
Emerging Trends for Radio-Immunotherapy in Rectal Cancer


1
Translational Research Center in Onco-Hematology, Department of Medicine, Faculty of Medicine, University of Geneva, 1205 Geneva, Switzerland
2
Swiss Cancer Center Léman, 1005 Lausanne, Switzerland
3
Department of Oncology, Geneva University Hospital, 1205 Geneva, Switzerland
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(6), 1374; https://doi.org/10.3390/cancers13061374
Submission received: 1 February 2021
/
Revised: 11 March 2021
/
Accepted: 13 March 2021
/
Published: 18 March 2021
(This article belongs to the Special Issue Combination Therapies in Cancers)
Abstract
:Simple Summary
Despite recent advances in understanding the molecular biology of tumors, obstacles still exist in the treatment of patients affected by rectal cancer. Recent evidence shows that ionizing radiation may have profound immunostimulatory effects, hinting at the possibility of exploiting radiotherapy to boost anti-tumor immunity. A bulk of work in pre-clinical tumor models have highlighted the potential benefit of this approach. Following these results, a few clinical trials are now evaluating the combination of radiotherapy and immune checkpoint inhibition. Remarkably, encouraging safety and toxicity profiles from these studies indicate that radio-immunotherapy combinations could represent a valid opportunity for rectal cancer patients. Yet, the biological and clinical impact of a radio-immunotherapy combination in rectal cancer remains unclear and further studies need to be performed to optimize the effect of these combinations.
Abstract
Rectal cancer is a heterogeneous disease at the genetic and molecular levels, both aspects having major repercussions on the tumor immune contexture. Whilst microsatellite status and tumor mutational load have been associated with response to immunotherapy, presence of tumor-infiltrating lymphocytes is one of the most powerful prognostic and predictive biomarkers. Yet, the majority of rectal cancers are characterized by microsatellite stability, low tumor mutational burden and poor T cell infiltration. Consequently, these tumors do not respond to immunotherapy and treatment largely relies on radiotherapy alone or in combination with chemotherapy followed by radical surgery. Importantly, pre-clinical and clinical studies suggest that radiotherapy can induce a complete reprograming of the tumor microenvironment, potentially sensitizing it for immune checkpoint inhibition. Nonetheless, growing evidence suggest that this synergistic effect strongly depends on radiotherapy dosing, fractionation and timing. Despite ongoing work, information about the radiotherapy regimen required to yield optimal clinical outcome when combined to checkpoint blockade remains largely unavailable. In this review, we describe the molecular and immune heterogeneity of rectal cancer and outline its prognostic value. In addition, we discuss the effect of radiotherapy on the tumor microenvironment, focusing on the mechanisms and benefits of its combination with immune checkpoint inhibitors.
1. Introduction
1.1. Epidemiology of Rectal Cancer
Colorectal cancer (CRC) is the fourth most common cancer worldwide, contributing to over two million new cases and 1 million cancer-related deaths in 2018 [1,2]. Of these, one-third is represented by rectal cancer (RC) [3]. Incidence rates of CRC have been steadily rising, particularly in developing countries, probably due to the westernization of lifestyle conditions and consequent exposure to risk factors (e.g., obesity, alcohol and limited physical activity) [4]. Additionally, non-modifiable predisposing factors include hereditary genetic alterations, sex and age. CRC indeed occurs more frequently in men than women and its incidence positively correlates with age [4]. In addition, germ-line mutations in the APC, BRCA, PTEN and MLH genes are responsible for inherited syndromes such as hereditary non-polyposis CRC and several polyposis conditions [5,6].
Despite the fact that the term CRC has long been used to identify tumors of the large intestine, growing evidence suggests that colon cancer (CC) and RC are distinct tumor entities [7]. Differences exist in terms of embryonic origin, anatomic location, microbiota, macroscopic and histopathological appearance and patient management [8]. While CC encompasses the large bowel, RC is restricted to the 15 cm of the distal colon between the sigmoid and the anal margin [9]. An accurate determination of the distance to the anal verge is crucial for patient stratification and the European Society for Medical Oncology (ESMO) classifies RC into upper (10–15cm), medium (5–10cm) and lower (<5 cm) third [10]. On the other hand, the Union for International Cancer Control (UICC) sets different cutoff values by 12–16 cm as high, 6–12 cm as mid and <6 cm as low RC [11]. Although CC incidence is greater than RC, in relation to the length, the rectum mucosa has a four times higher carcinogenic risk than the colon mucosa [8]. Consequently, different growing patterns have been observed, with the appearance of polypoid non-depressed lesions more frequently seen in CC than RC [12]. Based on venous drainage, metastatic dissemination in RC involves lungs and bones more frequently than for CC, whereas CC favors the liver [13]. Ultimately, treatment modalities differentially characterize localized CC and RC: the most common option for CC is upfront surgery followed by adjuvant chemotherapy, while neoadjuvant radiotherapy (RT) or chemo-radiotherapy (CRT) followed by surgery and, eventually, adjuvant chemotherapy is used in RC [14]. Whereas extensive literature is available for CRC, this review aims to spotlight RC biology and its treatment.
1.2. Molecular Pathogenesis of Rectal Tumors
RC arises from the malignant transformation of the epithelial cells lining the large intestine (Figure 1a) [15]. The first step of RC carcinogenesis involves the acquisition of genetic and epigenetic changes that promote proliferation [16]. These hyper-proliferative cells give rise to a benign adenoma, which may then evolve into carcinoma and spread to distant organs [17]. The latter occurs in approximately 20–30% of the cases [18], and 5-year survival for these patients is 20–40% [19]. In 1990, Vogelstein and colleagues elucidated the step-wise accumulation of genetic driver events leading the transition from adenoma to carcinoma [17]. This model describes the sequential inactivation of tumor suppressor genes (e.g., APC, SMAD4, and P53) and activation of oncogenic KRAS [20]. Thereafter, several genomic and epigenomic studies have contributed to a deeper understanding of the molecular pathogenesis of CRC at the gene-expression level and patients have been classified into two groups: microsatellite instability-high (MSI-H) and microsatellite stable (MSS). The MSI-H group accounts for 15% of the cases and is characterized by defects in the DNA mismatch repair program (dMMR), resulting frequently in high tumor mutational burden (TMB). Within this group, MSI-H sporadic CRC (67%) is caused by epigenetic inactivation of the MLH1 gene whereas the remaining 33% is caused by germline mutations in MMR genes (particularly MLH1 and MSH2) leading to hereditary non-polyposis CRC (HNPCC) [21]. The MSS group accounts for the remaining 85% of all CRCs and exhibits chromosomal instability (CIN), proficient DNA mismatch repair mechanisms (pMMR) and low TMB [22,23,24,25]. Notably, a gradient in dMMR/MSI-H has been observed in the right colon (22.3%) compared to left (4.6%) and the rectum (0.7%) [26]. Subtype analysis of the TCGA datasets revealed that, within the microsatellite classification, CC and RC have different immune microenvironments [27]. Integration of immune and transcriptomic profiling by the CRC Subtyping Consortium (CRCSC) established four consensus molecular subgroups (CMS) [28]. To date, CMS represent the best description of CRC heterogeneity (Figure 1b) [29], albeit other classifications have also been proposed [30,31]. The highly immunogenic CMS1 subtype (14%) is characterized by high TMB, dMMR/MSI-H and strong immune activation, the canonical CMS2 group (37%) displays WNT and MYC signaling activation and immune ignorance, the metabolic CMS3 class (13%) is distinguished for metabolic dysregulation, whereas the mesenchymal CMS4 subtype (23%) shows prominent transforming growth factor-β (TGFβ) activation, stromal invasion and angiogenesis. Interestingly, CMS profiles showed a decrease in CMS1 and CMS3 and an increase in CMS2 prevalence moving distally towards the rectum, while CMS4 remained relatively stable [32]. Despite recent advances in understanding the biological and molecular characteristics underpinning RC heterogeneity, the clinical significance of the CMS classification is still under evaluation [33].
2. Therapeutic Management of Rectal Cancer
2.1. Conventional Treatment Options
Surgical resection is the curative modality for the treatment of RC [34]. On top of surgery, management of RC patients largely relies on conventional methods such as chemotherapy and RT [35]. Local relapse represents a major surgical and therapeutic challenge for RC patients [36]. Because of the topographic position, innervation and vascular supply, the only curative possibility lies in an extensive surgery—abdominoperineal resection—removing the anus, the rectum, and part of the sigmoid colon along with the associated regional lymph nodes. The end of the remaining large intestine is brought out permanently as an opening, called colostomy, on the surface of the abdomen. This life-saving procedure is demanding for the surgeon and has a massive impact on the quality of life of RC patients.
Neo-adjuvant RT has been shown to reduce tumor burden, improve operative procedures and prevent local tumor recurrence [37,38,39,40]. For instance, short-course preoperative RT (SCPRT) showed a significant reduction in local recurrence compared to postoperative RT [40,41]. Similarly, preoperative CRT exhibited greater effectiveness and reduced morbidity than postoperative RT [42]. Remarkably, a lower recurrence rate was observed in patients who underwent surgery at least four weeks after RT compared to a shorter interval (≤1 week), providing the tumor adequate time to regress, allowing for tumor adaptive immune responses and enabling patients to recover from acute radiation toxicities [43,44]. Altogether, these studies contributed to the acceptance of neo-adjuvant RT and delayed surgery in clinical settings. Currently, European and American guidelines for the treatment of RC recommend five weeks neo-adjuvant CRT (45–50.4 Gy in 1.8–2 Gy/fraction, in association with fluoropyrimidine 5-fluorouracil (5-FU) radio-sensitization) followed by an 8- to 12-week treatment-free interval before radical surgery [45]. In addition to 5-FU, commonly used chemotherapies in the treatment of RC include oxaliplatin. Although they may have different mechanisms of action, these compounds share their anti-cancer effects through the disruption of DNA replication and transcription [46,47]. Alternatively, SCPRT of 25 Gy (5 × 5 Gy) with surgery taking place between 1 and 8 weeks after RT is being used [48].
Notably, RT can be administered to patients either as long-course treatment using conventional fractionation (1.8–2 Gy/fraction) or as short-course treatment using hypo-fractionation (>2 Gy/fraction) [49]. Indeed, the STOCKHOLM III study comparing long-course RT (25 × 2 Gy) with delayed surgery to short-course RT (5 × 5 Gy) and delayed surgery showed similar oncological results in terms of local recurrence, distant metastases, survival, and complications, indicating that these treatments are very similar [50]. Despite the concerns about late toxicity of hypo-fractionation, improved local tumor control and overall survival (OS) rates with minimal side effects led to the establishment of extreme hypo-fractionation regimens such as stereotactic body radiation therapy (SBRT). SBRT delivers high RT doses (≥8 Gy/fraction) in a few fractions [51], however it is not frequently used in the clinic. The biological bases for the selection of the fractionation regimen depends on the ability of the normal tissue to repair sublethal damage, tissue re-oxygenation, patients’ health conditions and concomitant therapies [52]. All these factors together determine the true biological effective dose (BED) delivered by a particular combination of dose per fraction and total dose to a particular tissue characterized by a specific α/β ratio, which reflects the sensitivity of the tissue to irradiation [53]. Consequently, the total RT dose cannot be directly compared between different fractionation regimens.
Two phase III clinical trials (POLISH and TROG 01.04) have reported a direct comparison between neo-adjuvant RT and CRT, suggesting that the two approaches are broadly comparable in their ability to lower the risk of local recurrence and so both are acceptable options [54,55]. In these trials, five-year recurrence rates for neo-adjuvant RT and CRT were 27% and 30% respectively, whereas OS rates were 74% and 70% [55]. In practice, the majority of localized RC patients are treated with SCPRT over CRT due to comparable outcomes, reduced toxicity and cost-effectiveness [56], while CRT or SCPRT with chemotherapy is favored for bigger tumors [9].
Pathological complete response (pCR) is defined as the absence of residual disease in the rectum and lymph nodes at the time of surgery [57]. In patients achieving pCR after neo-adjuvant treatment, a non-operative “watch and wait” management could be offered, hereby preserving the organ, its function and the patient’s quality of life. Up to 16% of patients receiving CRT reach pCR compared to 12% of those treated with SCPRT [48]. Patients undergoing chemotherapy before CRT or SCPRT showed increased pCR to 30–35% [58]. Similar results were observed when chemotherapy was applied after conventional neo-adjuvant RT and CRT treatments [59]. In some instances, patients achieving pCR showed lower risk of local or distant recurrence and improved survival compared to patients not achieving pCR [60], but in general there is no clear correlation between pCR and outcome. In fact, two international multicenter phase III trials (RAPIDO and PRODIGE 23) showed no survival benefit in any experimental arms where chemotherapy was applied either before or after neo-adjuvant RT or CRT compared to the conventional treatments [59,61]. Therefore, more efforts into exploring multimodality approaches should be invested.
With increasing insight into the molecular pathways governing tumor growth, the development of targeted therapies inhibiting these signals have gained attention, especially in the context of metastatic RC [62]. Despite the majority of RCs (>50%) being wild type for the RAS genes, targeted therapies against epithermal growth factor receptor (EGFR), which gene is mutated in a mutually exclusive manner with RAS, showed suboptimal activity in locally advanced RC patients [63]. On the other hand, in metastatic CRC settings, the combination of EGFR inhibitors with conventional chemotherapy showed improved progression-free survival (PFS) and OS [64,65,66]. Notably, 5% of CRCs carry a BRAF mutation. Remarkably, when combined with the RAF inhibitor encorafenib, which is part of the EGFR downstream signaling pathway, response rates to EGFR inhibition compared to chemotherapy alone increased from 2% to 30% and OS was improved from 5.4 months to 9 months, respectively [67]. Similarly, recent trials targeting HER2, which is a member of the EGFR superfamily, in combination with chemotherapy showed promising preliminary activity in metastatic CRC [68]. Nevertheless, anti-HER2 and anti-BRAF therapies have yet to be tested in localized RC settings. In addition to these studies, others investigated safety and efficacy of the anti-vascular endothelial growth factor (VEGF) monoclonal antibody bevacizumab with chemotherapy and CRT in CRC [69,70,71] and RC [70,71,72] showing opposite results. In particular, anti-VEGF antibody in combination with chemotherapy improved PFS and OS in metastatic CRC [69,70,71] whereas discouraging toxicity profiles were observed in phase I [70,71,72] and phase II [72] trials in locally advanced RC where anti-VEGF was combined with CRT. Despite the success of targeted therapies in advanced settings, RT followed by surgery remains the current standard-of-care for the treatment of localized RC patients.
