
Mycosis Fungoides and Sézary Syndrome: An Integrative Review of the Pathophysiology, Molecular Drivers, and Targeted Therapy

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Cutaneous T-Cell Lymphoma: Description of the Main Entities
1.1. Mycosis Fungoides
1.2. Mycosis Fungoides Variants
1.2.1. Folliculotropic MF
1.2.2. Pagetoid Reticulosis
1.2.3. Granulomatous Slack Skin (GSS)
1.3. Sézary Syndrome
1.4. Mycosis Fungoides/Sézary Syndrome Staging
1.5. Mycosis Fungoides/Sézary Syndrome Prognosis
2. A Landscape of Genomic Alterations in Mycosis Fungoides/Sézary Syndrome
3. A Malignant Network of Signaling Mechanisms Drives Mycosis Fungoides/Sézary Syndrome
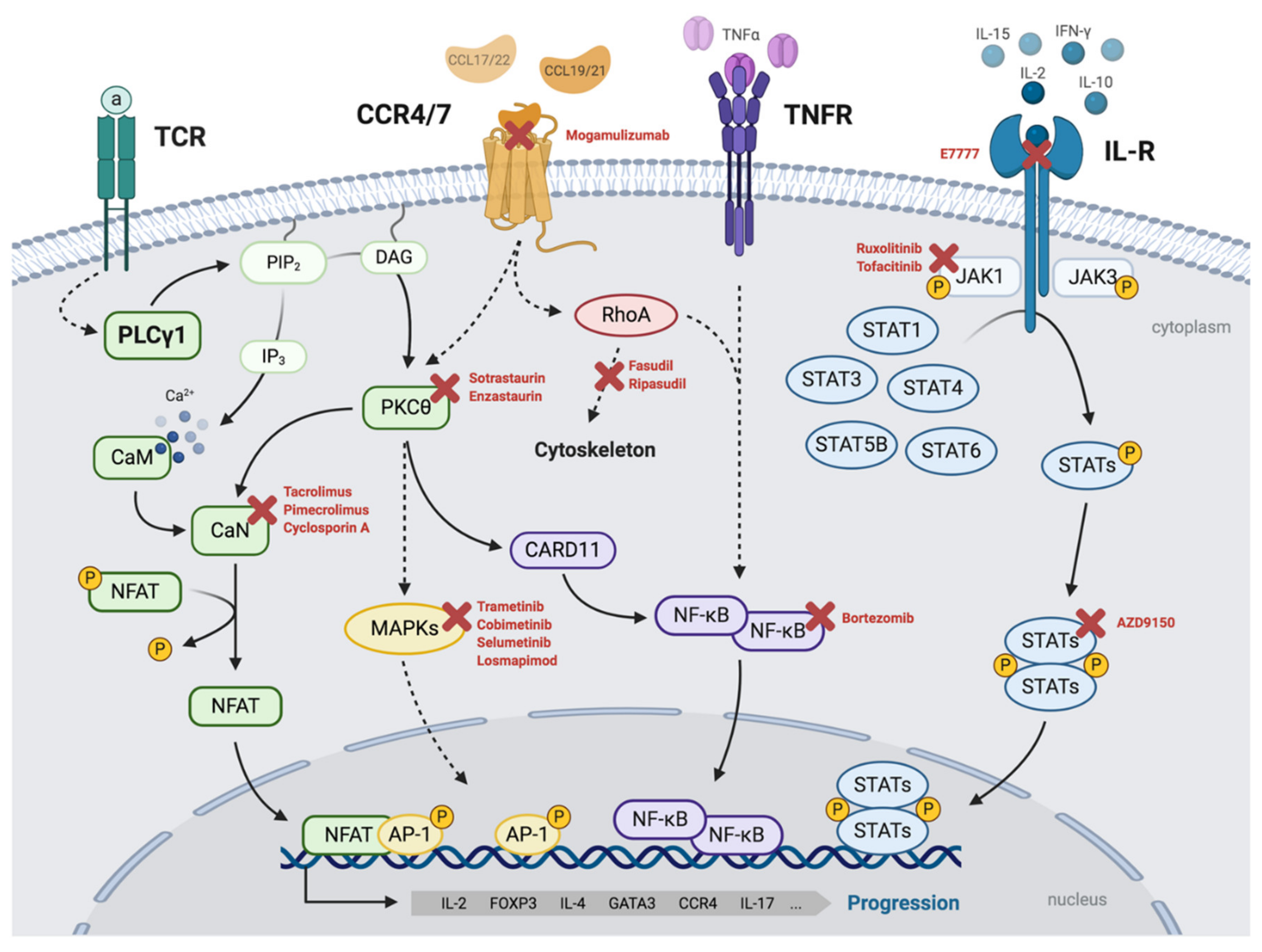
3.1. TCR/PLCγ1 Signaling
3.2. The PKC/NF-κB Axis
3.3. JAK/STAT Signaling
3.4. STAT3 Activation in Mycosis Fungoides/Sézary Syndrome
3.5. CCR4/CCR7 Signaling
4. Therapy for Mycosis Fungoides/Sézary Syndrome
4.1. Skin-Directed Therapies
4.1.1. Topical Corticosteroids
4.1.2. Topical Mechlorethamine
4.1.3. Ultraviolet Phototherapy
4.1.4. Total Skin Electron Beam Therapy
4.1.5. Localized Radiotherapy
4.2. Systemic Therapies
4.2.1. Retinoids
4.2.2. Interferon α
4.2.3. Extracorporeal Photopheresis
4.2.4. Chemotherapy
4.2.5. Allogenic Stem-Cell Transplantation
4.3. Targeted Therapies
4.3.1. Histone Deacetylase (HDAC) Inhibitors
4.3.2. Monoclonal Antibodies
4.3.3. Other Targeted Inhibitors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dobos, G.; de Masson, A.; Ram-Wolff, C.; Beylot-Barry, M.; Pham-Ledard, A.; Ortonne, N.; Ingen-Housz-Oro, S.; Battistella, M.; d’Incan, M.; Rouanet, J.; et al. Epidemiological Changes in Cutaneous Lymphomas: An Analysis of 8593 Patients from the French Cutaneous Lymphoma Registry. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.T.; Devesa, S.S.; Anderson, W.F.; Toro, J.R.; D’Incan, M.; Delaunay, M.; Vaillant, L.; Avril, M.F.; Bosq, J.; Wechsler, J.; et al. Cutaneous Lymphoma Incidence Patterns in the United States: A Population-Based Study of 3884 Cases. Blood 2009, 113, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Peñate, Y.; Servitje, O.; Machan, S.; Fernández-de-Misa, R.; Estrach, M.T.; Acebo, E.; Mitxelena, J.; Ramón, M.D.; Flórez, A.; Blanes, M.; et al. Registro de Linfomas Cutáneos Primarios de La AEDV: Primer Año de Funcionamiento. Actas Dermo-Sifiliográficas 2018, 109, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 Update of the WHO-EORTC Classification for Primary Cutaneous Lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissue: Lyon, France, 2017. [Google Scholar]
- Imam, M.H.; Shenoy, P.J.; Flowers, C.R.; Phillips, A.; Lechowicz, M.J. Incidence and Survival Patterns of Cutaneous T-Cell Lymphomas in the United States. Leuk. Lymphoma 2013, 54, 752–759. [Google Scholar] [CrossRef]
- Scarisbrick, J.J.; Quaglino, P.; Prince, H.M.; Papadavid, E.; Hodak, E.; Bagot, M.; Servitje, O.; Berti, E.; Ortiz-Romero, P.; Stadler, R.; et al. The PROCLIPI International Registry of Early-Stage Mycosis Fungoides Identifies Substantial Diagnostic Delay in Most Patients. Br. J. Dermatol. 2018, 181, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Pujol, R.M.; Gallardo, F.; Llistosella, E.; Blanco, A.; Bernadó, L.; Bordes, R.; Nomdedeu, J.F.; Servitje, O. Invisible Mycosis Fungoides: A Diagnostic Challenge. J. Am. Acad. Dermatol. 2002, 47, S168–S171. [Google Scholar] [CrossRef]
- Quaglino, P.; Pimpinelli, N.; Berti, E.; Calzavara-Pinton, P.; Alfonso Lombardo, G.; Rupoli, S.; Alaibac, M.; Bottoni, U.; Carbone, A.; Fava, P.; et al. Time Course, Clinical Pathways, and Long-Term Hazards Risk Trends of Disease Progression in Patients with Classic Mycosis Fungoides: A Multicenter, Retrospective Follow-up Study from the Italian Group of Cutaneous Lymphomas. Cancer 2012, 118, 5830–5839. [Google Scholar] [CrossRef]
- Van Doorn, R.; Scheffer, E.; Willemze, R. Follicular Mycosis Fungoides, a Distinct Disease Entity with or without Associated Follicular Mucinosis: A Clinicopathologic and Follow-up Study of 51 Patients. Arch. Dermatol. 2002, 138, 191–198. [Google Scholar] [CrossRef]
- van Santen, S.; Roach, R.E.J.; van Doorn, R.; Horvath, B.; Bruijn, M.S.; Sanders, C.J.G.; de Pooter, J.C.; van Rossum, M.M.; de Haas, E.R.M.; Veraart, J.C.J.M.; et al. Clinical Staging and Prognostic Factors in Folliculotropic Mycosis Fungoides. JAMA Dermatol. 2016, 152, 992–1000. [Google Scholar] [CrossRef] [Green Version]
- Willemze, R.; Jaffe, E.S.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Duncan, L.M.; Grange, F.; Harris, N.L.; et al. WHO-EORTC Classifcation for Cutaneous Lymphomas. Hematology 2005, 105, 3768–3785. [Google Scholar] [CrossRef]
- Vonderheid, E.C.; Bernengo, M.G.; Burg, G.; Duvic, M.; Heald, P.; Laroche, L.; Olsen, E.; Pittelkow, M.; Russell-Jones, R.; Takigawa, M.; et al. Update on Erythrodermic Cutaneous T-Cell Lymphoma: Report of the International Society for Cutaneous Lymphomas. J. Am. Acad. Dermatol. 2002, 46, 95–106. [Google Scholar] [CrossRef]
- Trotter, M.J.; Whittaker, S.J.; Orchard, G.E.; Smith, N.P. Cutaneous Histopathology of Sézary Syndrome: A Study of 41 Cases with a Proven Circulating T-Cell Clone. J. Cutan. Pathol. 1997, 24, 286–291. [Google Scholar] [CrossRef]
- Çetinözman, F.; Jansen, P.M.; Vermeer, M.H.; Willemze, R. Differential Expression of Programmed Death-1 (PD-1) in Sézary Syndrome and Mycosis Fungoides. Arch. Dermatol. 2012, 148, 1379–1385. [Google Scholar] [CrossRef] [Green Version]
- Fierro, M.T.; Comessatti, A.; Quaglino, P.; Ortoncelli, M.; Osella Abate, S.; Ponti, R.; Novelli, M.; Bernengo, M.G. Expression Pattern of Chemokine Receptors and Chemokine Release in Inflammatory Erythroderma and Sézary Syndrome. Dermatology 2006, 213, 284–292. [Google Scholar] [CrossRef]
- Narducci, M.G.; Scala, E.; Bresin, A.; Caprini, E.; Picchio, M.C.; Remotti, D.; Ragone, G.; Nasorri, F.; Frontani, M.; Arcelli, D.; et al. Skin Homing of Sézary Cells Involves SDF-1-CXCR4 Signaling and down-Regulation of CD26/Dipeptidylpeptidase IV. Blood 2006, 107, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Mourad, A.; Gniadecki, R. Overall Survival in Mycosis Fungoides: A Systematic Review and Meta-Analysis. J. Investig. Dermatol. 2020, 140, 495–497. [Google Scholar] [CrossRef]
