
Disrupting Mitochondrial Electron Transfer Chain Complex I Decreases Immune Checkpoints in Murine and Human Acute Myeloid Leukemic Cells

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. IncuCyte Assay
2.3. TMRE and MTDR Assay
2.4. ATP Content Assay
2.5. Cell Proliferation
2.6. Seahorse
2.7. Western Blot
2.8. Data Sets and the Gene Set Enrichment Analysis (GSEA)
2.9. Metabolomics
2.10. Transcriptomics
2.11. NAD+/NADH Ratio Assessment
2.12. PD-L1 and CD39 Expression Assessment
2.13. Statistical Analyses
2.14. Graphical Summaries
3. Results
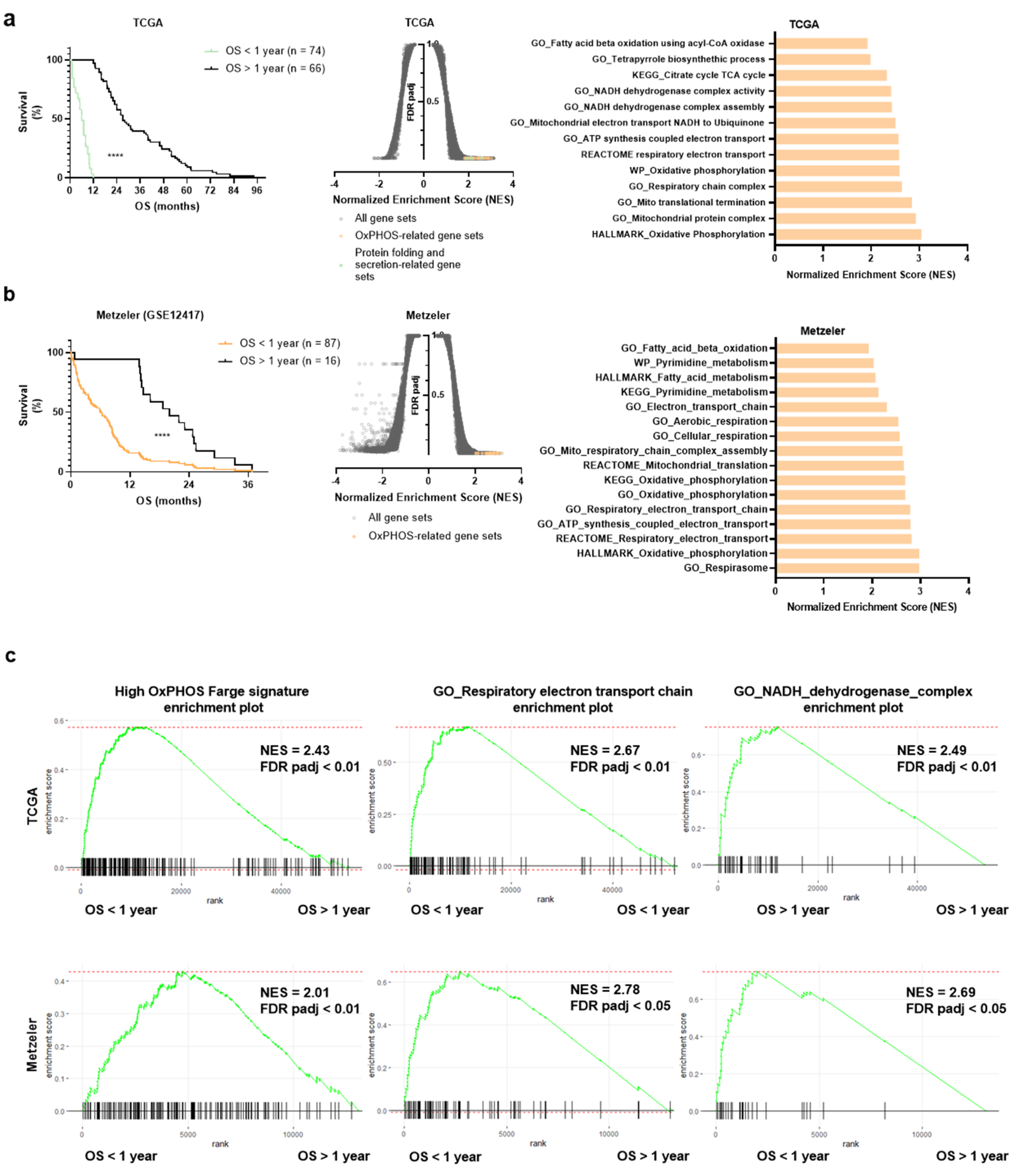
3.1. OxPHOS Phenotype and Mitochondrial Gene Signatures Are Enriched in AML Patients with a Shorter Survival
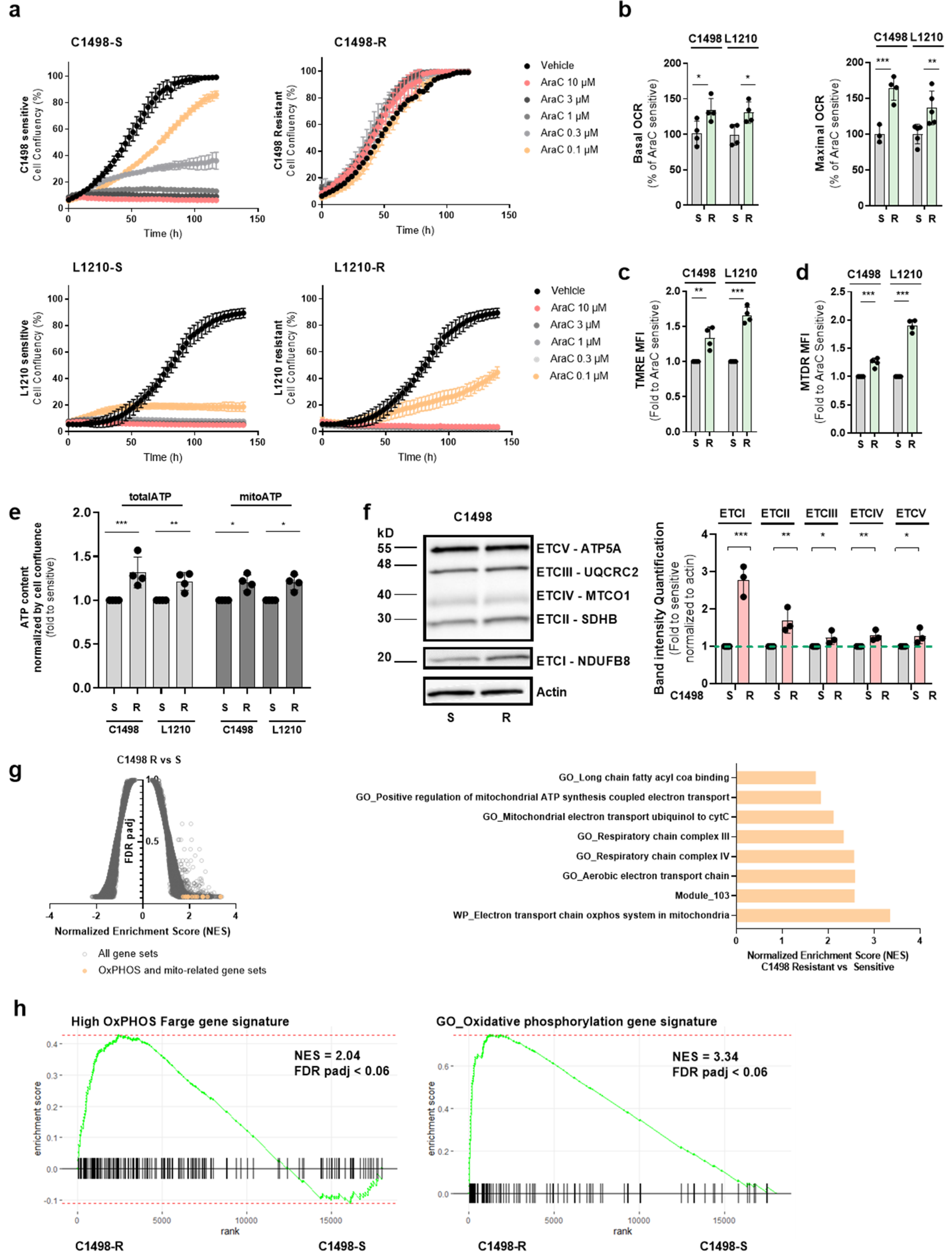
3.2. AraC-Tolerant Murine Leukemic Cells Exhibit a High OxPHOS Phenotype also Seen in Drug-Resistant Human AML Cells
3.3. EVT-701 Blocks OxPHOS by Inhibiting ETCI and Induces Metabolic Compensatory Reprogramming in Human and Murine AML Cells
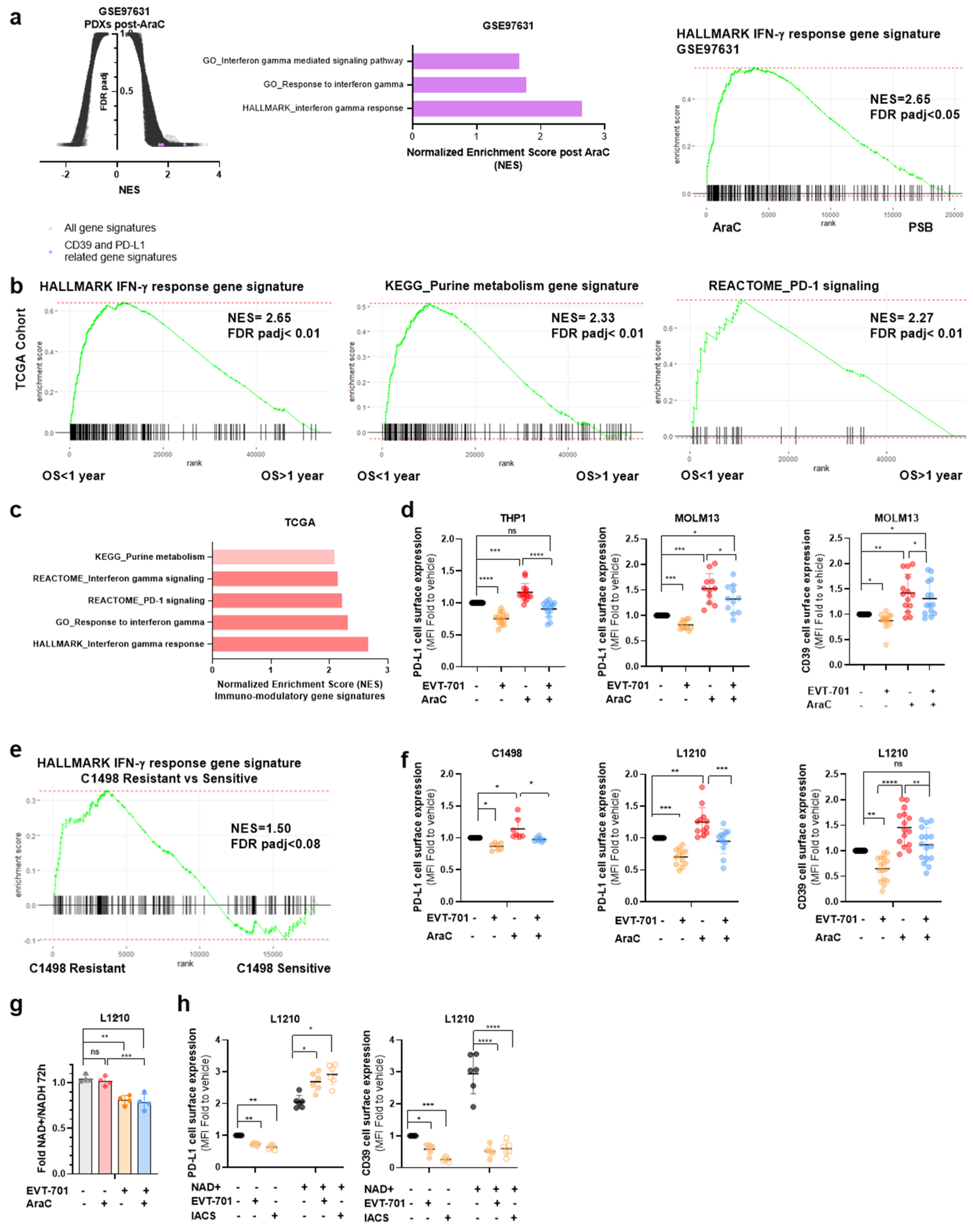