2.2. Checkpoint Inhibitor Therapy
Over the past decade, the rise of immunotherapy has revolutionized the way many cancers are treated [73]. Unlike conventional therapies, these novel anti-cancer treatment modalities focus on enhancing anti-tumor T cell functions rather than targeting tumor cells directly [74]. Cancer immunotherapies include therapeutic vaccines, chimeric antigen receptor (CAR) T cell infusions [75,76], immune modulators (e.g., cytokines) and monoclonal antibodies (e.g., immune checkpoint inhibitors (ICIs)) [77,78]. The latter, in particular, have shown substantial clinical activity in patients with advanced cancers, including melanoma, non-small-cell lung cancer and CRC [79,80,81]. Although several immune checkpoints have been discovered, monoclonal antibodies against three major targets (PD1, its ligand PD-L1, and CTLA-4) have been approved for the treatment of a wide variety of cancers [82]. Expression of PD-L1 by cancer cells leads to the inhibition of T cell proliferation and cytokine secretion while simultaneously reducing apoptosis in anti-inflammatory T regulatory cells (Tregs) [83]. Similarly, CTLA-4 promotes immune escape by increasing immune tolerance [84]. By blocking these immunosuppressive interactions, ICIs reinvigorate immune recognition of tumors. Despite these successes, only a minority of patients experience clinical benefits from immune checkpoint blockade. In metastatic CRC, ICIs showed remarkable results in dMMR/MSI-H patients [85]; however, these agents have not proved meaningful activity in pMMR/MSS CRC [86]. Indeed, two phase II trials (KEYNOTE-016 and KEYNOTE-164) evaluated safety and overall response rate (ORR) of the anti-PD1 antibody pembrolizumab in pre-treated patients with advanced dMMR/MSI-H CRC [87,88]. These studies revealed durable responses in dMMR/MSI-H CRC, whereas no effect was observed in pMMR/MSS CRC (ORR of 57% and 0%, respectively) [87,88]. Similarly, the CHECKMATE-142 trial testing the effect of another anti-PD1 agent, nivolumab, showed improved clinical outcome in dMMR/MSI-H CRCs [89]. These patients displayed disease control for 12 weeks or longer, with PFS and OS rates at 12 months of 50.4% and 73.4% respectively [89]. The KEYNOTE-177 study comparing the standard-of-care first-line chemotherapy treatment to pembrolizumab in dMMR/MSI-H patients showed superiority of pembrolizumab in term of PFS (16.5 months vs. 8.2 months for chemotherapy), response rate (44% vs. 33% for chemotherapy) and quality of life [85]. Following these data, the Food and Drug Administration (FDA) granted accelerated approval for the use of pembrolizumab and nivolumab in first- and second-line treatment, respectively, of unresectable or metastatic dMMR/MSI-H CRCs.
Further studies explored the combination of immunotherapies in dMMR/MSI-H metastatic CRC. Combining the anti-CTLA-4 agent ipilimumab and nivolumab led to 80% disease control rate (stable disease) at 12 weeks and PFS and OS rates at 12 months of 71% and 87% respectively [90], outperforming results published with first-line chemotherapy or immunotherapy with single agents. This combination has been approved by the FDA in second-line treatment of dMMR/MSI-H metastatic CRCs [91]. Similarly, the NICHE study tested the combination of ipilimumab and nivolumab in the neoadjuvant setting in dMMR/MSI-H or pMMR/MSS early-stage CRC [92]. Interestingly, 27% of patients with pMMR/MSS tumors displayed pathological responses ranging from partial to complete response, providing the first indication of a clinical benefit of immune checkpoint inhibition in pMMR/MSS patients [92]. In the context of RC specifically, a phase II clinical trial (iSCORE) is currently evaluating the combination of nivolumab and the anti-LAG3 antibody relatlimab in metastatic RAS/RAF wild type tumors (NCT03867799).
3. The Prognostic Value of the Tumor Microenvironment
The network of interactions established by immune cells, endothelial cells, stromal fibroblasts and matrix-associated molecules within the tumor and the surrounding tissues generates the tumor microenvironment (TME) [93]. There is substantial evidence recognizing the TME as a determining factor for disease progression, therapeutic responses and patient outcome [7,94]. While alterations to the TME composition during tumor development have been extensively described in other reviews [95,96], our manuscript will focus on discussing the prognostic and clinical value of these interactions in the light of advancing treatment options for RC patients.
3.1. The Immunoscore
The importance and decisive role of the immune microenvironment in determining tumor fate has been elegantly described in a recent study by Devaud et al., where the authors identified two opposing immune response profiles with either pro- or anti-tumor properties that establish at early stage of tumor development and determine tumor progression vs. rejection [97]. Phenotypic differences in T cell infiltration within the TME can be summarized into three categories: the “immune-inflamed/hot” phenotype, in which CD8+ T cells infiltrate the tumor; the “immune-excluded” phenotype, in which infiltrating CD8+ T cells accumulate in the tumor stroma and the “immune-desert/cold” phenotype, in which CD8+ T cells are low or absent from the tumor and the stroma [98,99]. As T cell infiltration is typically dictated by the presence of tumor-specific antigens (neoantigens), it is not surprising that dMMR/MSI-H tumors display a greater number of tumor-infiltrating lymphocytes (TILs) as compared to pMMR/MSS cancers [100]. The presence of TILs has been shown to correlate with improved outcomes in many cancers, including RC [101,102].
In line with these observations, Galon and colleagues demonstrated the prognostic value of CD3+ and CD8+ TIL densities in advanced CRC [103]. They conducted genomic analyses and in-situ immunohistochemistry, highlighting a positive correlation between the presence of markers for Th1 polarization and of cytotoxic and memory T cells (CD3, CD8, GZMB, and CD45RO) and a low incidence of tumor recurrence. Similarly, Tosolini et al. revealed that Th1-associated gene expression is linked to a positive clinical impact in CRC patients [104]. Following this work, an international validation study demonstrated that this “immunoscore” is an independent positive prognostic biomarker for recurrence in stage I-III CRC, outcompeting the MSI status [105,106]. Interestingly, CD8+ and CD3+ T cells but not FoxP3+ Tregs were found to correlate with frameshift mutations in dMMR/MSI tumors [107,108]. High CD8+ levels and CD8+/FoxP3+ TIL ratio inversely correlated with pathological stages [109,110], whereas a positive correlation was described between FoxP3 expression and nodal dissemination and disease burden [111]. Similarly, low CD3+/FoxP3+ TIL ratio predicted adverse outcomes [112]. Since the validation of the “immunoscore”, several teams have worked towards its refinement [30]. Interestingly, expression of PD-L1 in the tumor was found to be associated with a high “immunoscore” [113]. While the prognostic value of CD3+ and CD8+ T cells is well described, the significance of Tregs remains controversial. For instance, a study in CRC showed that a high infiltration rate of FoxP3+ Tregs at the tumor invasive front correlated with a significantly improved prognosis [114]. Later, Lin et al. identified two Tregs populations that were associated with distinct clinical outcomes, which could explain these contrasting results [115]. These findings were corroborated by Saito and colleagues, revealing that FoxP3high CD45RO− but not FoxP3low CD45RO− Tregs were associated with poor prognosis [116]. In addition, a detailed investigation of CD3+ T cells in CRC liver metastasis revealed the existence of distinct spatial distribution patterns of immune cells in relation to the tumor margin that were associated with specific prognostic outcomes [117]. For instance, a significant decreased in GZMB CD8+ T cell density at 20–30 μm from the tumor epithelium concomitant with an increase in CD163 macrophages correlated with improved OS [117]. On the contrary, high T cells and low CD163 macrophages, which were observed at the direct tumor border <10 μm, also correlated with improved OS [117]. Taken together, this study highlights the impact of T cell function and localization on clinical responses. In the light of the recent findings, further improvements are to be expected.
3.2. Other Immune Cells
In addition to T cells, other immune cell populations are commonly present in the TME in RC. Natural killer (NK) cell infiltration, in particular CD56bright cells, positively correlates with patient survival [118,119]. However, expression of inhibitory receptors was shown to impair NK cytotoxic function and was found to be associated with tumor progression [120]. The presence of mast cells has been associated with reduced survival and advanced pathological stages, especially in CRC liver metastasis [121,122]. Elevated neutrophil to lymphocyte ratio in the peripheral blood of advanced CRC patients has been correlated with poor clinical outcome [123], whereas the opposite has been shown for tumor eosinophil infiltration [124].
The prognostic significance of tumor-associated macrophages (TAMs) in RC remains controversial [125]. A bulk of studies indicate a positive association with survival [126,127,128], whereas others predict poor patient outcome [129,130]. Nearchou and colleagues applied automated image analysis and machine learning approaches to evaluate the prognostic significance of immune cell subpopulations and their spatial interactions in RC and found that a low CD68+/CD163+ cell ratio was significantly associated with improved DFS [131]. In contrast, Feng et al. and Yang et al. showed that high CD163+/CD68+ and CD206+/CD68+ ratios correlated with decreased relapse-free survival and OS in CRC [132,133]. Similarly, dendritic cells (DCs) exhibited positive as well as negative contributions to patients’ survival, indicative of the intrinsic heterogeneity and plasticity of these populations [134,135,136]. Finally, a high frequency of myeloid-derived suppressor cells (MDSCs) correlated with advanced stage and metastasis in the lymph nodes [137]. Interestingly, MDSCs isolated from the peripheral blood of CRC patients were able to inhibit T cell expansion in vitro [138]. Taken together, these studies show that a pro-inflammatory TME characterized by CD8+ T cell infiltration is associated with improved clinical outcomes in RC. In contrast, the immunosuppressive functions of other immune cells, such as MDSCs, immunosuppressive/M2 TAMs and Tregs, appear to play a major role in promoting tumor immune escape [139].
4. Biomarkers of Response to Immunotherapy
The positive correlation between the presence of TILs in the TME and good prognosis in RC further supports the idea that a treatment based on immunotherapy should provide clinical benefit [140]. Indeed, failure in achieving durable responses to immune checkpoint inhibition is often associated with scarce or absent T cell infiltration, indicative of an “immune-desert/cold” tumor microenvironment. The relationship between mutational load, microsatellite status and response to immunotherapy has been described in many solid cancers [141]. Along with the evidence that dMMR/MSI-H tumors are more responsive to ICIs, a significant association was predicted within the four CMS subtypes in CRC [142,143]. The highly immunogenic CMS1, characterized by elevated TMB, MSI and immune activation, correlated with good prognosis and was predicted to respond to checkpoint blockade. On the other hand, the immune ignorant CMS2 was shown to be associated with worse prognosis and was predicted to respond poorly to immunotherapy [142,143]. Although the microsatellite status is important, it is however not sufficient to guide immunotherapy responses. For instance, even within pMMR/MSS CRC, tumors with a high neoantigen load display increased T cell infiltration, which positively correlates with survival, suggesting that not all pMMR/MSS tumors are non-immunogenic [144]. Interestingly, the majority of these pMRR/MSS tumors show POLE mutations, which are known to lead to high TMB [145]. In this line, the pMMR/MSS RC patients who responded to the combination of PD1 and CTLA-4 checkpoint inhibitors showed high TMB, neoantigen load and TIL infiltration [92]. Furthermore, the authors found that CD8+ and PD1+ T cells were predictive of response [92]. Similarly, Llosa and colleagues reported that pMMR/MSS tumors responding to pembrolizumab showed an immune microenvironment resembling that of the dMMR/MSI-H tumors and were characterized by PD1+ CD8+ T cell infiltration and high levels of PD-L1 [146]. Nevertheless, in CRC, PD1/PD-L1 expression is poorly correlated with clinicopathological parameters such as tumor stage and grade [147] and the identification of more precise and reliable predictive biomarkers continues to be an unmet clinical need.
It appears increasingly clear that, in order to broaden the application of promising ICIs and develop an effective treatment strategy especially in the context of “non-inflamed/cold” tumors, novel therapeutic approaches aiming at reconditioning the TME by either promoting TIL activation and infiltration or by inhibiting immunosuppressive signals are required. To this end, cytotoxic agents such as chemotherapy and RT might affect the interplay between cancer cells and immune cells in the TME.
5. The Effect of Conventional Therapies on the Tumor Microenvironment
A bulk of pre-clinical work showed that modification of the TME can be achieved by chemotherapy, RT and/or targeted agents [148]. Recent evidence suggests that ionizing radiation can induce important immunomodulatory effects that may stimulate an in-situ vaccine phenomenon by reconditioning the TME and inducing T cell trafficking to the tumor [149]. Upon irradiation, cancer cells undergo immunogenic cell death that is associated with the release of damage associated molecular patterns (DAMPs), accumulation of cytosolic DNA, and upregulation of signals (e.g., IFN-γ, IL-1 and IL-6) that promote the recruitment of DCs, and expression of immunomodulatory genes including antigen presentation genes (Figure 2) [150,151].