- Olsen, E.; Vonderheid, E.; Pimpinelli, N.; Willemze, R.; Kim, Y.; Knobler, R.; Zackheim, H.; Duvic, M.; Estrach, T.; Lamberg, S.; et al. Revisions to the Staging and Classification of Mycosis Fungoides and Sézary Syndrome: A Proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Ca. Blood 2007, 110, 1713–1722. [Google Scholar] [CrossRef] [Green Version]
- Burg, G. Systemic Involvement in Mycosis Fungoides. Clin. Dermatol. 2015, 33, 563–571. [Google Scholar] [CrossRef]
- Scarisbrick, J.; Hodak, E.; Bagot, M.; Stranzenbach, R.; Stadler, R.; Ortiz-Romero, P.L.; Papadavid, E.; Evison, F.; Knobler, R.; Quaglino, P.; et al. Blood Classification and Blood Response Criteria in Mycosis Fungoides and Sézary Syndrome Using Flow Cytometry: Recommendations from the EORTC Cutaneous Lymphoma Task Force. Eur. J. Cancer 2018, 93, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Agar, N.S.; Wedgeworth, E.; Crichton, S.; Mitchell, T.J.; Cox, M.; Ferreira, S.; Robson, A.; Calonje, E.; Stefanato, C.M.; Wain, E.M.; et al. Survival Outcomes and Prognostic Factors in Mycosis Fungoides/Sézary Syndrome: Validation of the Revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer Staging Proposal. J. Clin. Oncol. 2010, 28, 4730–4739. [Google Scholar] [CrossRef]
- Pulitzer, M.; Myskowski, P.L.; Horwitz, S.M.; Querfeld, C.; Connolly, B.; Li, J.; Murali, R. Mycosis Fungoides with Large Cell Transformation: Clinicopathological Features and Prognostic Factors. Pathology 2014, 46, 610–616. [Google Scholar] [CrossRef] [Green Version]
- Scarisbrick, J.J.; Prince, H.M.; Vermeer, M.H.; Quaglino, P.; Horwitz, S.; Porcu, P.; Stadler, R.; Wood, G.S.; Beylot-Barry, M.; Pham-Ledard, A.; et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sézary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J. Clin. Oncol. 2015, 33, 3766–3773. [Google Scholar] [CrossRef]
- Mcgirt, L.Y.; Jia, P.; Baerenwald, D.A.; Duszynski, R.J.; Dahlman, K.B.; Zic, J.A.; Zwerner, J.P.; Hucks, D.; Dave, U.; Zhao, Z.; et al. Whole-Genome Sequencing Reveals Oncogenic Mutations in Mycosis Fungoides. Blood 2015, 126, 508–519. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ni, X.; Covington, K.R.; Yang, B.Y.; Shiu, J.; Zhang, X.; Xi, L.; Meng, Q.; Langridge, T.; Drummond, J.; et al. Genomic Profiling of Sézary Syndrome Identifies Alterations of Key T Cell Signaling and Differentiation Genes. Nat. Genet. 2015, 47, 1426–1434. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Goh, G.; Walradt, T.; Hong, B.S.; Bunick, C.G.; Chen, K.; Bjornson, R.D.; Maman, Y.; Wang, T.; Tordoff, J.; et al. Genomic Landscape of Cutaneous T Cell Lymphoma. Nat. Genet. 2015, 47, 1–11. [Google Scholar] [CrossRef]
- Prasad, A.; Rabionet, R.; Espinet, B.; Zapata, L.; Puiggros, A.; Melero, C.; Puig, A.; Sarria-Trujillo, Y.; Ossowski, S.; Garcia-Muret, M.P.; et al. Identification of Gene Mutations and Fusion Genes in Patients with Sézary Syndrome. J. Investig. Dermatol. 2016, 136, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma Genome Sequencing Reveals Frequent PREX2 Mutations. Nature 2012, 485, 502–506. [Google Scholar] [CrossRef]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef] [Green Version]
- González-Vela, M.d.C.; Curiel-Olmo, S.; Derdak, S.; Beltran, S.; Santibañez, M.; Martínez, N.; Castillo-Trujillo, A.; Gut, M.; Sánchez-Pacheco, R.; Almaraz, C.; et al. Shared Oncogenic Pathways Implicated in Both Virus-Positive and UV-Induced Merkel Cell Carcinomas. J. Investig. Dermatol. 2017, 137, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Vaqué, J.P.; Gómez-López, G.; Monsálvez, V.; Varela, I.; Martínez, N.; Pérez, C.; Domínguez, O.; Graña, O.; Rodríguez-Peralto, J.L.; Rodríguez-Pinilla, S.M.; et al. PLCG1 Mutations in Cutaneous T-Cell Lymphomas. Blood 2014, 123, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Liang, W.S.; Tembe, W.; Izatt, T.; Kruglyak, S.; Kiefer, J.A.; Cuyugan, L.; Zismann, V.; Legendre, C.; Pittelkow, M.R.; et al. Personalized Treatment of Sezary Syndrome by Targeting a Novel Ctla4:Cd28 Fusion. Mol. Genet. Genom. Med. 2015, 3, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Ungewickell, A.; Bhaduri, A.; Rios, E.; Reuter, J.; Lee, C.S.; Mah, A.; Zehnder, A.; Ohgami, R.; Kulkarni, S.; Armstrong, R.; et al. Genomic Analysis of Mycosis Fungoides and Sézary Syndrome Identifies Recurrent Alterations in TNFR2. Nat. Genet. 2015, 47, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.C.M.; Velusamy, T.; Chung, F.; Schaller, M.; Bailey, N.G.; Betz, B.L.; Miranda, R.N.; Porcu, P.; et al. Genomic Analyses Reveal Recurrent Mutations in Epigenetic Modifiers and the JAK-STAT Pathway in Sézary Syndrome. Nat. Commun. 2015, 6, 8470. [Google Scholar] [CrossRef]
- Pérez, C.; González-Rincón, J.; Onaindia, A.; Almaráz, C.; García-Díaz, N.; Pisonero, H.; Curiel-Olmo, S.; Gómez, S.; Cereceda, L.; Madureira, R.; et al. Mutated JAK Kinases and Deregulated STAT Activity Are Potential Therapeutic Targets in Cutaneous T-Cell Lymphoma. Haematologica 2015, 100, e450–e453. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Almeida, A.C.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.P.; Vermeer, M.H.; Rabadan, R.; Ferrando, A.; Palomero, T. The Mutational Landscape of Cutaneous T Cell Lymphoma and Sézary Syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef] [Green Version]
- Woollard, W.J.; Pullabhatla, V.; Lorenc, A.; Patel, V.M.; Butler, R.M.; Bayega, A.; Begum, N.; Bakr, F.; Dedhia, K.; Fisher, J.; et al. Candidate Driver Genes in Sézary Syndrome: Frequent Perturbations of Genes Involved in Genome Maintenance and DNA Repair. Blood 2016, 127, 3387–3397. [Google Scholar] [CrossRef] [Green Version]
