation | ||||||
| Cabozantinib and Atezolizumab NCT03170960 | Cabozantinib (see above), Atezolizumab: PD-L1 | Multiple tumors, including DTC that is locally advanced or metastatic | Dose escalation: maximum tolerated Dose Expansion: ORR | Phase I/II, dose escalation/expansion, open label, recruiting | Estimated End Date 12/2021 (clinicaltrials.gov) | N/A |
| Cabozantinib, Nivolumab, and Ipilimumab NCT03914300 | Cabozantinib (see above), Nivolumab: PD-1, Ipilimumab: CTLA-4 | DTC (RAI refractory progressive after one prior VEGFR therapy) | ORR | Phase II, single group assignment, open label, recruiting | Estimated End Date 1/2021 (clinicaltrials.gov) | N/A |
| Cediranib Maleate with or without Lenalidomide NCT01208051 | Cediranib: VEGFR, Lenalidomide: CRL4CRBN E3 ubiquitin ligase | DTC (unresectable progressive RAI refractory disease) | Phase I: Maximum tolerated dose Phase II: PFS | Phase I/II, parallel assignment, randomized, open label, active, not recruiting | Estimated End Date 2/2020 (clinicaltrials.gov) | N/A |
| Lenvatinib and Denosumab NCT03732495 | Lenvatinib (see above), Denosumab: RANKL | DTC (RAI resistant with bone metastasis) | Skeletal-related event-free (multiple) | Phase II, single group assignment, open label, recruiting | Estimated End Date 6/2022 (clinicaltrials.gov) | N/A |
| Lenvatinib and Pembrolizumab (PD-1 Inhibitor) NCT02973997 | Lenvatinib (see above), Pembrolizumab: (see above) | DTC (progressive RAI refractory disease) | ORR | Phase II, single group assignment, open label, active not recruiting | Estimated End Date 9/2022 (clinicaltrials.gov) | N/A |
Abbreviations: AE, adverse event; ATC, anaplastic thyroid cancer; CEA, carcinoembryonic antigen; CR, complete response; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; DCR, disease control rate; DTC, differentiated thyroid cancer; ECG, electrocardiogram; FDA, US Food and Drug Administration; FTC, follicular thyroid cancer; FGFR, fibroblast growth factor receptor; FTL-3, FMS-like receptor tyrosine kinase-3; HR, hazard ratio; MTC, medullary thyroid cancer; MTD, maximum tolerated dose; N/A, not available; ORR, objective response rate; OS, overall survival; PD, progressive disease; PD-1, programmed cell death protein 1; PDGFR, platelet-derived growth factor receptor PD; PFS, progression-free survival; PR, partial response; PRRT, peptide receptor radionuclide therapy; PTC, papillary thyroid cancer; RAI, radioactive iodine; SD, stable disease; SSTR, somatostatin receptor; TEAEs, treatment emergent adverse events; TKI, tyrosine kinase inhibitor; TRKI, tropomyosin receptor kinase inhibitor; RANK, receptor activator of nuclear factor kappa-Β ligand; RET, rearranged during transfection; VEGFR, vascular endothelial growth factor receptor. * For placebo-controlled studies, only the number for patients enrolled under the treatment arm is mentioned.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Al-Jundi, M.; Thakur, S.; Gubbi, S.; Klubo-Gwiezdzinska, J. Novel Targeted Therapies for Metastatic Thyroid Cancer—A Comprehensive Review. Cancers 2020, 12, 2104. https://doi.org/10.3390/cancers12082104
AMA Style
Al-Jundi M, Thakur S, Gubbi S, Klubo-Gwiezdzinska J. Novel Targeted Therapies for Metastatic Thyroid Cancer—A Comprehensive Review. Cancers. 2020; 12(8):2104. https://doi.org/10.3390/cancers12082104
Chicago/Turabian StyleAl-Jundi, Mohammad, Shilpa Thakur, Sriram Gubbi, and Joanna Klubo-Gwiezdzinska. 2020. "Novel Targeted Therapies for Metastatic Thyroid Cancer—A Comprehensive Review" Cancers 12, no. 8: 2104. https://doi.org/10.3390/cancers12082104
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.