5.1. Effect of Radiotherapy
In syngeneic mouse models of CRC, high dose irradiation (1 × 10 Gy and 1 × 30 Gy) promoted the expansion of effector CD8+ T cells and a loss of MDSCs via activation of antigen cross-presenting DCs, secretion of IFN-γ, and activation of CD4+ T cells expressing CD40L [152,153]. Altogether, these alterations resulted in improved tumor control in vivo [152,153]. On the other hand, high dose RT (1 × 10 Gy) was shown to increase the immunosuppressive function of Tregs in the TME via TGFβ and IL-33 signaling [154]. In RC patients, a decrease in the CD8+/FoxP3+ TIL ratio was observed after conventional as well as hypo-fractionated RT and positively correlated with OS [155]. These results are difficult to interpret and the prognostic and predictive significance of Tregs in RC remains controversial [115]. Furthermore, it appears increasingly clear that RT holds stimulatory as well as immunosuppressive traits [156], and that the fine balance between these properties dictate patient outcome. The effect of RT in promoting a pro- or an anti-inflammatory phenotype likely depends on dose, fractionation, and tumor type [157,158,159]. It has been hypothesized that high dose radiation is involved in the direct killing of primary tumor cells, which allows neoantigen release and T cell priming, whereas low dose radiation modulates the TME including at distant sites (abscopal effect) and enhances TIL infiltration, which leads to enhanced immune-cell recognition, tumor cell killing and antigen release [160]. Indeed, Dewan et al. showed that a single fraction of 20 Gy failed to inhibit tumor growth at a distant site compared to fractionated RT (3 × 8 Gy, 5 × 6 Gy) [161]. Remarkably, compared to conventional fractionated RT, hypo-fractionated RT further diminished the ratio of CD8+/FoxP3+ TILs in RC patients, indicating that differences can be observed even within fractionated regimens [155]. Grapin and colleagues showed that different fractionation schemes with similar BED (18 × 2 Gy, 3 × 8 Gy and 1 × 16.4 Gy) induced different lymphoid and myeloid responses as well as modulation of PD-L1 and TIGIT expression [162]. In particular, mice bearing subcutaneous CT26 colon tumors displayed the highest number of GZMB CD8+ T cells and M1/M2 TAM ratio concomitantly with the lowest CD8+/Treg ratio when irradiated with 3 × 8G y [162]. Contrary to the hypo-fractionated schemes, the conventional fractionated regimen (18 × 2 Gy) appeared to be the least effective [162]. Similarly, PD-L1 expression was found to be upregulated in the TME after high dose RT (1 × 12 Gy) in vitro and in vivo [163], while another pre-clinical study in syngeneic mouse model of CC demonstrated that 5 × 2 Gy RT leads to tumor cell expression of PD-L1 via IFN-γ production by infiltrating CD8+ T cells [164].
SCPRT (2 × 5 Gy) was also found to be associated with a shift in the polarization of TAMs towards an M1-like pro-inflammatory phenotype in vitro and in vivo [165]. Similarly, in another study, irradiated macrophages (5 × 2 Gy) showed a reduced anti-inflammatory profile, increased phagocytosis and unaltered pro-invasive and pro-angiogenic capacities [166]. Taken together, the studies presented above hints towards the possibility that hypo-fractionated RT might have a superior therapeutic potential compared to conventional fractionated RT. Nevertheless, these reports are not directly comparable as they investigated different fractionation regimens, which prevent us from drawing definitive conclusions. Therefore, the optimal radiation regimen (total dose and fractionation schedule) to stimulate an efficient anti-tumor immune response in RC remains unclear and additional work should focus on dissecting the exact kinetics and nature of how RT changes the TME.
5.2. Effect of Chemotherapy and CRT
The prognostic value of CD4+, CD8+ and FoxP3+ TILs in response to neoadjuvant chemotherapy and CRT has been extensively described [167,168,169]. Reduction in FoxP3+ Tregs and increased expression of MHC-I genes were observed in RC patients responding favorably to FOLFOX chemotherapy [170]. Similarly, in another study, FoxP3+ TILs correlated with poor therapeutic responses to FOLFOX chemotherapy alone or in combination with RT (25–50 Gy) [171]. Remarkably, in this study, neo-adjuvant CRT showed superior OS compared to chemotherapy alone, suggesting the importance of ionizing radiation in remodeling the TME [171]. In addition, a slight but not significant increase in the expression of CTLA-4 was observed upon CRT compared to chemotherapy, concomitantly to the expansion of FoxP3 Tregs [171]. These results are in line with previous reports where RT was shown to increase the proportion of Tregs, which may, in turn, induce the expression of CTLA-4 through various pathways [159,172]. On the other hand, a significant increase of CD3+ and CD8+ immune infiltrates in the TME of RC patients after neoadjuvant CRT was prognostic for the extent of tumor regression, distant metastasis rates and DFS [173]. In contrast, a multivariate analysis revealed that, while the total number of CD3+ and CD8+ TILs was significantly lower in RC after neo-adjuvant CRT compared to primarily resected cases, the level of GZMB+ CD8+ T cells was increased and positively correlated with tumor regression and lower recurrence [174]. These data suggest that the presence of GZMB+ CD8+ T cells in the TME may limit the occurrence of tumors and improve prognosis in RC patients. Alongside high levels of CD8+ TILs, the expression of immune checkpoint genes such as B7-H3 and B7-H5 was found upregulated upon CRT in RC patients and correlated with better outcome [175]. Similarly, high expression of PD-L1 in tumor nests was associated with favorable outcomes after neoadjuvant chemotherapy and CRT [171,176]. In addition, patients with high cyto-HMGB1 translocation and/or PD1+ TILs before treatment showed better disease control upon neo-adjuvant CRT [177]. Recent studies implied that high tumor PD-L1 expression is involved with the feedback mechanism caused by the induction of IFN-γ due to RT intervention [178,179]. Among other functions, IFN-γ has been shown to play an important role in the recruitment of T cells and expression of MHC-I through autocrine and paracrine signaling [180]. Taken together, RT has been shown to induce a two sided effect by recruiting immune cells into the tumor site and upregulating immune checkpoints. These results hint towards a potential therapeutic synergism between irradiation and immunotherapy.
6. Combining Radiotherapy with Immune Checkpoint Inhibitors
Combining chemotherapy with RT was shown to improve pCR in RC patients [181]. In addition, neo-adjuvant CRT has been shown to upregulate PD-L1 and promote T cell infiltration [182,183,184]. However, the combination of chemotherapy with RT often results in increased toxicity in normal tissues [185]. On the other hand, the potential of combining immunotherapy and RT has been increasingly investigated in many solid cancers, highlighting the benefit of such a combination [186,187,188]. The biological premise behind this strategy is that the release of immune-stimulating signals and neoantigens following RT will induce profound changes in the TME and promote anti-tumor immune responses that could be enhanced further by systemic immune-stimulating agents such as ICIs [189]. An overview of these alterations is outlined in Figure 2.
Deng et al. found that the combination of high dose RT (1 × 12 Gy) and anti–PD-L1 treatment induced tumor regression in a murine xenograft model of CRC [163]. In this work, radio-immunotherapy stimulated CD8+ T cell responses and reduced the local accumulation of MDSCs through TNF-α secretion [163]. This first study provided pre-clinical evidence of the promising effects of such a combination. Importantly, the effect of RT fractionation has been evaluated in the context of radio-immunotherapy in several studies. In a syngeneic mouse model of CC, Grapin and colleagues showed that, compared to 18 × 2 Gy and 1 × 16.4 Gy, 3 × 8 Gy irradiation induced the greatest increase in TILs and expression of PD-L1 and TIGIT and this protocol was even more effective in controlling tumor development when associated with anti-PD-L1 and anti-TIGIT treatment [162]. Similarly, combination of RT (3 × 8 Gy) with anti-CTLA-4 therapy induced tumor growth control in a MC38 xenograft model [161]. Using the same animal model, Morisada et al. showed that high dose hypo-fractionated RT (2 × 8 Gy) was superior to conventional RT (10 × 2 Gy) in enhancing anti-tumor immunity in combination with anti-PD1 treatment, highlighting the potential benefit of hypo-fractionated RT. On the other hand, two other pre-clinical studies using a CT26 xenograft model showed improved local tumor control, long-term survival, and protection against tumor re-challenge when 5 × 2 Gy RT was combined with anti-PD1 or anti-PD-L1 antibodies [164,190]. Nevertheless, the authors observed similar combinatorial activity between 5 × 2 Gy, 3 × 4 Gy and 1 × 7 Gy RT regimens even though the latter showed the poorest tumor control on its own [190]. Taken together, this study indicates that the degree to which radiation achieves local tumor control do not always predict its ability to synergize with ICIs.
Because of the differences in the dynamic progression of immunological responses upon RT and ICIs, it might be important to determine the most effective sequence of treatments [191]. Although a limited number of pre-clinical studies have been conducted on this topic, Dovedi et al. showed that anti-PD1 antibody should be administered simultaneously with RT compared to 7 days later in order to enhance its effect [164]. On the contrary, CTLA-4 inhibition was more effective when given 7 days prior to RT compared to day 1 and 5 after RT [192], probably due to the different mechanisms of action of these two immunotherapies. As CTLA-4 inhibits T cells in the early stages of the immunity cycle, it is plausible that CTLA-4 inhibition synergizes with the RT-induced enhancement of antigen-cross presentation and T cell priming by pre-conditioning the lymph node microenvironment and, subsequently, boosting T cell activation and expansion. On the other hand, PD1 acts directly on the TME; therefore, blockade of the PD1 axis promotes the activation of T cells that infiltrated the tumor site upon RT intervention [193]. In line with these observations, pre-clinical in vivo data highlighted a trajectory of immune effects such as reduction in the infiltration of MDSCs, TAMs and Tregs, and an increase in CD8+ T cells alongside the expression of HLA, CEA, MUC-1 and ICAM-1 reaching a peak within 8–15 days after irradiation [153,194,195]. Although optimal sequencing of RT and ICIs is not yet determined, these studies suggest that, in order to benefit from the highest levels of CD8+ T cells, anti-PD1 antibodies should be administered together with RT [191]. On the other hand, treatment with anti-CTLA-4 can be effective even before RT by reconditioning the lymph node microenvironment and diminishing Tregs [192].
Many clinical trials exploring the synergistic effect of combining RT with checkpoint blockade in solid cancers are ongoing [186,187,188]. In this line, a few trials are evaluating the combination of RT and immune checkpoint inhibition in RC (e.g., NCT02298946 [196], NCT02948348 [197], NCT04124601, NCT04262687, NCT04558684), but always in combination with chemotherapy. A brief summary of the relevant radio-immunotherapy clinical trials in RC is presented in Table 1. Encouraging safety and toxicity profiles from these studies indicate that radio-immunotherapy combinations could represent a valid opportunity for RC patients. For instance, in the VOLTAGE trial (NCT02948348) where patients received CRT followed by nivolumab, only mild toxicity was reported, with 7.7% of patients experiencing immune-related grade 3/4 side effects [197]. Moreover, 30% of patients with locally advanced pMMR/MSS RC reached pCR compared to 60% of dMMR/MSI-H RCs [197]. After these encouraging results, a few other trials have begun investigating the effect of such combination without the addition of chemotherapy. Remarkably, the combination of ipilimumab with SBRT (50 Gy in 4 fractions or 60 Gy in 10 fractions) has shown to induce an increase in peripheral CD8+ T cells and CD8+/CD4+ T cell ratio, indicating a systemic immune activation [198]. Likewise, the authors observed an increased proportion of CD8+ T cells expressing 4-1BB and PD1 in the tumors after treatment, which positively correlated with clinical benefit [198]. These data confirm the pre-clinical evidence on the effects of RT in remodeling the TME and are suggestive of additional immune checkpoint targets may help optimize treatment efficacy. Lastly, the combination of nivolumab and ipilimumab with RT is currently under investigation in CRC and pancreatic cancer (NCT03104439), whereas a phase II clinical trial (NCT04109755) is studying the impact of combining pembrolizumab with SCPRT in the neo-adjuvant treatment of localized pMMR/MSS RC. The results of these future clinical trials will shed light on the future of radio-immunotherapy in the treatment of RC.
7. Conclusions
In conclusion, the results from these first clinical and pre-clinical studies combining RT with ICIs are promising. Nevertheless, in the absence of a comprehensive understanding of the biological effect of dose/time/fractionation factors and their contribution to patient outcome, combination therapies and treatment regimens remain largely empirical. Therefore, more efforts should be directed towards designing rational and robust pre-clinical studies dissecting these aspects. A better understanding of the dynamic interaction between the immune and cancer cells and how this interaction changes after therapeutic interventions will help defining the optimal radio-immunotherapy combination to achieve the best clinical results. This may also include the addition of a third agent. For instance, Son et al. showed that injection of immature DCs in a CC xenograft model potentiated anti-tumor responses of anti-CTLA-4 therapy with RT [199]. In addition, the combination of RT with IL-12/GM-CSF and anti-PD-L1 antibody enhanced tumor regression and accumulation of CD8+ T cells and tumor-associated neutrophils at the primary and metastatic site in vivo [200]. Lastly, radiation combined with macrophage depletion was shown to promote adaptive immunity and potentiate checkpoint blockade [201]. While further studies need to be performed to optimize the effect of these treatment combinations, radio-immunotherapy offers an exciting new therapeutic modality for RC.
Author Contributions
Manuscript conceptualization, C.C., V.D. and T.K.; original draft preparation and manuscript writing, C.C. and T.K.; review and editing, V.D. and T.K.; critical inputs, V.D.; supervision, V.D and T.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors thank Laurence Zulianello for iconographic support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 1 January 2021).