- Izykowska, K.; Przybylski, G.K.; Gand, C.; Braun, F.C.; Grabarczyk, P.; Kuss, A.W.; Olek-Hrab, K.; Torres, A.N.B.; Vermeer, M.H.; Zoutman, W.H.; et al. Genetic Rearrangements Result in Altered Gene Expression and Novel Fusion Transcripts in Sézary Syndrome. Oncotarget 2017, 8, 39627–39639. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Yang, J.; Wenzel, A.T.; Ramachandran, A.; Lee, W.J.; Daniels, J.C.; Kim, J.; Martinez-Escala, E.; Amankulor, N.; Pro, B.; et al. Genomic Analysis of 220 CTCLs Identifies a Novel Recurrent Gain-of-Function Alteration in RLTPR (p.Q575E). Blood 2017, 130, 1430–1440. [Google Scholar] [CrossRef]
- Bastidas Torres, A.N.; Cats, D.; Mei, H.; Szuhai, K.; Willemze, R.; Vermeer, M.H.; Tensen, C.P. Genomic Analysis Reveals Recurrent Deletion of JAK-STAT Signaling Inhibitors HNRNPK and SOCS1 in Mycosis Fungoides. Genes Chromosomes Cancer 2018, 57, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Pérez, C.; Mondéjar, R.; García-Díaz, N.; Cereceda, L.; León, A.; Montes, S.; Durán Vian, C.; Pérez Paredes, M.G.; González-Morán, A.; Alegre de Miguel, V.; et al. Advanced-stage Mycosis Fungoides. Role of STAT3, NFKB and NFAT Pathways. Br. J. Dermatol. 2019, 182, 147–155. [Google Scholar] [CrossRef]
- Iyer, A.; Hennessey, D.; O’Keefe, S.; Patterson, J.; Wang, W.; Wong, G.K.S.; Gniadecki, R. Branched Evolution and Genomic Intratumor Heterogeneity in the Pathogenesis of Cutaneous T-Cell Lymphoma. Blood Adv. 2020, 4, 2489–2500. [Google Scholar] [CrossRef]
- Chang, L.W.; Patrone, C.C.; Yang, W.; Rabionet, R.; Gallardo, F.; Espinet, B.; Sharma, M.K.; Girardi, M.; Tensen, C.P.; Vermeer, M.H.; et al. An Integrated Data Resource for Genomic Analysis of Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2018, 138, 2681–2683. [Google Scholar] [CrossRef]
- Lee, C.S.; Ungewickell, A.; Bhaduri, A.; Qu, K.; Webster, D.E.; Armstrong, R.; Weng, W.K.; Aros, C.J.; Mah, A.; Chen, R.O.; et al. Transcriptome Sequencing in Sézary Syndrome Identifies Sézary Cell and Mycosis Fungoides-Associated IncRNAs and Novel Transcripts. Blood 2012, 120, 3288–3297. [Google Scholar] [CrossRef] [Green Version]
- Litvinov, I.V.; Tetzlaff, M.T.; Thibault, P.; Gangar, P.; Moreau, L.; Watters, A.K.; Netchiporouk, E.; Pehr, K.; Prieto, V.G.; Rahme, E.; et al. Gene Expression Analysis in Cutaneous T-Cell Lymphomas (CTCL) Highlights Disease Heterogeneity and Potential Diagnostic and Prognostic Indicators. OncoImmunology 2017, 6, e1306618. [Google Scholar] [CrossRef]
- Buus, T.B.; Willerslev-Olsen, A.; Fredholm, S.; Blümel, E.; Nastasi, C.; Gluud, M.; Hu, T.; Lindahl, L.M.; Iversen, L.; Fogh, H.; et al. Single-Cell Heterogeneity in Sézary Syndrome. Blood Adv. 2018, 2, 2115–2126. [Google Scholar] [CrossRef] [Green Version]
- Borcherding, N.; Voigt, A.P.; Liu, V.; Link, B.K.; Zhang, W.; Jabbari, A. Single-Cell Profiling of Cutaneous T-Cell Lymphoma Reveals Underlying Heterogeneity Associated with Disease Progression. Clin. Cancer Res. 2019, 25, 2996–3005. [Google Scholar] [CrossRef] [Green Version]
- Gaydosik, A.M.; Tabib, T.; Geskin, L.J.; Bayan, C.A.; Conway, J.F.; Lafyatis, R.; Fuschiotti, P. Single-Cell Lymphocyte Heterogeneity in Advanced Cutaneous T-Cell Lymphoma Skin Tumors. Clin. Cancer Res. 2019, 25, 4443–4454. [Google Scholar] [CrossRef] [Green Version]
- Iyer, A.; Hennessey, D.; O’Keefe, S.; Patterson, J.; Wang, W.; Salopek, T.; Wong, G.K.S.; Gniadecki, R. Clonotypic Heterogeneity in Cutaneous T-Cell Lymphoma (Mycosis Fungoides) Revealed by Comprehensive Whole-Exome Sequencing. Blood Adv. 2019, 3, 1175–1184. [Google Scholar] [CrossRef] [Green Version]
- Tracey, L.; Villuendas, R.; Dotor, A.M.; Spiteri, I.; Ortiz, P.; García, J.F.; Rodríguez Peralto, J.L.; Lawler, M.; Piris, M.A. Mycosis Fungoides Shows Concurrent Deregulation of Multiple Genes Involved in the TNF Signaling Pathway: An Expression Profile Study. Blood 2003, 102, 1042–1050. [Google Scholar] [CrossRef] [Green Version]
- De Masson, A.; O’Malley, J.T.; Elco, C.P.; Garcia, S.S.; Divito, S.J.; Lowry, E.L.; Tawa, M.; Fisher, D.C.; Devlin, P.M.; Teague, J.E.; et al. High-Throughput Sequencing of the T Cell Receptor b Gene Identifies Aggressive Early-Stage Mycosis Fungoides. Sci. Transl. Med. 2018, 10, eaar5894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcover, A.; Alar, B.; Di Bartolo, V. Cell Biology of T Cell Receptor Expression and Regulation. Annu. Rev. Immunol. 2018, 36, 103–125. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Zheng, L.; Lin, J.; Zhang, B.; Zhu, Y.; Li, N.; Xie, S.; Wang, Y.; Gao, N.; Huang, Z. Structural Basis of Assembly of the Human T Cell Receptor–CD3 Complex. Nature 2019, 573, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Koretzky, G.; Schatzman, R.C.; Kadlecek, T. Functional Activation of the T-Cell Antigen Receptor Induces Tyrosine Phosphorylation of Phospholipase C-Gamma 1. Proc. Natl. Acad. Sci. USA 1991, 88, 5484–5488. [Google Scholar] [CrossRef] [Green Version]
- Brownlie, R.J.; Zamoyska, R. T Cell Receptor Signalling Networks: Branched, Diversified and Bounded. Nat. Rev. Immunol. 2013, 13, 257–269. [Google Scholar] [CrossRef]
- Wilcox, R.; Wada, D.A.; Ziesmer, S.C.; Elsawa, S.F.; Comfere, N.I.; Dietz, A.B.; Novak, A.J.; Witzig, T.E.; Feldman, A.L.; Pittelkow, M.R.; et al. Monocytes Promote Tumor Cell Survival in T-Cell Lymphoproliferative Disorders and Are Impaired in Their Ability to Differentiate into Mature Dendritic Cells. Blood 2009, 114, 2936–2944. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, R. A Three-Signal Model of T-Cell Lymphoma Pathogenesis. Am. J. Hematol. 2016, 91, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.M.; Flanagan, C.E.; Martins, M.; Jones, C.L.; Butler, R.M.; Woollard, W.J.; Bakr, F.S.; Yoxall, A.; Begum, N.; Katan, M.; et al. Frequent and Persistent PLCG1 Mutations in Sézary Cells Directly Enhance PLCγ1 Activity and Stimulate NFκB, AP-1, and NFAT Signaling. J. Investig. Dermatol. 2020, 140, 380–389. [Google Scholar] [CrossRef]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S.J. Amplification and Overexpression of JUNB Is Associated with Primary Cutaneous T-Cell Lymphomas. Blood 2003, 101, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Orchard, G.; Mitchell, T.J.; Oyama, N.; Russell-Jones, R.; Vermeer, M.H.; Willemze, R.; Van Doorn, R.; Tensen, C.P.; Young, B.D.; et al. A Genomic and Expression Study of AP-1 in Primary Cutaneous T-Cell Lymphoma: Evidence for Dysregulated Expression of JUNB and JUND in MF and SS. J. Cutan. Pathol. 2008, 35, 899–910. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Kim, L.J.; Walters, R.D.; Drullinger, L.F.; Lively, T.N.; Kugel, J.F.; Goodrich, J.A. The C-Terminal Region of Human NFATc2 Binds CJun to Synergistically Activate Interleukin-2 Transcription. Mol. Immunol. 2010, 47, 2314–2322. [Google Scholar] [CrossRef] [Green Version]