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; McSorley, S.T.; Park, J.H.; Roxburgh, C.S.D.; Edwards, J.; Horgan, P.G.; McMillan, D.C. The relationship between right-sided tumour location, tumour microenvironment, systemic inflammation, adjuvant therapy and survival in patients undergoing surgery for colon and rectal cancer. Br. J. Cancer 2018, 118, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Paschke, S.; Jafarov, S.; Staib, L.; Kreuser, E.D.; Maulbecker-Armstrong, C.; Roitman, M.; Holm, T.; Harris, C.C.; Link, K.H.; Kornmann, M. Are Colon and Rectal Cancer Two Different Tumor Entities? A Proposal to Abandon the Term Colorectal Cancer. Int. J. Mol. Sci. 2018, 19, 2577. [Google Scholar] [CrossRef] [Green Version]
- Glynne-Jones, R.; Wyrwicz, L.; Tiret, E.; Brown, G.; Rodel, C.; Cervantes, A.; Arnold, D.; Committee, E.G. Rectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv22–iv40. [Google Scholar] [CrossRef]
- Schmoll, H.J.; Van Cutsem, E.; Stein, A.; Valentini, V.; Glimelius, B.; Haustermans, K.; Nordlinger, B.; van de Velde, C.J.; Balmana, J.; Regula, J.; et al. ESMO Consensus Guidelines for management of patients with colon and rectal cancer. a personalized approach to clinical decision making. Ann. Oncol. 2012, 23, 2479–2516. [Google Scholar] [CrossRef] [PubMed]
- Attenberger, U.I.; Winter, J.; Harder, F.N.; Burkholder, I.; Dinter, D.; Kaltschmidt, S.; Kienle, P.; Schoenberg, S.O.; Hofheinz, R. Height of Rectal Cancer: A Comparison between Rectoscopic and Different MRI Measurements. Gastroenterol. Res. Pract. 2020, 2020, 2130705. [Google Scholar] [CrossRef]
- Konishi, K.; Fujii, T.; Boku, N.; Kato, S.; Koba, I.; Ohtsu, A.; Tajiri, H.; Ochiai, A.; Yoshida, S. Clinicopathological differences between colonic and rectal carcinomas: Are they based on the same mechanism of carcinogenesis? Gut 1999, 45, 818–821. [Google Scholar] [CrossRef]
- Qiu, M.; Hu, J.; Yang, D.; Cosgrove, D.P.; Xu, R. Pattern of distant metastases in colorectal cancer: A SEER based study. Oncotarget 2015, 6, 38658–38666. [Google Scholar] [CrossRef] [Green Version]
- Aleman, B.M.; Bartelink, H.; Gunderson, L.L. The current role of radiotherapy in colorectal cancer. Eur. J. Cancer 1995, 31A, 1333–1339. [Google Scholar] [CrossRef]
- Fazeli, M.S.; Keramati, M.R. Rectal cancer: A review. Med. J. Islam Repub. Iran. 2015, 29, 171. [Google Scholar]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-C.; Lee, Y.-L.; Chuang, J.-P.; Lee, J.-C. Differences in Survival between Colon and Rectal Cancer from SEER Data. PLoS ONE 2013, 8, e78709. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arends, J.W. Molecular interactions in the Vogelstein model of colorectal carcinoma. J. Pathol. 2000, 190, 412–416. [Google Scholar] [CrossRef]
- Kawakami, H.; Zaanan, A.; Sinicrope, F.A. Microsatellite instability testing and its role in the management of colorectal cancer. Curr. Treat. Options Oncol. 2015, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, J.P. CpG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Tabernero, J. Molecular dissection of microsatellite instable colorectal cancer. Cancer Discov. 2013, 3, 502–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087 e2073. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; Weinberg, B.A.; Xiu, J.; El-Deiry, W.S.; Hwang, J.J.; Gatalica, Z.; Philip, P.A.; Shields, A.F.; Lenz, H.-J.; Marshall, J.L. Comparative molecular analyses of left-sided colon, right-sided colon, and rectal cancers. Oncotarget 2017, 8, 86456. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Zhang, J.; Luo, P. Crosstalk between the MSI Status and Tumor Microenvironment in Colorectal Cancer. Front. Immunol. 2020, 11, 2039. [Google Scholar] [CrossRef]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Pedrosa, L.; Esposito, F.; Thomson, T.M.; Maurel, J. The Tumor Microenvironment in Colorectal Cancer Therapy. Cancers 2019, 11, 1172. [Google Scholar] [CrossRef] [Green Version]
- Hamada, T.; Soong, T.R.; Masugi, Y.; Kosumi, K.; Nowak, J.A.; da Silva, A.; Mu, X.J.; Twombly, T.S.; Koh, H.; Yang, J.; et al. TIME (Tumor Immunity in the MicroEnvironment) classification based on tumor CD274 (PD-L1) expression status and tumor-infiltrating lymphocytes in colorectal carcinomas. Oncoimmunology 2018, 7, e1442999. [Google Scholar] [CrossRef] [Green Version]
- Loree, J.M.; Pereira, A.A.L.; Lam, M.; Willauer, A.N.; Raghav, K.; Dasari, A.; Morris, V.K.; Advani, S.; Menter, D.G.; Eng, C.; et al. Classifying Colorectal Cancer by Tumor Location Rather than Sidedness Highlights a Continuum in Mutation Profiles and Consensus Molecular Subtypes. Clin. Cancer Res. 2018, 24, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooi, J.K.; Wirapati, P.; Asher, R.; Lee, C.K.; Savas, P.; Price, T.J.; Townsend, A.; Hardingham, J.; Buchanan, D.; Williams, D.; et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: Molecular analysis of the AGITG MAX clinical trial. Ann. Oncol. 2018, 29, 2240–2246. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal. Transduct Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Quirke, P.; Durdey, P.; Dixon, M.F.; Williams, N.S. Local recurrence of rectal adenocarcinoma due to inadequate surgical resection. Histopathological study of lateral tumour spread and surgical excision. Lancet 1986, 2, 996–999. [Google Scholar] [CrossRef]
- Camma, C.; Giunta, M.; Fiorica, F.; Pagliaro, L.; Craxi, A.; Cottone, M. Preoperative radiotherapy for resectable rectal cancer—A meta-analysis. JAMA J. Am. Med. Assoc. 2000, 284, 1008–1015. [Google Scholar] [CrossRef]
- Sauer, R.; Liersch, T.; Merkel, S.; Fietkau, R.; Hohenberger, W.; Hess, C.; Becker, H.; Raab, H.R.; Villanueva, M.T.; Witzigmann, H.; et al. Preoperative versus postoperative chemoradiotherapy for locally advanced rectal cancer: Results of the German CAO/ARO/AIO-94 randomized phase III trial after a median follow-up of 11 years. J. Clin. Oncol. 2012, 30, 1926–1933. [Google Scholar] [CrossRef]
- van Gijn, W.; Marijnen, C.A.; Nagtegaal, I.D.; Kranenbarg, E.M.; Putter, H.; Wiggers, T.; Rutten, H.J.; Pahlman, L.; Glimelius, B.; van de Velde, C.J.; et al. Preoperative radiotherapy combined with total mesorectal excision for resectable rectal cancer: 12-Year follow-up of the multicentre, randomised controlled TME trial. Lancet Oncol. 2011, 12, 575–582. [Google Scholar] [CrossRef]
- Swedish Rectal Cancer, T.; Cedermark, B.; Dahlberg, M.; Glimelius, B.; Pahlman, L.; Rutqvist, L.E.; Wilking, N. Improved survival with preoperative radiotherapy in resectable rectal cancer. N. Engl. J. Med. 1997, 336, 980–987. [Google Scholar] [CrossRef]
- Folkesson, J.; Birgisson, H.; Pahlman, L.; Cedermark, B.; Glimelius, B.; Gunnarsson, U. Swedish Rectal Cancer Trial: Long lasting benefits from radiotherapy on survival and local recurrence rate. J. Clin. Oncol. 2005, 23, 5644–5650. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.; Becker, H.; Hohenberger, W.; Rödel, C.; Wittekind, C.; Fietkau, R.; Martus, P.; Tschmelitsch, J.; Hager, E.; Hess, C.F.; et al. Preoperative versus Postoperative Chemoradiotherapy for Rectal Cancer. N. Engl. J. Med. 2004, 351, 1731–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Fang, C.; Huang, L.; Fan, C.; Wang, C.; Yang, L.; Li, Y.; Zhou, Z. Short-course radiotherapy with immediate or delayed surgery in rectal cancer: A meta-analysis. Int. J. Surg. 2018, 56, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Glynne-Jones, R.; Hall, M.; Nagtegaal, I.D. The optimal timing for the interval to surgery after short course preoperative radiotherapy (5 × 5 Gy) in rectal cancer—Are we too eager for surgery? Cancer Treat. Rev. 2020, 90, 102104. [Google Scholar] [CrossRef] [PubMed]
- Alliance, N.G. Preoperative Radiotherapy and Chemoradiotherapy for Rectal Cancer; National Institute for Health and Care Excellence (UK): London, UK, 2020. [Google Scholar]
- Arango, D.; Wilson, A.J.; Shi, Q.; Corner, G.A.; Arañes, M.J.; Nicholas, C.; Lesser, M.; Mariadason, J.M.; Augenlicht, L.H. Molecular mechanisms of action and prediction of response to oxaliplatin in colorectal cancer cells. Br. J. Cancer 2004, 91, 1931–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Pettersson, D.; Lorinc, E.; Holm, T.; Iversen, H.; Cedermark, B.; Glimelius, B.; Martling, A. Tumour regression in the randomized Stockholm III Trial of radiotherapy regimens for rectal cancer. Br. J. Surg. 2015, 102, 972–978; discussion 978. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.; Marta, G.N. Hypofractionated and hyper-hypofractionated radiation therapy in postoperative breast cancer treatment. Rev. Assoc. Med. Bras. (1992) 2020, 66, 1301–1306. [Google Scholar] [CrossRef]
- Erlandsson, J.; Holm, T.; Pettersson, D.; Berglund, A.; Cedermark, B.; Radu, C.; Johansson, H.; Machado, M.; Hjern, F.; Hallbook, O.; et al. Optimal fractionation of preoperative radiotherapy and timing to surgery for rectal cancer (Stockholm III): A multicentre, randomised, non-blinded, phase 3, non-inferiority trial. Lancet Oncol. 2017, 18, 336–346. [Google Scholar] [CrossRef]
- Petrelli, F.; Comito, T.; Barni, S.; Pancera, G.; Scorsetti, M.; Ghidini, A. Stereotactic body radiotherapy for colorectal cancer liver metastases: A systematic review. Radiother. Oncol. 2018, 129, 427–434. [Google Scholar] [CrossRef]
- Halperin, E.C.; Wazer, D.E.; Perez, C.A.; Brady, L.W. Perez & Brady’s Principles and Practice of Radiation Oncology; Wolters Kluwer: Philadelphia, PA, USA, 2019. [Google Scholar]
- Jones, B.; Dale, R.G.; Deehan, C.; Hopkins, K.I.; Morgan, D.A.L. The Role of Biologically Effective Dose (BED) in Clinical Oncology. Clin. Oncol. 2001, 13, 71–81. [Google Scholar] [CrossRef]
- Bujko, K.; Nowacki, M.P.; Nasierowska-Guttmejer, A.; Michalski, W.; Bebenek, M.; Kryj, M. Long-term results of a randomized trial comparing preoperative short-course radiotherapy with preoperative conventionally fractionated chemoradiation for rectal cancer. Br. J. Surg. 2006, 93, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Ngan, S.Y.; Burmeister, B.; Fisher, R.J.; Solomon, M.; Goldstein, D.; Joseph, D.; Ackland, S.P.; Schache, D.; McClure, B.; McLachlan, S.A.; et al. Randomized trial of short-course radiotherapy versus long-course chemoradiation comparing rates of local recurrence in patients with T3 rectal cancer: Trans-Tasman Radiation Oncology Group trial 01.04. J. Clin. Oncol. 2012, 30, 3827–3833. [Google Scholar] [CrossRef]
- Tseng, M.; Soon, Y.Y.; Vellayappan, B.; Ho, F.; Tey, J. Radiation therapy for rectal cancer. J. Gastrointest. Oncol. 2019, 10, 1238–1250. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Cercek, A.; Goodman, K.A.; Hajj, C.; Weisberger, E.; Segal, N.H.; Reidy-Lagunes, D.L.; Stadler, Z.K.; Wu, A.J.; Weiser, M.R.; Paty, P.B.; et al. Neoadjuvant chemotherapy first, followed by chemoradiation and then surgery, in the management of locally advanced rectal cancer. J. Natl. Compr. Cancer Netw. 2014, 12, 513–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hospers, G. Short-course radiotherapy followed by chemotherapy before TME in locally advanced rectal cancer: The randomized RAPIDO trial. ASCO Virtual Scientific Program. J. Clin. Oncol. 2020, 38, 4006. [Google Scholar] [CrossRef]
- Maas, M.; Nelemans, P.J.; Valentini, V.; Das, P.; Rödel, C.; Kuo, L.J.; Calvo, F.A.; García-Aguilar, J.; Glynne-Jones, R.; Haustermans, K.; et al. Long-term outcome in patients with a pathological complete response after chemoradiation for rectal cancer: A pooled analysis of individual patient data. Lancet Oncol. 2010, 11, 835–844. [Google Scholar] [CrossRef]
- Conroy, T. Total neoadjuvant therapy with mFOLFIRINOX versus preoperative chemoradiation in patients with locally advanced rectal cancer: Final results of PRODIGE 23 phase III trial, a UNICANCER GI trial. ASCO Virtual Scientific Program. J. Clin. Oncol. 2020, 38, 4007. [Google Scholar] [CrossRef]
- Willett, C.G.; Duda, D.G.; Czito, B.G.; Bendell, J.C.; Clark, J.W.; Jain, R.K. Targeted therapy in rectal cancer. Oncology (Williston Park) 2007, 21, 1055–1065. [Google Scholar]
- Dewdney, A.; Cunningham, D.; Tabernero, J.; Capdevila, J.; Glimelius, B.; Cervantes, A.; Tait, D.; Brown, G.; Wotherspoon, A.; Gonzalez de Castro, D.; et al. Multicenter randomized phase II clinical trial comparing neoadjuvant oxaliplatin, capecitabine, and preoperative radiotherapy with or without cetuximab followed by total mesorectal excision in patients with high-risk rectal cancer (EXPERT-C). J. Clin. Oncol. 2012, 30, 1620–1627. [Google Scholar] [CrossRef] [Green Version]
- Zeuli, M.; Gelibter, A.; Nardoni, C.; Gamucci, T.; Gabriele, A.; Pollera, C.F.; Costanzo, F.D.; Signorelli, C.; Ferraresi, V.; Cognetti, F. A feasibility study of gefitinib in association with capecitabine (CAP) and oxaliplatin (OXA) as first-line treatment in patients with advanced colorectal cancer (ACRC). J. Clin. Oncol. 2004, 22, 3748. [Google Scholar] [CrossRef]
- Fisher, G.A.; Kuo, T.; Cho, C.D.; Halsey, J.; Jambalos, C.N.; Schwartz, E.J.; Robert, R.V.; Advani, R.H.; Wakelee, H.A. A phase II study of gefitinib in combination with FOLFOX-4 (IFOX) in patients with metastatic colorectal cancer. J. Clin. Oncol. 2004, 22, 3514. [Google Scholar] [CrossRef]
- Meyerhardt, J.A.; Zhu, A.X.; Enzinger, P.C.; Ryan, D.P.; Clark, J.W.; Kulke, M.H.; Earle, C.C.; Vincitore, M.; Michelini, A.; Sheehan, S.; et al. Phase II study of capecitabine, oxaliplatin, and erlotinib in previously treated patients with metastastic colorectal cancer. J. Clin. Oncol. 2006, 24, 1892–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopetz, S.; McDonough, S.L.; Morris, V.K.; Lenz, H.-J.; Magliocco, A.M.; Atreya, C.E.; Diaz, L.A.; Allegra, C.J.; Wang, S.E.; Lieu, C.H.; et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG 1406). J. Clin. Oncol. 2017, 35, 520. [Google Scholar] [CrossRef] [Green Version]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Giantonio, B.J.; Levy, D.E.; O’Dwyer, P.J.; Meropol, N.J.; Catalano, P.J.; Benson, A.B., 3rd. A phase II study of high-dose bevacizumab in combination with irinotecan, 5-fluorouracil, leucovorin, as initial therapy for advanced colorectal cancer: Results from the Eastern Cooperative Oncology Group study E2200. Ann. Oncol. 2006, 17, 1399–1403. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Kabbinavar, F.; Hurwitz, H.I.; Fehrenbacher, L.; Meropol, N.J.; Novotny, W.F.; Lieberman, G.; Griffing, S.; Bergsland, E. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J. Clin. Oncol. 2003, 21, 60–65. [Google Scholar] [CrossRef]
- Borg, C.; Mantion, G.; Boudghène, F.; Mornex, F.; Ghiringhelli, F.; Adenis, A.; Azria, D.; Balosso, J.; Ben Abdelghani, M.; Bachet, J.B.; et al. Efficacy and Safety of Two Neoadjuvant Strategies With Bevacizumab in MRI-Defined Locally Advanced T3 Resectable Rectal Cancer: Final Results of a Randomized, Noncomparative Phase 2 INOVA Study. Clin. Colorectal. Cancer 2019, 18, 200–208.e201. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvistborg, P.; Philips, D.; Kelderman, S.; Hageman, L.; Ottensmeier, C.; Joseph-Pietras, D.; Welters, M.J.; van der Burg, S.; Kapiteijn, E.; Michielin, O.; et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci. Transl. Med. 2014, 6, 254ra128. [Google Scholar] [CrossRef]
- Diaz, L.A.; Le, D.T.; Yoshino, T.; André, T.; Bendell, J.C.; Rosales, M.; Kang, S.P.; Lam, B.; Jäger, D. KEYNOTE-177: Phase 3, open-label, randomized study of first-line pembrolizumab (Pembro) versus investigator-choice chemotherapy for mismatch repair-deficient (dMMR) or microsatellite instability-high (MSI-H) metastatic colorectal carcinoma (mCRC). J. Clin. Oncol. 2018, 36, TPS877. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Cutsem, E.V.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.-J.; Lonardi, S.; Zagonel, V.; Cutsem, E.V.; Limon, M.L.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; Garcia-Alfonso, P.; Neyns, B.; et al. Nivolumab plus low-dose ipilimumab as first-line therapy in microsatellite instability-high/DNA mismatch repair deficient metastatic colorectal cancer: Clinical update. J. Clin. Oncol. 2020, 38, 11. [Google Scholar] [CrossRef]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; Van den Berg, J.G.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020, 26, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal. Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Peddareddigari, V.G.; Wang, D.; Dubois, R.N. The tumor microenvironment in colorectal carcinogenesis. Cancer Microenviron. 2010, 3, 149–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colangelo, T.; Polcaro, G.; Muccillo, L.; D’Agostino, G.; Rosato, V.; Ziccardi, P.; Lupo, A.; Mazzoccoli, G.; Sabatino, L.; Colantuoni, V. Friend or foe? The tumour microenvironment dilemma in colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Trimaglio, G.; Tilkin-Mariamé, A.-F.; Feliu, V.; Lauzéral-Vizcaino, F.; Tosolini, M.; Valle, C.; Ayyoub, M.; Neyrolles, O.; Vergnolle, N.; Rombouts, Y.; et al. Colon-specific immune microenvironment regulates cancer progression versus rejection. OncoImmunology 2020, 9, 1790125. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [Green Version]
- Bever, K.M.; Le, D.T. An Expanding Role for Immunotherapy in Colorectal Cancer. J. Natl. Compr. Cancer Netw. 2017, 15, 401–410. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Teng, F.; Meng, X.; Kong, L.; Mu, D.; Zhu, H.; Liu, S.; Zhang, J.; Yu, J. Tumor-infiltrating lymphocytes, forkhead box P3, programmed death ligand-1, and cytotoxic T lymphocyte-associated antigen-4 expressions before and after neoadjuvant chemoradiation in rectal cancer. Transl. Res. 2015, 166, 721–732. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pages, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Maby, P.; Tougeron, D.; Hamieh, M.; Mlecnik, B.; Kora, H.; Bindea, G.; Angell, H.K.; Fredriksen, T.; Elie, N.; Fauquembergue, E.; et al. Correlation between Density of CD8+ T-cell Infiltrate in Microsatellite Unstable Colorectal Cancers and Frameshift Mutations: A Rationale for Personalized Immunotherapy. Cancer Res. 2015, 75, 3446–3455. [Google Scholar] [CrossRef] [Green Version]
- Tougeron, D.; Maby, P.; Elie, N.; Fauquembergue, E.; Le Pessot, F.; Cornic, M.; Sabourin, J.C.; Michel, P.; Frebourg, T.; Latouche, J.B. Regulatory T lymphocytes are associated with less aggressive histologic features in microsatellite-unstable colorectal cancers. PLoS ONE 2013, 8, e61001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Chikazawa, N.; Tasaka, T.; Wada, J.; Yamasaki, A.; Kitaura, Y.; Sozaki, M.; Tanaka, M.; Onishi, H.; Morisaki, T.; et al. Intratumoral CD8(+) T/FOXP3(+) cell ratio is a predictive marker for survival in patients with colorectal cancer. Cancer Immunol. Immunother. 2010, 59, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Saito, K.; Shiiba, K.; Ohuchi, A.; Saigenji, K.; Nagura, H.; Ohtani, H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998, 58, 3491–3494. [Google Scholar] [PubMed]
- Liu, Z.; Huang, Q.; Liu, G.; Dang, L.; Chu, D.; Tao, K.; Wang, W. Presence of FOXP3(+)Treg cells is correlated with colorectal cancer progression. Int. J. Clin. Exp. Med. 2014, 7, 1781–1785. [Google Scholar] [PubMed]
- Sinicrope, F.A.; Rego, R.L.; Ansell, S.M.; Knutson, K.L.; Foster, N.R.; Sargent, D.J. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology 2009, 137, 1270–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, Y.S.; Pyo, J.S. Clinicopathological significance and prognostic role of tumor-infiltrating lymphocytes in colorectal cancer. Int. J. Biol. Markers 2019, 34, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Ling, A.; Edin, S.; Wikberg, M.L.; Öberg, Å.; Palmqvist, R. The intratumoural subsite and relation of CD8(+) and FOXP3(+) T lymphocytes in colorectal cancer provide important prognostic clues. Br. J. Cancer 2014, 110, 2551–2559. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.C.; Mahalingam, J.; Chiang, J.M.; Su, P.J.; Chu, Y.Y.; Lai, H.Y.; Fang, J.H.; Huang, C.T.; Chiu, C.T.; Lin, C.Y. Activated but not resting regulatory T cells accumulated in tumor microenvironment and correlated with tumor progression in patients with colorectal cancer. Int. J. Cancer 2013, 132, 1341–1350. [Google Scholar] [CrossRef]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Berthel, A.; Zoernig, I.; Valous, N.A.; Kahlert, C.; Klupp, F.; Ulrich, A.; Weitz, J.; Jaeger, D.; Halama, N. Detailed resolution analysis reveals spatial T cell heterogeneity in the invasive margin of colorectal cancer liver metastases associated with improved survival. Oncoimmunology 2017, 6, e1286436. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Y.; Dai, Y.; Cheng, J.-N.; Gong, Z.; Feng, Y.; Sun, C.; Jia, Q.; Zhu, B. Immune Landscape of Colorectal Cancer Tumor Microenvironment from Different Primary Tumor Location. Front. Immunol. 2018, 9, 1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sconocchia, G.; Eppenberger, S.; Spagnoli, G.C.; Tornillo, L.; Droeser, R.; Caratelli, S.; Ferrelli, F.; Coppola, A.; Arriga, R.; Lauro, D.; et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. Oncoimmunology 2014, 3, e952197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocca, Y.S.; Roberti, M.P.; Arriaga, J.M.; Amat, M.; Bruno, L.; Pampena, M.B.; Huertas, E.; Loria, F.S.; Pairola, A.; Bianchini, M.; et al. Altered phenotype in peripheral blood and tumor-associated NK cells from colorectal cancer patients. Innate Immun. 2013, 19, 76–85. [Google Scholar] [CrossRef]
- Malfettone, A.; Silvestris, N.; Saponaro, C.; Ranieri, G.; Russo, A.; Caruso, S.; Popescu, O.; Simone, G.; Paradiso, A.; Mangia, A. High density of tryptase-positive mast cells in human colorectal cancer: A poor prognostic factor related to protease-activated receptor 2 expression. J. Cell Mol. Med. 2013, 17, 1025–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Ichikawa, Y.; Nakagawa, K.; Kumamoto, T.; Mori, R.; Matsuyama, R.; Takeda, K.; Ota, M.; Tanaka, K.; Tamura, T.; et al. High infiltration of mast cells positive to tryptase predicts worse outcome following resection of colorectal liver metastases. BMC Cancer 2015, 15, 840. [Google Scholar] [CrossRef] [Green Version]
- Pine, J.K.; Morris, E.; Hutchins, G.G.; West, N.P.; Jayne, D.G.; Quirke, P.; Prasad, K.R. Systemic neutrophil-to-lymphocyte ratio in colorectal cancer: The relationship to patient survival, tumour biology and local lymphocytic response to tumour. Br. J. Cancer 2015, 113, 204–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prizment, A.E.; Vierkant, R.A.; Smyrk, T.C.; Tillmans, L.S.; Lee, J.J.; Sriramarao, P.; Nelson, H.H.; Lynch, C.F.; Thibodeau, S.N.; Church, T.R.; et al. Tumor eosinophil infiltration and improved survival of colorectal cancer patients: Iowa Women’s Health Study. Mod. Pathol. 2016, 29, 516–527. [Google Scholar] [CrossRef]
- Norton, S.E.; Dunn, E.T.; McCall, J.L.; Munro, F.; Kemp, R.A. Gut macrophage phenotype is dependent on the tumor microenvironment in colorectal cancer. Clin. Transl. Immunol. 2016, 5, e76. [Google Scholar] [CrossRef] [PubMed]
- Koelzer, V.H.; Canonica, K.; Dawson, H.; Sokol, L.; Karamitopoulou-Diamantis, E.; Lugli, A.; Zlobec, I. Phenotyping of tumor-associated macrophages in colorectal cancer: Impact on single cell invasion (tumor budding) and clinicopathological outcome. Oncoimmunology 2016, 5, e1106677. [Google Scholar] [CrossRef] [Green Version]