- Meller, N.; Altman, A.; Isakov, N. New Perspectives on PKCθ, a Member of the Novel Subfamily of Protein Kinase C. Stem. Cells 1998, 16, 178–192. [Google Scholar] [CrossRef]
- Altman, A.; Kong, K.-F. Protein Kinase C Enzymes in the Hematopoietic and Immune Systems. Annu. Rev. Immunol. 2016, 34, 511–538. [Google Scholar] [CrossRef]
- Monks, C.R.F.; Kupfer, H.; Tamir, I.; Barlow, A.; Kupfer, A. Selective Modulation of Protein Kinase C-θ during T-Cell Activation. Nature 1997, 385, 83–86. [Google Scholar] [CrossRef]
- Isakov, N.; Altman, A. Protein Kinase Cθ in T Cell Activation. Annu. Rev. Immunol 2002, 20, 761–794. [Google Scholar] [CrossRef]
- Kong, K.F.; Yokosuka, T.; Canonigo-Balancio, A.J.; Isakov, N.; Saito, T.; Altman, A. A Motif in the V3 Domain of the Kinase PKC-θ Determines Its Localization in the Immunological Synapse and Functions in T Cells via Association with CD28. Nat. Immunol. 2011, 12, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chuang, H.-C.; Li, J.-P.; Tan, T.-H. Regulation of PKC-θ Function by Phosphorylation in T Cell Receptor Signaling. Front. Immunol. 2012, 3, 197. [Google Scholar] [CrossRef] [Green Version]
- Coudronniere, N.; Villalba, M.; Englund, N.; Altman, A. NF-ΚB Activation Induced by T Cell Receptor/CD28 Costimulation Is Mediated by Protein Kinase C-Theta. Proc. Natl. Acad. Sci. USA 2000, 97, 3394–3399. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; You, Y.; Case, S.M.; McAllister-Lucas, L.M.; Wang, L.; DiStefano, P.S.; Nuñez, G.; Bertin, J.; Lin, X. A Requirement for CARMA1 in TCR-Induced NF-ΚB Activation. Nat. Immunol. 2002, 3, 830–835. [Google Scholar] [CrossRef]
- Oh, H.; Ghosh, S. NF-ΚB: Roles and Regulation in Different CD4+ T-Cell Subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Verstrepen, L.; Bekaert, T.; Chau, T.L.; Tavernier, J.; Chariot, A.; Beyaert, R. TLR-4, IL-1R and TNF-R Signaling to NF-ΚB: Variations on a Common Theme. Cell. Mol. Life Sci. 2008, 65, 2964–2978. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-ΚB Signaling in Inflammation. Signal. Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, K.; Guo, B.; Pomerantz, J.L.; Bandaranayake, A.D.; Moreno-García, M.E.; Ovechkina, Y.L.; Rawlings, D.J. Phosphorylation of the CARMA1 Linker Controls NF-ΚB Activation. Immunity 2005, 23, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Mao, R.; Yang, J. NF-ΚB and STAT3 Signaling Pathways Collaboratively Link Inflammation to Cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E.T.; Silvennoinen, O.; O’Shea, J.J. The Janus Kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef] [Green Version]
- Goswami, R.; Kaplan, M.H. STAT Transcription Factors in T Cell Control of Health and Disease. Int. Rev. Cell Mol. Biol. 2017, 331, 123–180. [Google Scholar] [CrossRef]
- Calò, V.; Migliavacca, M.; Bazan, V.; Macaluso, M.; Buscemi, M.; Gebbia, N.; Russo, A. STAT Proteins: From Normal Control of Cellular Events to Tumorigenesis. J. Cell. Physiol. 2003, 197, 157–168. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E. STATs: Transcriptional Control and Biological Impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Lim, C.; Cao, X. Structure, Function, and Regulation of STAT Proteins. Mol. Biosyst. 2006, 2, 536–550. [Google Scholar] [CrossRef]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 Phosphorylates Histone H3Y41 and Excludes HP1α from Chromatin. Nature 2009. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Dutta, P.; Tsurumi, A.; Li, J.; Wang, J.; Land, H.; Li, W.X. Unphosphorylated STAT5A Stabilizes Heterochromatin and Suppresses Tumor Growth. Proc. Natl. Acad. Sci. USA 2013, 110, 10213–10218. [Google Scholar] [CrossRef] [Green Version]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and Consequences of Jak-STAT Signaling in the Immune System. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu. Rev. Immunol. 2017, 35, 533–550. [Google Scholar] [CrossRef]
- Shahmarvand, N.; Nagy, A.; Shahryari, J.; Ohgami, R.S. Mutations in the Signal Transducer and Activator of Transcription Family of Genes in Cancer. Cancer Sci. 2018, 109, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Eriksen, K.W.; Kaltoft, K.; Mikkelsen, G.; Nielsen, M.; Zhang, Q.; Geisler, C.; Nissen, M.H.; Röpke, C.; Wasik, M.A.; Ødum, N. Constitutive STAT3-Activation in Sezary Syndrome: Tyrphostin AG490 Inhibits STAT3-Activation, Interleukin-2 Receptor Expression and Growth of Leukemic Sezary Cells. Leukemia 2001, 15, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Sommer, V.H.; Clemmensen, O.J.; Nielsen, O.; Wasik, M.; Lovato, P.; Brender, C.; Eriksen, K.W.; Woetmann, A.; Kaestel, C.G.; Nissen, M.H.; et al. In Vivo Activation of STAT3 in Cutaneous T-Cell Lymphoma. Evidence for an Antiapoptotic Function of STAT3. Leukemia 2004, 18, 1288–1295. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, R.C.T.; Jones, C.L.; Tosi, I.; Caesar, J.A.; Whittaker, S.J.; Mitchell, T.J. Constitutive Activation of STAT3 in Sézary Syndrome Is Independent of SHP-1. Leukemia 2012, 26, 323–331. [Google Scholar] [CrossRef]
- Talpur, R.; Bassett, R.; Duvic, M. Prevalence and Treatment of Staphylococcus Aureus Colonization in Patients with Mycosis Fungoides and Sézary Syndrome. Br. J. Dermatol. 2008, 159, 105–112. [Google Scholar] [CrossRef]