- Forssell, J.; Oberg, A.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res. 2007, 13, 1472–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edin, S.; Wikberg, M.L.; Dahlin, A.M.; Rutegård, J.; Öberg, Å.; Oldenborg, P.A.; Palmqvist, R. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS ONE 2012, 7, e47045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.C.; Chen, J.S.; Lee, C.H.; Chang, J.J.; Shieh, Y.S. Intratumoral macrophage counts correlate with tumor progression in colorectal cancer. J. Surg. Oncol. 2010, 102, 242–248. [Google Scholar] [CrossRef]
- Trumpi, K.; Frenkel, N.; Peters, T.; Korthagen, N.M.; Jongen, J.M.J.; Raats, D.; van Grevenstein, H.; Backes, Y.; Moons, L.M.; Lacle, M.M.; et al. Macrophages induce “budding” in aggressive human colon cancer subtypes by protease-mediated disruption of tight junctions. Oncotarget 2018, 9, 19490–19507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nearchou, I.P.; Gwyther, B.M.; Georgiakakis, E.C.T.; Gavriel, C.G.; Lillard, K.; Kajiwara, Y.; Ueno, H.; Harrison, D.J.; Caie, P.D. Spatial immune profiling of the colorectal tumor microenvironment predicts good outcome in stage II patients. NPJ Digit. Med. 2020, 3, 71. [Google Scholar] [CrossRef]
- Feng, Q.; Chang, W.; Mao, Y.; He, G.; Zheng, P.; Tang, W.; Wei, Y.; Ren, L.; Zhu, D.; Ji, M.; et al. Tumor-associated Macrophages as Prognostic and Predictive Biomarkers for Postoperative Adjuvant Chemotherapy in Patients with Stage II Colon Cancer. Clin. Cancer Res. 2019, 25, 3896–3907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Wei, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Dou, R.; Xiong, B. Elevated CD163(+)/CD68(+) Ratio at Tumor Invasive Front is Closely Associated with Aggressive Phenotype and Poor Prognosis in Colorectal Cancer. Int. J. Biol. Sci. 2019, 15, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Nagorsen, D.; Voigt, S.; Berg, E.; Stein, H.; Thiel, E.; Loddenkemper, C. Tumor-infiltrating macrophages and dendritic cells in human colorectal cancer: Relation to local regulatory T cells, systemic T-cell response against tumor-associated antigens and survival. J. Transl. Med. 2007, 5, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadabayev, A.R.; Sandel, M.H.; Menon, A.G.; Morreau, H.; Melief, C.J.; Offringa, R.; van der Burg, S.H.; Janssen-van Rhijn, C.; Ensink, N.G.; Tollenaar, R.A.; et al. Dendritic cells in colorectal cancer correlate with other tumor-infiltrating immune cells. Cancer Immunol. Immunother. 2004, 53, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Sandel, M.H.; Dadabayev, A.R.; Menon, A.G.; Morreau, H.; Melief, C.J.; Offringa, R.; van der Burg, S.H.; Janssen-van Rhijn, C.M.; Ensink, N.G.; Tollenaar, R.A.; et al. Prognostic value of tumor-infiltrating dendritic cells in colorectal cancer: Role of maturation status and intratumoral localization. Clin. Cancer Res. 2005, 11, 2576–2582. [Google Scholar] [CrossRef] [Green Version]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wang, Z.; Wu, L.; Zhang, M.; Li, W.; Ding, J.; Zhu, J.; Wei, H.; Zhao, K. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS ONE 2013, 8, e57114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, E.; Verschoor, C.P.; Ma, G.W.; Pawelec, G. Relationships Between Immune Landscapes, Genetic Subtypes and Responses to Immunotherapy in Colorectal Cancer. Front. Immunol. 2020, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; de Reynies, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautes-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [Green Version]
- Mondaca, S.; Yaeger, R. Genetics of rectal cancer and novel therapies: Primer for radiologists. Abdom. Radiol. (Ny) 2019, 44, 3743–3750. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; Freeman-Mills, L.; Rayner, E.; Glaire, M.; Briggs, S.; Vermeulen, L.; Fessler, E.; Medema, J.P.; Boot, A.; Morreau, H.; et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 2016, 1, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Llosa, N.J.; Luber, B.; Tam, A.J.; Smith, K.N.; Siegel, N.; Awan, A.H.; Fan, H.; Oke, T.; Zhang, J.; Domingue, J.; et al. Intratumoral Adaptive Immunosuppression and Type 17 Immunity in Mismatch Repair Proficient Colorectal Tumors. Clin. Cancer Res. 2019, 25, 5250–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Xue, R.; Pan, C. Prognostic and clinicopathological value of PD-L1 in colorectal cancer: A systematic review and meta-analysis. Onco Targets Ther. 2019, 12, 3671–3682. [Google Scholar] [CrossRef] [PubMed]
- van den Ende, T.; van den Boorn, H.G.; Hoonhout, N.M.; van Etten-Jamaludin, F.S.; Meijer, S.L.; Derks, S.; de Gruijl, T.D.; Bijlsma, M.F.; van Oijen, M.G.H.; van Laarhoven, H.W.M. Priming the tumor immune microenvironment with chemo(radio)therapy: A systematic review across tumor types. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188386. [Google Scholar] [CrossRef]
- Lhuillier, C.; Rudqvist, N.P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharm. 2018, 9, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prise, K.M.; Schettino, G.; Folkard, M.; Held, K.D. New insights on cell death from radiation exposure. Lancet Oncol. 2005, 6, 520–528. [Google Scholar] [CrossRef]
- Gupta, A.; Probst, H.C.; Vuong, V.; Landshammer, A.; Muth, S.; Yagita, H.; Schwendener, R.; Pruschy, M.; Knuth, A.; van den Broek, M. Radiotherapy Promotes Tumor-Specific Effector CD8+ T Cells via Dendritic Cell Activation. J. Immunol. 2012, 189, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filatenkov, A.; Baker, J.; Mueller, A.M.; Kenkel, J.; Ahn, G.O.; Dutt, S.; Zhang, N.; Kohrt, H.; Jensen, K.; Dejbakhsh-Jones, S.; et al. Ablative Tumor Radiation Can Change the Tumor Immune Cell Microenvironment to Induce Durable Complete Remissions. Clin. Cancer Res. 2015, 21, 3727–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muroyama, Y.; Nirschl, T.R.; Kochel, C.M.; Lopez-Bujanda, Z.; Theodros, D.; Mao, W.; Carrera-Haro, M.A.; Ghasemzadeh, A.; Marciscano, A.E.; Velarde, E.; et al. Stereotactic Radiotherapy Increases Functionally Suppressive Regulatory T Cells in the Tumor Microenvironment. Cancer Immunol. Res. 2017, 5, 992–1004. [Google Scholar] [CrossRef] [Green Version]
- Mirjolet, C.; Charon-Barra, C.; Ladoire, S.; Arbez-Gindre, F.; Bertaut, A.; Ghiringhelli, F.; Leroux, A.; Peiffert, D.; Borg, C.; Bosset, J.F.; et al. Tumor lymphocyte immune response to preoperative radiotherapy in locally advanced rectal cancer: The LYMPHOREC study. Oncoimmunology 2018, 7, e1396402. [Google Scholar] [CrossRef]
- Monjazeb, A.M.; Schalper, K.A.; Villarroel-Espindola, F.; Nguyen, A.; Shiao, S.L.; Young, K. Effects of Radiation on the Tumor Microenvironment. Semin. Radiat. Oncol. 2020, 30, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cao, Y.; Markelc, B.; Kaeppler, J.; Vermeer, J.A.; Muschel, R.J. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J. Clin. Investig. 2019, 129, 4224–4238. [Google Scholar] [CrossRef] [PubMed]
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 2019, 178, 933–948. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, B.; Liu, S.; Zhang, A.; Wu, T.; Zhao, Y. 2-Gy whole-body irradiation significantly alters the balance of CD4+ CD25- T effector cells and CD4+ CD25+ Foxp3+ T regulatory cells in mice. Cell Mol. Immunol. 2010, 7, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Menon, H.; Chen, D.; Ramapriyan, R.; Verma, V.; Barsoumian, H.B.; Cushman, T.R.; Younes, A.I.; Cortez, M.A.; Erasmus, J.J.; de Groot, P.; et al. Influence of low-dose radiation on abscopal responses in patients receiving high-dose radiation and immunotherapy. J. Immunother. Cancer 2019, 7, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [Green Version]
- Grapin, M.; Richard, C.; Limagne, E.; Boidot, R.; Morgand, V.; Bertaut, A.; Derangere, V.; Laurent, P.A.; Thibaudin, M.; Fumet, J.D.; et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: A promising new combination. J. Immunother. Cancer 2019, 7, 160. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stary, V.; Wolf, B.; Unterleuthner, D.; List, J.; Talic, M.; Laengle, J.; Beer, A.; Strobl, J.; Stary, G.; Dolznig, H.; et al. Short-course radiotherapy promotes pro-inflammatory macrophages via extracellular vesicles in human rectal cancer. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Teresa Pinto, A.; Laranjeiro Pinto, M.; Patrícia Cardoso, A.; Monteiro, C.; Teixeira Pinto, M.; Filipe Maia, A.; Castro, P.; Figueira, R.; Monteiro, A.; Marques, M.; et al. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci. Rep. 2016, 6, 18765. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Nirei, T.; Sunami, E.; Nagawa, H.; Kitayama, J. Density of CD4(+) and CD8(+) T lymphocytes in biopsy samples can be a predictor of pathological response to chemoradiotherapy (CRT) for rectal cancer. Radiat. Oncol. 2011, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- McCoy, M.J.; Hemmings, C.; Miller, T.J.; Austin, S.J.; Bulsara, M.K.; Zeps, N.; Nowak, A.K.; Lake, R.A.; Platell, C.F. Low stromal Foxp3+ regulatory T-cell density is associated with complete response to neoadjuvant chemoradiotherapy in rectal cancer. Br. J. Cancer 2015, 113, 1677–1686. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Ge, X.; He, J.; Cheng, Y.; Wang, Z.; Wang, J.; Sun, L. The prognostic value of tumor-infiltrating lymphocytes in colorectal cancer differs by anatomical subsite: A systematic review and meta-analysis. World J. Surg. Oncol. 2019, 17, 85. [Google Scholar] [CrossRef] [Green Version]
- Roxburgh, C.S.; Shia, J.; Vakiani, E.; Daniel, T.; Weiser, M.R. Potential immune priming of the tumor microenvironment with FOLFOX chemotherapy in locally advanced rectal cancer. Oncoimmunology 2018, 7, e1435227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Bai, W.; Tong, X.; Bu, P.; Xu, J.; Xi, Y. Correlation between tumor microenvironment-associated factors and the efficacy and prognosis of neoadjuvant therapy for rectal cancer. Oncol. Lett. 2019, 17, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Billiard, F.; Buard, V.; Benderitter, M.; Linard, C. Abdominal gamma-radiation induces an accumulation of function-impaired regulatory T cells in the small intestine. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Wu, M.L.; Huang, K.C.; Huang, I.P.; Chung, Y.L. The Effects of Neoadjuvant Treatment on the Tumor Microenvironment in Rectal Cancer: Implications for Immune Activation and Therapy Response. Clin. Colorectal. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Jarosch, A.; Sommer, U.; Bogner, A.; Reissfelder, C.; Weitz, J.; Krause, M.; Folprecht, G.; Baretton, G.B.; Aust, D.E. Neoadjuvant radiochemotherapy decreases the total amount of tumor infiltrating lymphocytes, but increases the number of CD8+/Granzyme B+ (GrzB) cytotoxic T-cells in rectal cancer. Oncoimmunology 2018, 7, e1393133. [Google Scholar] [CrossRef] [Green Version]
- Yasui, K.; Kondou, R.; Iizuka, A.; Miyata, H.; Tanaka, E.; Ashizawa, T.; Nagashima, T.; Ohshima, K.; Urakami, K.; Kusuhara, M.; et al. Effect of preoperative chemoradiotherapy on the immunological status of rectal cancer patients. J. Radiat. Res. 2020, 61, 766–775. [Google Scholar] [CrossRef]
- Chen, T.W.; Huang, K.C.; Chiang, S.F.; Chen, W.T.; Ke, T.W.; Chao, K.S.C. Prognostic relevance of programmed cell death-ligand 1 expression and CD8+ TILs in rectal cancer patients before and after neoadjuvant chemoradiotherapy. J. Cancer Res. Clin. Oncol. 2019, 145, 1043–1053. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chiang, S.F.; Ke, T.W.; Chen, T.W.; Lan, Y.C.; You, Y.S.; Shiau, A.C.; Chen, W.T.; Chao, K.S.C. Cytosolic high-mobility group box protein 1 (HMGB1) and/or PD-1+ TILs in the tumor microenvironment may be contributing prognostic biomarkers for patients with locally advanced rectal cancer who have undergone neoadjuvant chemoradiotherapy. Cancer Immunol. Immunother. 2018, 67, 551–562. [Google Scholar] [CrossRef]
- Chiang, S.-F.; Huang, C.-Y.; Ke, T.-W.; Chen, T.-W.; Lan, Y.-C.; You, Y.-S.; Chen, W.T.-L.; Chao, K.S.C. Upregulation of tumor PD-L1 by neoadjuvant chemoradiotherapy (neoCRT) confers improved survival in patients with lymph node metastasis of locally advanced rectal cancers. Cancer Immunol. Immunother. 2019, 68, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Droeser, R.A.; Hirt, C.; Viehl, C.T.; Frey, D.M.; Nebiker, C.; Huber, X.; Zlobec, I.; Eppenberger-Castori, S.; Tzankov, A.; Rosso, R.; et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur. J. Cancer 2013, 49, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerard, J.P.; Conroy, T.; Bonnetain, F.; Bouche, O.; Chapet, O.; Closon-Dejardin, M.T.; Untereiner, M.; Leduc, B.; Francois, E.; Maurel, J.; et al. Preoperative radiotherapy with or without concurrent fluorouracil and leucovorin in T3-4 rectal cancers: Results of FFCD 9203. J. Clin. Oncol. 2006, 24, 4620–4625. [Google Scholar] [CrossRef] [PubMed]
- Ogura, A.; Akiyoshi, T.; Yamamoto, N.; Kawachi, H.; Ishikawa, Y.; Mori, S.; Oba, K.; Nagino, M.; Fukunaga, Y.; Ueno, M. Pattern of programmed cell death-ligand 1 expression and CD8-positive T-cell infiltration before and after chemoradiotherapy in rectal cancer. Eur. J. Cancer 2018, 91, 11–20. [Google Scholar] [CrossRef]
- Teng, F.; Mu, D.; Meng, X.; Kong, L.; Zhu, H.; Liu, S.; Zhang, J.; Yu, J. Tumor infiltrating lymphocytes (TILs) before and after neoadjuvant chemoradiotherapy and its clinical utility for rectal cancer. Am. J. Cancer Res. 2015, 5, 2064–2074. [Google Scholar]
- Hecht, M.; Buttner-Herold, M.; Erlenbach-Wunsch, K.; Haderlein, M.; Croner, R.; Grutzmann, R.; Hartmann, A.; Fietkau, R.; Distel, L.V. PD-L1 is upregulated by radiochemotherapy in rectal adenocarcinoma patients and associated with a favourable prognosis. Eur. J. Cancer 2016, 65, 52–60. [Google Scholar] [CrossRef]
- Mierzwa, M.L.; Nyati, M.K.; Morgan, M.A.; Lawrence, T.S. Recent advances in combined modality therapy. Oncologist 2010, 15, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.M.; Fyles, A.; Nguyen, L.T.; Neel, B.G.; Sacher, A.; Rottapel, R.; Wang, B.X.; Ohashi, P.S.; Sridhar, S.S. Phase I study of local radiation and tremelimumab in patients with inoperable locally recurrent or metastatic breast cancer. Oncotarget 2019, 10, 2947–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahig, H.; Aubin, F.; Stagg, J.; Gologan, O.; Ballivy, O.; Bissada, E.; Nguyen-Tan, F.P.; Soulières, D.; Guertin, L.; Filion, E.; et al. Phase I/II trial of Durvalumab plus Tremelimumab and stereotactic body radiotherapy for metastatic head and neck carcinoma. BMC Cancer 2019, 19, 68. [Google Scholar] [CrossRef]
- Kim, H.J.; Chang, J.S.; Roh, M.R.; Oh, B.H.; Chung, K.Y.; Shin, S.J.; Koom, W.S. Effect of Radiotherapy Combined with Pembrolizumab on Local Tumor Control in Mucosal Melanoma Patients. Front. Oncol. 2019, 9, 835. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Wang, X.; Soh, H.; Seyedin, S.; Cortez, M.A.; Krishnan, S.; Massarelli, E.; Hong, D.; Naing, A.; Diab, A.; et al. Combining radiation and immunotherapy: A new systemic therapy for solid tumors? Cancer Immunol. Res. 2014, 2, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovedi, S.J.; Cheadle, E.J.; Popple, A.L.; Poon, E.; Morrow, M.; Stewart, R.; Yusko, E.C.; Sanders, C.M.; Vignali, M.; Emerson, R.O.; et al. Fractionated Radiation Therapy Stimulates Antitumor Immunity Mediated by Both Resident and Infiltrating Polyclonal T-cell Populations when Combined with PD-1 Blockade. Clin. Cancer Res. 2017, 23, 5514–5526. [Google Scholar] [CrossRef] [Green Version]
- Garelli, E.; Rittmeyer, A.; Putora, P.M.; Glatzer, M.; Dressel, R.; Andreas, S. Abscopal effect in lung cancer: Three case reports and a concise review. Immunotherapy 2019, 11, 1445–1461. [Google Scholar] [CrossRef] [PubMed]
- Young, K.H.; Baird, J.R.; Savage, T.; Cottam, B.; Friedman, D.; Bambina, S.; Messenheimer, D.J.; Fox, B.; Newell, P.; Bahjat, K.S.; et al. Optimizing Timing of Immunotherapy Improves Control of Tumors by Hypofractionated Radiation Therapy. PLoS ONE 2016, 11, e0157164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Filatenkov, A.; Baker, J.; Strober, S. Disruption of evasive immune cell microenvironment in tumors reflects immunity induced by radiation therapy. Oncoimmunology 2016, 5, e1072673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, S.R.; Ardiani, A.; Kwilas, A.; Hodge, J.W. Radiation-induced survival responses promote immunogenic modulation to enhance immunotherapy in combinatorial regimens. Oncoimmunology 2014, 3, e28643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floudas, C.S.; Brar, G.; Mabry-Hrones, D.; Duffy, A.G.; Wood, B.; Levy, E.; Krishnasamy, V.; Fioravanti, S.; Bonilla, C.M.; Walker, M.; et al. A Pilot Study of the PD-1 Targeting Agent AMP-224 Used With Low-Dose Cyclophosphamide and Stereotactic Body Radiation Therapy in Patients with Metastatic Colorectal Cancer. Clin. Colorectal. Cancer 2019, 18, e349–e360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, T.; Bando, H.; Tsukada, Y.; Inamori, K.; Yuki, S.; Komatsu, Y.; Homma, S.; Uemura, M.; Kato, T.; Kotani, D.; et al. Voltage: Investigator-initiated clinical trial of nivolumab monotherapy and subsequent radical surgery following preoperative chemoradiotherapy in patients with microsatellite stable locally advanced rectal cancer. J. Clin. Oncol. 2019, 37, 3606. [Google Scholar] [CrossRef]
- Tang, C.; Welsh, J.W.; de Groot, P.; Massarelli, E.; Chang, J.Y.; Hess, K.R.; Basu, S.; Curran, M.A.; Cabanillas, M.E.; Subbiah, V.; et al. Ipilimumab with Stereotactic Ablative Radiation Therapy: Phase I Results and Immunologic Correlates from Peripheral T Cells. Clin. Cancer Res. 2017, 23, 1388–1396. [Google Scholar] [CrossRef] [Green Version]
- Son, C.H.; Bae, J.H.; Shin, D.Y.; Lee, H.R.; Choi, Y.J.; Jo, W.S.; Ho Jung, M.; Kang, C.D.; Yang, K.; Park, Y.S. CTLA-4 blockade enhances antitumor immunity of intratumoral injection of immature dendritic cells into irradiated tumor in a mouse colon cancer model. J. Immunother. 2014, 37, 1–7. [Google Scholar] [CrossRef]
- Wu, C.-J.; Tsai, Y.-T.; Chang, C.-C.; Lee, I.-J.; Wu, P.-Y.; Tao, M.-H. Efficacy of combining radiotherapy and immunotherapy for metastatic colorectal cancer and the modulation of tumor microenvironment. J. Immunol. 2017, 198, 79.15. [Google Scholar]
- Jones, K.I.; Tiersma, J.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Buzzelli, J.; Im, J.H.; Muschel, R.J. Radiation combined with macrophage depletion promotes adaptive immunity and potentiates checkpoint blockade. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
Figure 1.
The rectal tumor microenvironment. (a) Illustration representing the anatomy of the large intestine and the development of a tumor localized in the rectum. (b) The four consensus molecular subtypes and their specific stroma-immune microenvironments. CMS1 cluster displays strong immune activation with high levels of CD8+ T cells, CD4+ T cells, γδ T cells, activated dendritic cells (DCs), natural killer (NK) cells and M1 macrophages alongside high expression of cytokines, PD1, PD-L1 and MHC-I. CMS2 shows an immune-desert microenvironment characterized by a few immune cells and poor expression of PD1, PD-L1, LAG-3 and CTLA-4. CMS3 is distinguished by metabolic dysregulation, infiltration of Th17 cells, naive B and T cells, and expression of MHC-I, PD1 and PD-L1. CMS4 exhibits high angiogenesis activity, expression of TGF-β, and infiltration of CD8+ T cells, CD4+ T cells, Tregs, M2 macrophages, monocytes, eosinophils and resting DCs.
Figure 1.
The rectal tumor microenvironment. (a) Illustration representing the anatomy of the large intestine and the development of a tumor localized in the rectum. (b) The four consensus molecular subtypes and their specific stroma-immune microenvironments. CMS1 cluster displays strong immune activation with high levels of CD8+ T cells, CD4+ T cells, γδ T cells, activated dendritic cells (DCs), natural killer (NK) cells and M1 macrophages alongside high expression of cytokines, PD1, PD-L1 and MHC-I. CMS2 shows an immune-desert microenvironment characterized by a few immune cells and poor expression of PD1, PD-L1, LAG-3 and CTLA-4. CMS3 is distinguished by metabolic dysregulation, infiltration of Th17 cells, naive B and T cells, and expression of MHC-I, PD1 and PD-L1. CMS4 exhibits high angiogenesis activity, expression of TGF-β, and infiltration of CD8+ T cells, CD4+ T cells, Tregs, M2 macrophages, monocytes, eosinophils and resting DCs.

Figure 2.
Tumor microenvironment after radiotherapy and immune checkpoint Inhibition. Upon radiotherapy, cancer cells undergo immunogenic cell death that is associated with the release of damage associated molecular patterns (DAMPs), neoantigens and pro-inflammatory cytokines (e.g., IFN-γ, IL-1 and IL-6), which promote the expression of immunomodulatory genes including antigen presentation genes and lead to the recruitment and activation of dendritic cells (DCs). Following migration to the lymph node, DCs are involved in priming and activation of T lymphocytes, which are then recruited into the tumor site alongside other immune cells. Additional modifications in the tumor microenvironment (TME) upon radiotherapy (RT) treatment include shift in macrophage phenotype towards M1, modulation of the tumor vasculature and alteration of the cell metabolism. Altogether these events enhance the recognition and killing of tumor cells. Besides inducing de novo inflammation, RT is also responsible for increasing the expression of immune checkpoints such as PD1, PD-L1 and CTLA-4. In this context, the application of immune checkpoint inhibitors might further add to the ongoing adaptive anti-tumor immunity. Taken together, the effect of RT on the TME could evoke the transition from CMS2-like TME to CMS1-like TME.
Figure 2.
Tumor microenvironment after radiotherapy and immune checkpoint Inhibition. Upon radiotherapy, cancer cells undergo immunogenic cell death that is associated with the release of damage associated molecular patterns (DAMPs), neoantigens and pro-inflammatory cytokines (e.g., IFN-γ, IL-1 and IL-6), which promote the expression of immunomodulatory genes including antigen presentation genes and lead to the recruitment and activation of dendritic cells (DCs). Following migration to the lymph node, DCs are involved in priming and activation of T lymphocytes, which are then recruited into the tumor site alongside other immune cells. Additional modifications in the tumor microenvironment (TME) upon radiotherapy (RT) treatment include shift in macrophage phenotype towards M1, modulation of the tumor vasculature and alteration of the cell metabolism. Altogether these events enhance the recognition and killing of tumor cells. Besides inducing de novo inflammation, RT is also responsible for increasing the expression of immune checkpoints such as PD1, PD-L1 and CTLA-4. In this context, the application of immune checkpoint inhibitors might further add to the ongoing adaptive anti-tumor immunity. Taken together, the effect of RT on the TME could evoke the transition from CMS2-like TME to CMS1-like TME.


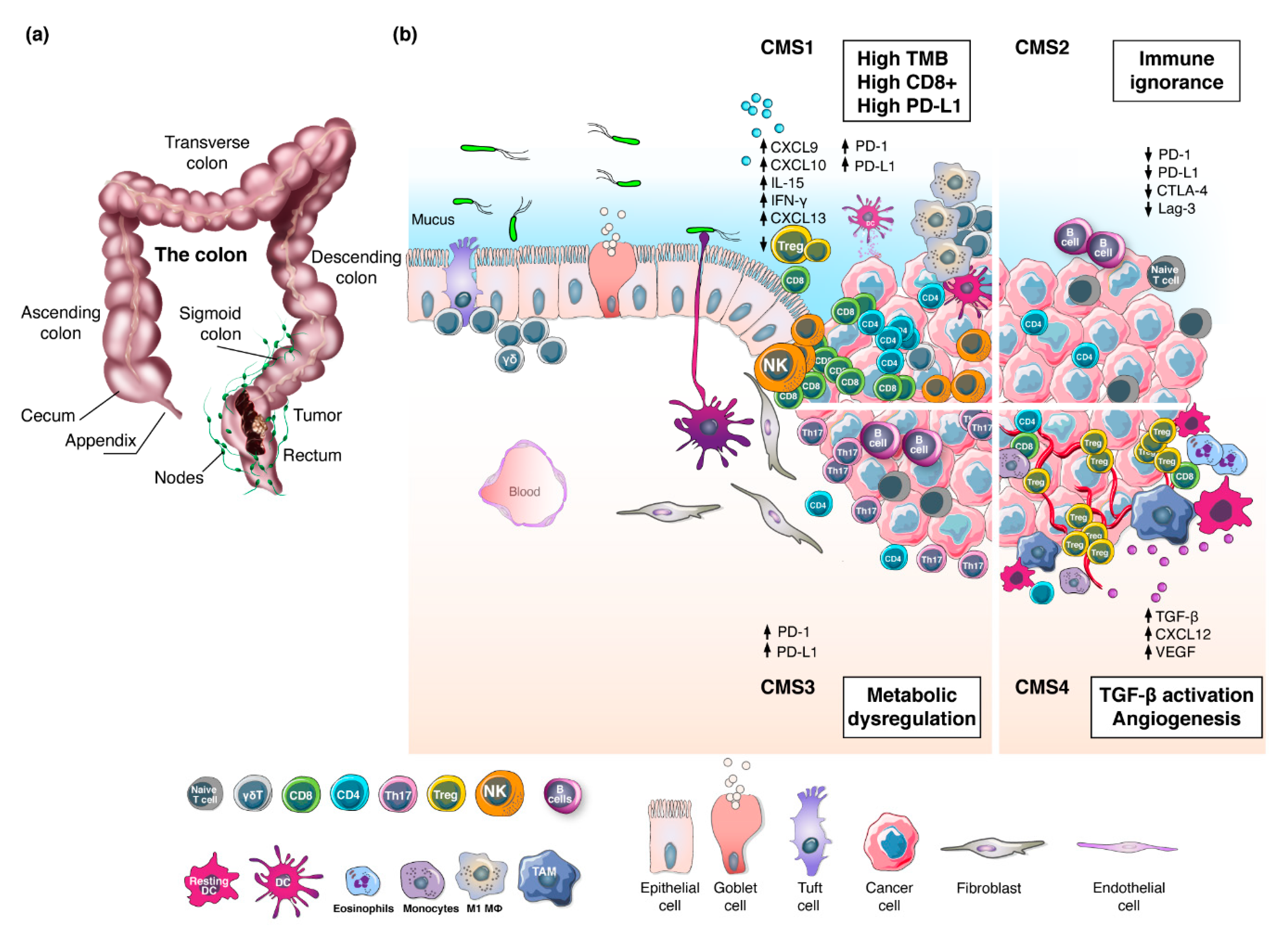{kind=link}
{kind=link}
Table 1.
Summary of the current clinical trials in the field of radio-immunotherapy in rectal cancer.
Table 1.