- Willerslev-Olsen, A.; Krejsgaard, T.; Lindahl, L.M.; Litvinov, I.V.; Fredholm, S.; Petersen, D.L.; Nastasi, C.; Gniadecki, R.; Mongan, N.P.; Sasseville, D.; et al. Staphylococcal Enterotoxin A (SEA) Stimulates STAT3 Activation and IL-17 Expression in Cutaneous T-Cell Lymphoma. Blood 2016, 127, 1287–1296. [Google Scholar] [CrossRef]
- Willerslev-Olsen, A.; Buus, T.B.; Nastasi, C.; Blümel, E.; Gluud, M.; Bonefeld, C.M.; Geisler, C.; Lindahl, L.M.; Vermeer, M.; Wasik, M.A.; et al. Staphylococcus Aureus Enterotoxins Induce FOXP3 in Neoplastic T Cells in Sézary Syndrome. Blood Cancer J. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Jackow, C.M.; Cather, J.C.; Hearne, V.; Asano, A.T.; Musser, J.M.; Duvic, M. Association of Erythrodermic Cutaneous T-Cell Lymphoma, Superantigen- Positive Staphylococcus Aureus, and Oligoclonal T-Cell Receptor Vβ Gene Expansion. Blood 1997, 89, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Emge, D.A.; Bassett, R.L.; Duvic, M.; Huen, A.O. Methicillin-Resistant Staphylococcus Aureus (MRSA) Is an Important Pathogen in Erythrodermic Cutaneous T-Cell Lymphoma (CTCL) Patients. Arch. Dermatol. Res. 2020, 312, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Farber, D.L.; Yudanin, N.A.; Restifo, N.P. Human Memory T Cells: Generation, Compartmentalization and Homeostasis. Nat. Rev. Immunol. 2014, 14, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Förster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and Its Ligands: Balancing Immunity and Tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef]
- Kohout, T.A.; Nicholas, S.L.; Perry, S.J.; Reinhart, G.; Junger, S.; Scott Struthers, R. Differential Desensitization, Receptor Phosphorylation,-Arrestin Recruitment, and ERK1/2 Activation by the Two Endogenous Ligands for the CC Chemokine Receptor 7. J. Biol. Chem. 2004, 279, 23214–23222. [Google Scholar] [CrossRef] [Green Version]
- Al-Haidari, A.A.; Syk, I.; Jirström, K.; Thorlacius, H. CCR4 Mediates CCL17 (TARC)-Induced Migration of Human Colon Cancer Cells via RhoA/Rho-Kinase Signaling. Int. J. Colorectal Dis. 2013, 28, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Jean-Charles, P.Y.; Kaur, S.; Shenoy, S.K. G Protein-Coupled Receptor Signaling Through β-Arrestin-Dependent Mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 142–158. [Google Scholar] [CrossRef]
- Lin, R.; Choi, Y.H.; Zidar, D.A.; Walker, J.K.L. β-Arrestin-2-Dependent Signaling Promotes CCR4-Mediated Chemotaxis of Murine T-Helper Type 2 Cells. Am. J. Respir. Cell Mol. Biol. 2018, 58, 745–755. [Google Scholar] [CrossRef]
- Cronshaw, D.G.; Kouroumalis, A.; Parry, R.; Webb, A.; Brown, Z.; Ward, S.G. Evidence That Phospholipase C-Dependent, Calcium-Independent Mechanisms Are Required for Directional Migration of T Lymphocytes in Response to the CCR4 Ligands CCL17 and CCL22. J. Leukoc. Biol. 2006, 79, 1369–1380. [Google Scholar] [CrossRef]
- Shannon, L.A.; Calloway, P.A.; Welch, T.P.; Vines, C.M. CCR7/CCL21 Migration on Fibronectin Is Mediated by Phospholipase Cγ1 and ERK1/2 in Primary T Lymphocytes. J. Biol. Chem. 2010, 285, 38781–38787. [Google Scholar] [CrossRef] [Green Version]
- Trautinger, F.; Eder, J.; Assaf, C.; Bagot, M.; Cozzio, A.; Dummer, R.; Gniadecki, R.; Klemke, C.-D.; Ortiz-Romero, P.L.; Papadavid, E.; et al. European Organisation for Research and Treatment of Cancer Consensus Recommendations for the Treatment of Mycosis Fungoides/Sézary Syndrome—Update 2017. Eur. J. Cancer 2017, 77, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Willemze, R.; Hodak, E.; Zinzani, P.L.; Specht, L.; Ladetto, M. Primary Cutaneous Lymphomas: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv30–iv40. [Google Scholar] [CrossRef]
- Amitay-Laish, I.; Prag-Naveh, H.; Dalal, A.; Pavlovsky, L.; Feinmesser, M.; Hodak, E. Treatment of Early Folliculotropic Mycosis Fungoides with Special Focus on Psoralen plus Ultraviolet A. Acta Derm. Venereol. 2018, 98, 951–955. [Google Scholar] [CrossRef] [Green Version]
- Mielke, V.; Wolff, H.H.; Winzer, M.; Sterry, W. Localized and Disseminated Pagetoid Reticulosis: Diagnostic Immunophenotypical Findings. Arch. Dermatol. 1989, 125, 402–406. [Google Scholar] [CrossRef]
- Clarijs, M.; Poot, F.; Laka, A.; Pirard, C.; Bourlond, A. Granulomatous Slack Skin: Treatment with Extensive Surgery and Review of the Literature. Dermatology 2003, 206, 393–397. [Google Scholar] [CrossRef]
- Zackheim, H.S.; Kashani-Sabet, M.; Amin, S. Topical Corticosteroids for Mycosis Fungoides. Arch. Dermatol. 1998, 134, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Lessin, S.R.; Duvic, M.; Guitart, J.; Pandya, A.G.; Strober, B.E.; Olsen, E.A.; Hull, C.M.; Knobler, E.H.; Rook, A.H.; Kim, E.J.; et al. Topical Chemotherapy in Cutaneous T-Cell Lymphoma. JAMA Dermatol. 2013, 149, 25. [Google Scholar] [CrossRef] [Green Version]
- Olsen, E.A.; Hodak, E.; Anderson, T.; Carter, J.B.; Henderson, M.; Cooper, K.; Lim, H.W. Guidelines for Phototherapy of Mycosis Fungoides and Sézary Syndrome: A Consensus Statement of the United States Cutaneous Lymphoma Consortium. J. Am. Acad. Dermatol. 2016, 74, 27–58. [Google Scholar] [CrossRef]
- Lee, E.; Koo, J.; Berger, T. UVB Phototherapy and Skin Cancer Risk: A Review of the Literature. Int. J. Dermatol. 2005, 44, 355–360. [Google Scholar] [CrossRef]
- Querfeld, C.; Rosen, S.T.; Kuze, T.M.; Kirby, K.A.; Henry, H.; Roenigk, J.; Prinz, B.M.; Guitart, J. Long-Term Follow-up of Patients With Early-Stage Cutaneous T-Cell Lymphoma Who Achieved Complete Remission With Psoralen Plus UV-A Monotherapy. Arch. Dermatol. 2005, 141, 305–311. [Google Scholar] [CrossRef]
- Georgakopoulos, I.; Papadavid, E.; Platoni, K.; Dilvoi, M.; Patatoukas, G.; Kypraiou, E.; Nikolaou, V.; Efstathopoulos, E.; Kelekis, N.; Kouloulias, V. Clinical Application of Total Skin Electron Beam (TSEB) Therapy for the Management of T Cell Cutaneous Lymphomas. The Evolving Role of Low Dose (12 Gy) Treatment Schedule. Clin. Transl. Radiat. Oncol. 2018, 15, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.; Scarisbrick, J.; Frew, J.; Irwin, C.; Grieve, R.; Humber, C.; Kuciejewska, A.; Bayne, S.; Weatherhead, S.; Child, F.; et al. The Results of Low-Dose Total Skin Electron Beam Radiation Therapy (TSEB) in Patients With Mycosis Fungoides From the UK Cutaneous Lymphoma Group. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Micaily, B.; Miyamoto, C.; Kantor, G.; Lessin, S.; Rook, A.; Brady, L.; Goodman, R.; Vonderheid, E.C. Radiotherapy for Unilesional Mycosis Fungoides. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 361–364. [Google Scholar] [CrossRef]
- Claudy, A.L.; Rouchouse, B.; Boucheron, S.; Lepetit, J.C. Treatment of Cutaneous Lymphoma with Etretinate. Br. J. Dermatol. 1983, 109, 49–56. [Google Scholar] [CrossRef]
- Molin, L.; Thomsen, K.; Volden, G.; Aronsson, A.; Hammar, H.; Hellbe, L.; Wantzin, G.L.; Roupe, G. Oral Retinoids in Mycosis Fungoides and Sézary Syndrome: A Comparison of Isotretinoin and Etretinate. A Study from the Scandinavian Mycosis Fungoides Group. Acta Derm.-Venereol. 1987, 67, 232–236. [Google Scholar] [PubMed]
- Duvic, M.; Hymes, K.; Heald, P.; Breneman, D.; Martin, A.G.; Myskowski, P.; Crowley, C.; Yocum, R.C. Bexarotene Is Effective and Safe for Treatment of Refractory Advanced-Stage Cutaneous T-Cell Lymphoma: Multinational Phase II–III Trial Results. J. Clin. Oncol. 2001, 19, 2456–2471. [Google Scholar] [CrossRef]
- Whittaker, S.; Ortiz, P.; Dummer, R.; Ranki, A.; Hasan, B.; Meulemans, B.; Gellrich, S.; Knobler, R.; Stadler, R.; Karrasch, M. Efficacy and Safety of Bexarotene Combined with Psoralen-Ultraviolet A (PUVA) Compared with PUVA Treatment Alone in Stage IB-IIA Mycosis Fungoides: Final Results from the EORTC Cutaneous Lymphoma Task Force Phase III Randomized Clinical Trial (NCT00056056). Br. J. Dermatol. 2012, 167, 678–687. [Google Scholar] [CrossRef]
- Olsen, E.A. Interferon in the Treatment of Cutaneous T-Cell Lymphoma. Dermatol. Ther. 2003, 16, 311–321. [Google Scholar] [CrossRef]
- Kuzel, T.M.; Roenigk, H.H.; Samuelson, E.; Herrmann, J.J.; Hurria, A.; Rademaker, A.W.; Rosen, S.T. Effectiveness of Interferon Alfa-2a Combined with Phototherapy for Mycosis Fungoides and the Sézary Syndrome. J. Clin. Oncol. 1995, 13, 257–263. [Google Scholar] [CrossRef]
- Schiller, M.; Tsianakas, A.; Sterry, W.; Dummer, R.; Hinke, A.; Nashan, D.; Stadler, R. Dose-Escalation Study Evaluating Pegylated Interferon Alpha-2a in Patients with Cutaneous T-Cell Lymphoma. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1841–1847. [Google Scholar] [CrossRef] [PubMed]
- Knobler, R.; Arenberger, P.; Arun, A.; Assaf, C.; Bagot, M.; Berlin, G.; Bohbot, A.; Calzavara-Pinton, P.; Child, F.; Cho, A.; et al. European Dermatology Forum—Updated Guidelines on the Use of Extracorporeal Photopheresis 2020—Part 1. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 2693–2716. [Google Scholar] [CrossRef]
- Knobler, R.; Berlin, G.; Calzavara-Pinton, P.; Greinix, H.; Jaksch, P.; Laroche, L.; Ludvigsson, J.; Quaglino, P.; Reinisch, W.; Scarisbrick, J.; et al. Guidelines on the Use of Extracorporeal Photopheresis. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1–37. [Google Scholar] [CrossRef] [Green Version]
- Kaye, F.J.; Bunn, P.A.; Steinberg, S.M.; Stocker, J.L.; Ihde, D.C.; Fischmann, A.B.; Glatstein, E.J.; Schechter, G.P.; Phelps, R.M.; Foss, F.M.; et al. A Randomized Trial Comparing Combination Electron-Beam Radiation and Chemotherapy with Topical Therapy in the Initial Treatment of Mycosis Fungoides. New Engl. J. Med. 1989, 321, 1784–1790. [Google Scholar] [CrossRef]
- Zackheim, H.S.; Kashani-Sabet, M.; McMillan, A. Low-Dose Methotrexate to Treat Mycosis Fungoides: A Retrospective Study in 69 Patients. J. Am. Acad. Dermatol. 2003, 49, 873–878. [Google Scholar] [CrossRef]
- Bunn, P.A.; Hoffmann, S.J.; Golitz, L.E.; Aeling, J.L. Systemic Therapy of Cutaneous T-Cell Lymphomas (Mycosis Fungoides and the Sezary Syndrome). Ann. Intern. Med. 1994, 121, 592–602. [Google Scholar] [CrossRef]
- Dummer, R.; Quaglino, P.; Becker, J.C.; Hasan, B.; Karrasch, M.; Whittaker, S.; Morris, S.; Weichenthal, M.; Stadler, R.; Bagot, M.; et al. Prospective International Multicenter Phase II Trial of Intravenous Pegylated Liposomal Doxorubicin Monochemotherapy in Patients with Stage IIB, IVA, or IVB Advanced Mycosis Fungoides: Final Results from EORTC 21012. J. Clin. Oncol. 2012, 30, 4091–4097. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Kim, Y.H.; Foss, F.; Zain, J.M.; Myskowski, P.L.; Lechowicz, M.J.; Fisher, D.C.; Shustov, A.R.; Nancy, L.B.; Maria, L.D.; et al. Identification of an Active, Well-Tolerated Dose of Pralatrexate in Patients with Relapsed or Refractory Cutaneous T-Cell Lymphoma. Blood 2012, 119, 4115–4122. [Google Scholar] [CrossRef] [Green Version]
- Winkelmann, R.K.; Diaz-Perez, J.L.; Buechner, S.A. The Treatment of Sézary Syndrome. J. Am. Acad. Dermatol. 1984, 10, 1000–1004. [Google Scholar] [CrossRef]
- Duarte, R.F.; Boumendil, A.; Onida, F.; Gabriel, I.; Arranz, R.; Arcese, W.; Poiré, X.; Kobbe, G.; Narni, F.; Cortelezzi, A.; et al. Long-Term Outcome of Allogeneic Hematopoietic Cell Transplantation for Patients with Mycosis Fungoides and Sézary Syndrome: A European Society for Blood and Marrow Transplantation Lymphoma Working Party Extended Analysis. J. Clin. Oncol. 2014, 32, 3347–3348. [Google Scholar] [CrossRef]
- Isufi, I.; Seropian, S.; Gowda, L.; Wilson, L.D.; Roberts, K.; Girardi, M.; Perreault, S.; Foss, F. Outcomes for Allogeneic Stem Cell Transplantation in Refractory Mycosis Fungoides and Primary Cutaneous Gamma Delta T Cell Lymphomas. Leuk. Lymphoma 2020, 61, 2955–2961. [Google Scholar] [CrossRef]
- Lopez, A.T.; Bates, S.; Geskin, L. Current Status of HDAC Inhibitors in Cutaneous T-Cell Lymphoma. Am. J. Clin. Dermatol. 2018, 19, 805–819. [Google Scholar] [CrossRef]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIB Multicenter Trial of Vorinostat in Patients with Persistent, Progressive, or Treatment Refractory Cutaneous t-Cell Lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [Green Version]