Summary of the current clinical trials in the field of radio-immunotherapy in rectal cancer.
| Short Course Radiotherapy | |||||||||||
| NCT Number | Phase | Clinical Stage | Microsatellite Status | Arm | Neo-Adjuvant Treatment Sequence | Time to Surgery | Adjuvant Treatment | Nb of Patients | Primary Outcome | ||
| Pre-Radiotherapy Treatment | Radiotherapy Treatment | Post-Radiotherapy Treatment | |||||||||
| NCT04663763 | II | T3–4 and/or N+ | MSS | A | - | scRT (5 × 5 Gy) | 4 cycles of CAPOX and anti-PD1 antibody (Sintilimab) | 1 week after the end of neoadjuvant therapy | 4 cycles of CAPOX | 32 | pCR rate |
| MSI-H | B | - | scRT (5 × 5 Gy) | 4 cycles of CAPOX and PD1 antibody (Sintilimab) | 1 week after the end of neoadjuvant therapy | 4 cycles of CAPOX | 8 | pCR rate | |||
| NCT04518280 | II R* | T3–4 and/or N+ | MSS | A | 2 cycles of CAPOX and anti-PD1 antibody (Toripalimab) | scRT (5 × 5 Gy) | 4 cycles of CAPOX and anti-PD1 antibody (Toripalimab) | 2–4 weeks after the end of neoadjuvant therapy | - | 65 | pCR rate |
| B | - | scRT (5 × 5 Gy) | 6 cycles of CAPOX and anti-PD1 antibody (Toripalimab) | 2–4 weeks after the end of neoadjuvant therapy | - | 65 | pCR rate | ||||
| NCT04558684 | II | >T2N0 or low T2N0 | - | - | - | scRT (5 × 5 Gy) | 6 cycles of CAPOX and anti-PD1 antibody (Camrelizumab) | 8 (+/−4) weeks after the end of neoadjuvant therapy | - | 30 | cCR rate |
| NCT04621370 PRIME-RT study | II R* | cT3b+, N+, EMVI+ | - | A | Anti-PD-L1 antibody (Durvalumab) week prior radiotherapy | scRT (5 × 5 Gy) | 6 cycles of FOLFOX and anti-PD-L1 antibody (Durvalumab) | 3–5 weeks after the end of neoadjuvant therapy | - | 24 | pCR rate |
| - | B | Anti-PD-L1 antibody (Durvalumab) week prior radiotherapy | capecitabine radiosensitized NACRT (50Gy) | 4 cycles of FOLFOX and anti-PD-L1 antibody (Durvalumab) | 3–5 weeks after the end of neoadjuvant therapy | - | 24 | pCR rate | |||
| NCT04109755 PEMREC study | II | cT3–T4 N0 or cT any and N1-2 | MSS | - | scRT (5 × 5 Gy) with anti-PD1 antibody (Pembrolizumab) | 3 cycles of anti-PD1 antibody (Pembrolizumab) | 3 weeks after the end of neoadjuvant therapy | - | 25 | TRG | |
| NCT04231552 | II | cT3–4 or N+ | - | - | scRT (5 × 5 Gy) | 2 cycles of CAPOX and anti-PD1 antibody (Camrelizumab) | n.a | - | 30 | pCR rate | |
| NCT03503630 | II | cT2 N1–3, cT3 N0–3 | - | - | scRT (5 × 5 Gy) | 6 cycles of FOLFOX and anti-PD-L1 antibody (COMPOUND 2055269) | 2–3 weeks after the end of neoadjuvant therapy | - | 44 | pCR | |
| NCT04503694 REGINA study | II | Intermediate risk MRI-defined rectal cancer | - | 2 cycles anti-PD1 antibody (Nivolumab) + regorafenib | scRT (5 × 5 Gy) | 3 cycles anti-PD1 antibody (Nivolumab) + regorafenib | 7–8 weeks after the end of scRT therapy | - | 60 | pCR | |
| NCT04636008 | Ib | ≥cT2 | MSI-H/dMMR | - | scRT (5 × 5 Gy) | 3 cycles of anti-PD1 antibody (Sintilimab) | 1–2 weeks after the end of neoadjuvant therapy | - | 20 | TRAE rate | |
| Long Course Chemo-Radiotherapy | |||||||||||
| NCT Number | Phase | Clinical Stage | Microsatellite Status | Arm | Neo-Adjuvant Treatment Sequence | Time to Surgery | Adjuvant Treatment | Nb of Patients | Primary Outcome | ||
| Pre-Radiotherapy treatment | Radiotherapy Treatment | Post-Radiotherapy Treatment | |||||||||
| NCT04411537 | II | T3–4 and/or N+ | MSS | 2 cycles of anti-PD1 antibody | Capecitabine plus irinotecan radiosensitized NACRT (50 Gy) | 3 cycles of anti-PD1 antibody | 1–2 weeks after the end of neoadjuvant therapy | 6 cycles of XELOX | 50 | pCR rate | |
| NCT04411524 | II | T3–4 and/or N+ | MSI-H | 2 cycles of anti-PD1 antibody | Capecitabine plus irinotecan radiosensitized NACRT (50 Gy) | 3 cycles of anti-PD1 antibody | 1–2 weeks after the end of neoadjuvant therapy | 6 cycles of XELOX | 50 | pCR rate | |
| NCT03854799 AVANA study | II | cN+, cT4, high risk cT3 | - | - | Capecitabine radiosensitized NACRT (50.4 Gy) with anti-PD-L1 antibody (Avelumab) | - | 8–10 weeks after the end of neoadjuvant therapy | - | 101 | pCR rate | |
| NCT04357587 | II | Stage II or stage III or olimetastatic | dMMR | - | Capecitabine radiosensitized NACRT (50 Gy) with PD1 antibody (Pembrolizumab) | 1 cycle of anti-PD1 antibody (Pembrolizumab) | n.a | - | 10 | Rate of AE Feasability | |
| NCT03921684 | II | T3–4 N0 or TX N+ | - | - | Capecitabine radiosensitized NACRT (50.4 Gy) | 6 cycles of FOLFOX and anti-PD-1 antibody (Nivolumab) | 4 weeks after the end of neoadjuvant therapy | - | 29 | pCR rate TRAE | |
| NCT02921256 | II | Stage II or stage III | - | C | 8 cycles of FOLFOX | Capecitabine radiosensitized NACRT (50 Gy) with anti-PD1 antibody (Pembrolizumab) | 5 cycles of anti-PD1 antibody (Pembrolizumab) | n.a | - | >100 | Change in NAR score |
| NCT03127007 R-IMMUNE study | Ib/II R* | Stage II or stage III | - | A | - | 5-FU radiosensitized NACRT (50 Gy) with anti-PD-L1 antibody (Atezolizumab) | 3 cycles of anti-PD-L1 antibody (Atezolizumab) | 3 weeks after the end of neoadjuvant therapy | - | 54 | pCR rate AE rate |
| - | B | - | 5-FU radiosensitized NACRT (50 Gy) | - | 10 weeks after the end of neoadjuvant therapy | - | |||||
| NCT04443543 | II | T2–4 and/or N+ | MSS | A | - | Capecitabine plus irinotecan radiosensitized NACRT (50 Gy) | XELIRI or FOLFIRINOX adaptive number of cycles and regimen | No surgery for those in cCR | - | 222 | cCR rate |
| MSI-H/dMMR | B | - | Capecitabine plus irinotecan radiosensitized NACRT (50 Gy) | 3 cycles of anti-PD1 antibody (Tislelizumab) | No surgery for those in cCR | - | NA | cCR rate | |||
| NCT04017455 TARZAN study | II | Intermediate risk rectal cancer or low risk distal rectal cancer | - | - | Radiotherapy | 3 cycles of anti-VEGF antibody (bevacizumab) combined with anti-PD-L1 antibody (Atezolizumab) | 3 weeks after the end of neoadjuvant therapy | - | 38 | cCR rate ncCR rate | |
| NCT04124601 CHINOREC Study | II R* | NA | - | A | - | Capecitabine radiosensitized NACRT (50 Gy) | - | n.a | - | 80 | TRAE rate |
| NA | - | B | - | Capecitabine radiosensitized NACRT (50 Gy) | Anti-CTLA-4 antibody (Ipilimumab) on day 7 and anti-PD1 antibody (Nivolumab) on day 14, 28 and 42) | n.a | - | ||||
| NCT04293419 DUREC study | II | High risk MRI-defined rectal cancer | - | 6 cycles of FOLFOX + 4 cycles anti-PD-L1 (Durvalumab) | Capecitabine radiosensitized NACRT (50.4 Gy) + anti-PD-L1 (Durvalumab) | 2 cycles anti-PD-L1 (Durvalumab) | 2–6 weeks after the end of neoadjuvant therapy | - | 58 | pCR rate | |
| NCT03102047 FR-2 study | II | Stage II–IV rectal cancer | MSS | - | Capecitabine radiosensitized NACRT (50.4 Gy) | 4 cycles anti-PD-L1 (Durvalumab) | 1–4 weeks after the end of neoadjuvant therapy | - | 47 | NAR | |
| NCT02948348 VOLTAGE trial | Ib/II | T3 and T4, N any | - | - | Capecitabine radiosensitized NACRT (50.4 Gy) | 5 cycles anti-PD1 antibody (Nivolumab) | 2 weeks after the end of neoadjuvant therapy | - | 50 | pCR rate | |
| NCT03299660 | II | T3bN1-N2M0, T3c/dN0-N2M0, T4N0-N2M0 | - | - | Capecitabine/5FU radiosensitized NACRT (50.4 Gy) | 4 cycles of anti-PD-L1 antibody (Avelumab) | 8–10 weeks after the end of neoadjuvant therapy | - | 45 | pCR rate | |
| NCT04083365 PANDORA study | II | cT3/4N0/M0 or Tx N1-2/M0 | - | - | Capecitabine radiosensitized NACRT (50.4 Gy) | 3 cycles of anti-PD-L1 antibody (Avelumab) | 1–2 weeks after the end of neoadjuvant therapy | - | 60 | pCR rate | |
| Only Immune Checkpoint Inhibitors | |||||||||||
| NCT Number | Phase | Clinical Stage | Microsatellite Status | Arm | Neo-Adjuvant Treatment Sequence | Time to Surgery | Adjuvant Treatment | Nb of Patients | Primary Outcome | ||
| Pre-Radiotherapy Treatment | Radiotherapy Treatment | Post-Radiotherapy Treatment | |||||||||
| NCT04643041 BASKET study | II | T × N × M0 | MSI-H/dMMR | 6 cycles of anti-PD1 antibody | No radiotherapy | - | No surgery | - | 47 | 1 year DFS rate | |
| Other Immunotherapies | |||||||||||
| NCT Number | Phase | Clinical Stage | Microsatellite Status | Arm | Neo-Adjuvant Treatment Sequence | Time to Surgery | Adjuvant Treatment | Nb of Patients | Primary Outcome | ||
| Pre-Radiotherapy Treatment | Radiotherapy Treatment | Post-RadiotherApy treatment | |||||||||
| NCT03300544 | I | cT3–4, N+ | - | Four injections of T-VEC with 2 cycle of FOLFOX | Capecitabine radiosensitized NACRT (50.4 Gy) | - | 8–12 weeks after the end of neoadjuvant therapy | 21 | DLT | ||
| NCT02688712 ExIST Study | II | Stage II or stage III | - | TGFβ Type I Receptor Inhibitor (LY2157299) for 15 days | Capecitabine radiosensitized NACRT (50.4 Gy) with (LY2157299) 15 days after start | - | 6–10 weeks after the end of neoadjuvant therapy | 50 | pCR rate | ||
| NCT04130854 INNATE study | II R* | cT4 or within 3mm of MR fascia | - | A | - | CD-40 agonist antibody with scRT (5 × 5 Gy) | 6 cycles of FOLFOX and CD-40 agonist antibody (APX005M) | n.a | 58 | pCR rate | |
| - | B | - | scRT (5 × 5 Gy) | 6 cycles of FOLFOX | n.a | ||||||
| NCT03916510 CEDAR study | I | cT3mrf+ or N+ or low tumors | - | Three enadenotucirev loading doses in weeks 1-2 | Capecitabine radiosensitized NACRT (50.4 Gy) +/− Enadenotucirev | +/− Enadenotucirev | n.a | 30 | DLT MRI-TRG | ||
| NCT04304209 | II R* | cT3–4N0M0 or cT×N+M0 | dMMR or MSI-H | A | 4 cycles of anti-PD1 antibody (Sintilimab) | No Radiotherapy | 4 cycles of anti-PD1 antibody (Sintilimab) +/− CAPEOX chemotherapy | Surgery or watch and wait | 195 | pCR | |
| pMMR/MSS/MSI-L | B1 | 4 cycles of anti-PD1 antibody (Sintilimab) | Capecitabine radiosensitized NACRT (50.4 Gy) + CAPEOX | - | Surgery or watch and wait | ||||||
| B2 | - | Capecitabine radiosensitized NACRT (50.4 Gy) + CAPEOX | - | Surgery or watch and wait | |||||||
AE: adverse event; CAPOX (also called XELOX): capecitabine and oxaliplatin; cCR: clinical complete response; DLT: dose limiting toxicities; dMMR: deficient mismatch mechanisms of repair; FOLFOX: leucovorin, 5-FU and oxaliplatin; FOLFIRINOX: leucovorin, 5-FU, irinotecan and oxaliplatin; IMRT DT: 50Gy in 25 fractions; MR: mesorectal; MRF: MR fascia; MRI: magnetic resonance imaging; MSI-H: microsatellite instability high; MSS: microsatellite stable; NACRT: neoadjuvant chemo-radiotherapy; NAR: neoadjuvant rectal cancer; ncCR: near-complete response rate; pCR: pathological complete response; R*: randomized; scRT: short course radiotherapy (5 × 5 Gy); TME: total mesorectal excision; TRAE: treatment-related adverse event; TRG: tumor regression grade; XELIRI: irinotecan and capecitabine; n.a.: information not available.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Corrò, C.; Dutoit, V.; Koessler, T. Emerging Trends for Radio-Immunotherapy in Rectal Cancer. Cancers 2021, 13, 1374. https://doi.org/10.3390/cancers13061374
AMA Style
Corrò C, Dutoit V, Koessler T. Emerging Trends for Radio-Immunotherapy in Rectal Cancer. Cancers. 2021; 13(6):1374. https://doi.org/10.3390/cancers13061374
Chicago/Turabian StyleCorrò, Claudia, Valérie Dutoit, and Thibaud Koessler. 2021. "Emerging Trends for Radio-Immunotherapy in Rectal Cancer" Cancers 13, no. 6: 1374. https://doi.org/10.3390/cancers13061374
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.