- Duvic, M.; Bates, S.E.; Piekarz, R.; Eisch, R.; Kim, Y.H.; Lerner, A.; Robak, T.; Samtsov, A.; Becker, J.C.; McCulloch, W.; et al. Responses to Romidepsin in Patients with Cutaneous T-Cell Lymphoma and Prior Treatment with Systemic Chemotherapy. Leuk. Lymphoma 2018, 59, 880–887. [Google Scholar] [CrossRef]
- Prince, H.M.; Dickinson, M. Romidepsin for Cutaneous T-Cell Lymphoma. Clin. Cancer Res. 2012, 18, 3509–3515. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Pan, B.; O’Connor, O.A. Brentuximab Vedotin. Clin. Cancer Res. 2013, 19, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Onaindia, A.; Martínez, N.; Montes-Moreno, S.; Almaraz, C.; Rodríguez-Pinilla, S.M.; Cereceda, L.; Revert, J.B.; Ortega, C.; Tardio, A.; González, L.; et al. CD30 Expression by B and T Cells: A Frequent Finding in Angioimmunoblastic T-Cell Lymphoma and Peripheral T-Cell Lymphoma-Not Otherwise Specified. Am. J. Surg. Pathol. 2016, 40, 378–385. [Google Scholar] [CrossRef]
- Prince, H.M.; Kim, Y.H.; Horwitz, S.M.; Dummer, R.; Scarisbrick, J.; Quaglino, P.; Zinzani, P.L.; Wolter, P.; Sanches, J.A.; Ortiz-Romero, P.L.; et al. Brentuximab Vedotin or Physician’s Choice in CD30-Positive Cutaneous T-Cell Lymphoma (ALCANZA): An International, Open-Label, Randomised, Phase 3, Multicentre Trial. Lancet 2017, 390, 555–566. [Google Scholar] [CrossRef]
- Kim, Y.H.; Tavallaee, M.; Sundram, U.; Salva, K.A.; Wood, G.S.; Li, S.; Rozati, S.; Nagpal, S.; Krathen, M.; Reddy, S.; et al. Phase II Investigator-Initiated Study of Brentuximab Vedotin in Mycosis Fungoides and Sézary Syndrome with Variable CD30 Expression Level: A Multi-Institution Collaborative Project. J. Clin. Oncol. 2015, 33, 3750–3758. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Ishida, T.; Utsunomiya, A.; Inagaki, A.; Yano, H.; Komatsu, H.; Iida, S.; Imada, K.; Uchiyama, T.; Akinaga, S.; et al. Defucosylated Humanized Anti-CCR4 Monoclonal Antibody KW-0761 as a Novel Immunotherapeutic Agent for Adult T-Cell Leukemia/Lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 1520–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.J.; Clark, R.A.; Watanabe, R.; Kupper, T.S. Sézary Syndrome and Mycosis Fungoides Arise from Distinct T-Cell Subsets: A Biologic Rationale for Their Distinct Clinical Behaviors. Blood 2010, 116, 767–771. [Google Scholar] [CrossRef]
- Kim, Y.H.; Bagot, M.; Pinter-Brown, L.; Rook, A.H.; Porcu, P.; Horwitz, S.M.; Whittaker, S.; Tokura, Y.; Vermeer, M.H.; Zinzani, P.L.; et al. Mogamulizumab versus Vorinostat in Previously Treated Cutaneous T-Cell Lymphoma (MAVORIC): An International, Open-Label, Randomised, Controlled Phase 3 Trial. Lancet Oncol. 2018, 19. [Google Scholar] [CrossRef]
- Wilcox, R.A. Mogamulizumab: 2 Birds, 1 Stone. Blood 2015, 125, 1847–1848. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Ishida, T.; Masaki, A.; Murase, T.; Yonekura, K.; Tashiro, Y.; Tokunaga, M.; Utsunomiya, A.; Ito, A.; Kusumoto, S.; et al. CCR4 Mutations Associated with Superior Outcome of Adult T-Cell Leukemia/Lymphoma under Mogamulizumab Treatment. Blood 2018, 132, 758–761. [Google Scholar] [CrossRef] [Green Version]
- Wartewig, T.; Kurgyis, Z.; Keppler, S.; Pechloff, K.; Hameister, E.; Öllinger, R.; Maresch, R.; Buch, T.; Steiger, K.; Winter, C.; et al. Erratum: PD-1 Is a Haploinsufficient Suppressor of T Cell Lymphomagenesis. Nature 2018. [Google Scholar] [CrossRef] [Green Version]
- Klemke, C.D.; Booken, N.; Weiss, C.; Nicolay, J.P.; Goerdt, S.; Felcht, M.; Géraud, C.; Kempf, W.; Assaf, C.; Ortonne, N.; et al. Histopathological and Immunophenotypical Criteria for the Diagnosis of Sézary Syndrome in Differentiation from Other Erythrodermic Skin Diseases: A European Organisation for Research and Treatment of Cancer (EORTC) Cutaneous Lymphoma Task Force Study of 9. Br. J. Dermatol. 2015. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Rook, A.H.; Porcu, P.; Foss, F.; Moskowitz, A.J.; Shustov, A.; Shanbhag, S.; Sokol, L.; Fling, S.P.; Ramchurren, N.; et al. Pembrolizumab in Relapsed and Refractory Mycosis Fungoides and Sézary Syndrome: A Multicenter Phase II Study. J. Clin. Oncol. 2020, 38, 20–28. [Google Scholar] [CrossRef]
- Bagot, M.; Porcu, P.; Marie-Cardine, A.; Battistella, M.; William, B.M.; Vermeer, M.; Whittaker, S.; Rotolo, F.; Ram-Wolff, C.; Khodadoust, M.S.; et al. IPH4102, a First-in-Class Anti-KIR3DL2 Monoclonal Antibody, in Patients with Relapsed or Refractory Cutaneous T-Cell Lymphoma: An International, First-in-Human, Open-Label, Phase 1 Trial. Lancet Oncol. 2019, 20, 1160–1170. [Google Scholar] [CrossRef]
- Oka, T.; Miyagaki, T. Novel and Future Therapeutic Drugs for Advanced Mycosis Fungoides and Sézary Syndrome. Front. Med. 2019, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.M.; Koch, R.; Porcu, P.; Oki, Y.; Moskowitz, A.; Perez, M.; Myskowski, P.; Officer, A.; Jaffe, J.D.; Morrow, S.N.; et al. Activity of the PI3K-δ,g Inhibitor Duvelisib in a Phase 1 Trial and Preclinical Models of T-Cell Lymphoma. Blood 2018, 131, 888–898. [Google Scholar] [CrossRef]
- Witzig, T.E.; Reeder, C.; Han, J.J.; LaPlant, B.; Stenson, M.; Tun, H.W.; Macon, W.; Ansell, S.M.; Habermann, T.M.; Inwards, D.J.; et al. The MTORC1 Inhibitor Everolimus Has Antitumor Activity in Vitro and Produces Tumor Responses in Patients with Relapsed T-Cell Lymphoma. Blood 2015, 126, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Guenther, L.; Lynde, C.; Poulin, Y. Off-Label Use of Topical Calcineurin Inhibitors in Dermatologic Disorders. J. Cutan. Med. Surg. 2019, 23, 27S–34S. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.B.; Fleischmann, R.; Hall, S.; Wilkinson, B.; Bradley, J.D.; Gruben, D.; Koncz, T.; Krishnaswami, S.; Wallenstein, G.V.; Zang, C.; et al. Tofacitinib versus Methotrexate in Rheumatoid Arthritis. New Engl. J. Med. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Freitas, R.M.; da Costa Maranduba, C.M. Myeloproliferative Neoplasms and the JAK/STAT Signaling Pathway: An Overview. Rev. Bras. Hematol. Hemoter. 2015, 37, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Olsen, E.A.; Kornacki, D.; Sun, K.; Hordinsky, M.K. Ruxolitinib Cream for the Treatment of Patients with Alopecia Areata: A 2-Part, Double-Blind, Randomized, Vehicle-Controlled Phase 2 Study. J. Am. Acad. Dermatol. 2020, 82, 412–419. [Google Scholar] [CrossRef]
- Rosmarin, D.; Pandya, A.G.; Lebwohl, M.; Grimes, P.; Hamzavi, I.; Gottlieb, A.B.; Butler, K.; Kuo, F.; Sun, K.; Ji, T.; et al. Ruxolitinib Cream for Treatment of Vitiligo: A Randomised, Controlled, Phase 2 Trial. Lancet 2020, 396, 110–120. [Google Scholar] [CrossRef]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 Antisense Oligonucleotide AZD9150 in a Subset of Patients with Heavily Pretreated Lymphoma: Results of a Phase 1b Trial. J. Immunother. Cancer 2018, 6. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Stage | T | N | M | B | Median OS (Years) | 5 Year OS a (%) | 5 Year DSS (%) | |
|---|---|---|---|---|---|---|---|---|
| Early | IA | 1 | 0 | 0 | 0–1 | |||
| Limited patches or plaques <10% skin surface | No nodal involvement | No visceral involvement | <1000 atypical cells | 35.5 | N/S | 98 | ||
| IB | 2 | 0 | 0 | 0–1 | ||||
| Patches or plaques ≥10% skin surface | No nodal involvement | No visceral involvement | <1000 atypical cells | 21.5 | 86 | 89 | ||
| IIA | 1–2 | 1–2 | 0 | 0–1 | ||||
| Any patches or plaques | Aggregates of atypical cells | No visceral involvement | <1000 atypical cells | 15.8 | N/S | 89 | ||
| Advanced | IIB | 3 | 0–2 | 0 | 0–1 | |||
| Tumoral lesions | No involvement or aggregates of atypical cells | No visceral involvement | <1000 atypical cells | 4.7 | 62 | 56 | ||
| III | 4 | 0–2 | 0 | 0–1 | ||||
| Erythroderma | No involvement or aggregates of atypical cells | No visceral involvement | <1000 atypical cells | 4.7 | N/S | 56 | ||
| IIIA | 4 | 0–2 | 0 | 0 | ||||
| Erythroderma | No involvement or aggregates of atypical cells | No visceral involvement | <250 atypical cells | 4.7 | 60 | 54 | ||
| IIIB | 4 | 0–2 | 0 | 1 | ||||
| Erythroderma | No involvement or aggregates of atypical cells | No visceral involvement | 250–1000 atypical cells | 3.4 | 54 | 48 | ||
| IVA1 | 1–4 | 0–2 | 0 | 2 | ||||
| Any skin involvement | No involvement or aggregates of atypical cells | No visceral involvement | >1000 atypical cells + clonality | 3.8 | 52 | 41 | ||
| IVA2 | 1–4 | 3 | 0 | 0–2 | ||||
| Any skin involvement | Partial or complete effacement of nodal architecture | No visceral involvement | <1000 or >1000 atypical cells + clonality | 2.1 | 34 | 23 | ||
| IVB | 1–4 | 0–3 | 1 | 0–2 | ||||
| Any skin involvement | Any nodal involvement | Visceral involvement | <1000 or >1000 atypical cells + clonality | 1.4 | 23 | 18 | ||
| Authors | Ref. | Number of Samples | WGS | WES | TS | RNA-Seq | scRNA-Seq | Highlighted Contribution | |
|---|---|---|---|---|---|---|---|---|---|
| MF | SS | ||||||||
| Lee et al., 2012 | [45] | 24 | 3 | 27 | SS-associated lncRNAs | ||||
| Vaqué et al., 2014 | [32] | 45 | 8 | 53 | PLCG1, JAK mutants, and NFAT activation (IHC) | ||||
| Sekulic et al., 2015 | [33] | 1 | 1 | 1 | CTLA4:CD28 gene fusion | ||||
| McGirt et al., 2015 | [25] | 30 | 5 | 25 | JAK mut. and JAK inhibitors; C > T | ||||
| Ungewickell et al., 2015 | [34] | 41 | 32 | 11 | 73 | TNFR2 mut. and recurrent CTLA4:CD28 gene fusion | |||
| Choi et al., 2015 | [27] | 40 | 2 | 40 | CNVs as drivers (STAT5B and PRKCQ amplification); C > T | ||||
| Kiel et al., 2015 | [35] | 66 | 6 | 66 | JAK1, 3/STAT3, 5B and ARID1A mut. | ||||
| Pérez et al., 2015 | [36] | 35 | JAK mut. and JAK inhibitors | ||||||
| Da Silva Almeida et al., 2015 | [37] | 8 | 25 | 42 | Mutational landscape: CARD11 and cGKIβ mut. | ||||
| Wang et al., 2015 | [26] | 37 | 37 | 37 | 32 | TCR signaling and IL32, IL2RG expression; C > T | |||
| Prasad et al., 2016 | [28] | 12 | 12 | 10 | ITPR1, 2, PKHD1L1 and DSC1 mut. and fusion genes; C > T | ||||
| Woollard et al., 2016 | [38] | 101 | 10 | 101 | POT1 mut., BRCA2 del. and PRKCQ, STAT3/5B amp; C > T | ||||
| Izykowska et al., 2017 | [39] | 9 | 9 | TOX and MYC amp. and deregulated expression | |||||
| Litvinov et al., 2017 | [46] | 181 | 181 a | Single-cell heterogeneity and transcriptional signatures for prognosis | |||||
| Park et al., 2017 | [40] | 220 b | RLTPR (NF-κB), CSNK1A1 and RHOA mut. | ||||||
| Chang et al., 2018 | [44] | 18 c | 121 c | Mutually exclusive mut. within the NF-κB pathway | |||||
| Bastidas Torres et al., 2018 | [40] | 9 | 9 | 8 | HNRNPK and SOCS1 del. (JAK/STAT pathway) | ||||
| Buus et al., 2018 | [47] | 11 | 11 | Single-cell heterogeneity and surface marker expression | |||||
| Perez et al., 2019 | [41] | 95 | 95 | P-STAT3 (IHC) in advanced vs. initial MFs | |||||
| Borcherding et al., 2019 | [48] | 1 | 1 | Intra-patient heterogeneity | |||||
| Gaydosik et al., 2019 | [49] | 5 | 5 | Patient-specific heterogeneity and clustering | |||||
| Iyer et al., 2019 | [50] | 27 | 10 d | 27 | Clonotypic heterogeneity | ||||
| Iyer et al., 2020 | [43] | 31 | 31 | Divergent evolution of cancer subclones | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Díaz, N.; Piris, M.Á.; Ortiz-Romero, P.L.; Vaqué, J.P. Mycosis Fungoides and Sézary Syndrome: An Integrative Review of the Pathophysiology, Molecular Drivers, and Targeted Therapy. Cancers 2021, 13, 1931. https://doi.org/10.3390/cancers13081931
García-Díaz N, Piris MÁ, Ortiz-Romero PL, Vaqué JP. Mycosis Fungoides and Sézary Syndrome: An Integrative Review of the Pathophysiology, Molecular Drivers, and Targeted Therapy. Cancers. 2021; 13(8):1931. https://doi.org/10.3390/cancers13081931
Chicago/Turabian StyleGarcía-Díaz, Nuria, Miguel Ángel Piris, Pablo Luis Ortiz-Romero, and José Pedro Vaqué. 2021. "Mycosis Fungoides and Sézary Syndrome: An Integrative Review of the Pathophysiology, Molecular Drivers, and Targeted Therapy" Cancers 13, no. 8: 1931. https://doi.org/10.3390/cancers13081931