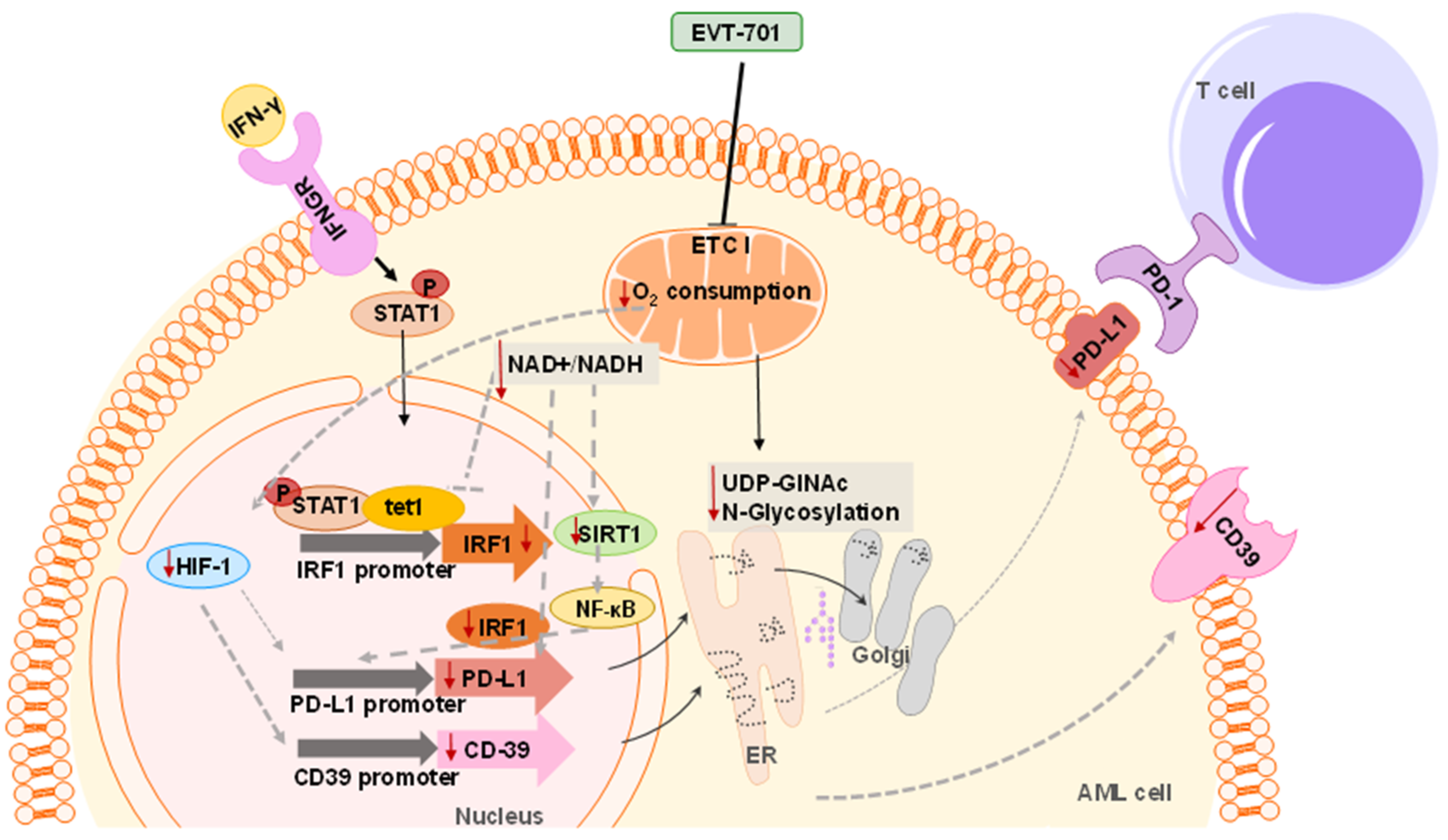
3.4. EVT-701 Decreases the Expression of Immune Checkpoint Markers in Murine and Human Leukemic Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, M.; Wang, F.; Loghavi, S.; Bueso-Ramos, C.; Gumbs, C.; Little, L.; Song, X.; Zhang, J.; Kadia, T.; Borthakur, G.; et al. Late relapse in acute myeloid leukemia (AML): Clonal evolution or therapy-related leukemia? Blood Cancer J. 2019, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin targets the metabolic Achilles heel of human pancreatic cancer stem cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.; Granter, S.R.; Widlund, H.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Ricci, J.-E.; Chiche, J. Metabolic reprogramming of non-Hodgkin’s B-cell lymphomas and potential therapeutic strategies. Front. Oncol. 2018, 8, 556. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Vivas-García, Y.; Falletta, P.; Liebing, J.; Louphrasitthiphol, P.; Feng, Y.; Chauhan, J.; Scott, D.; Glodde, N.; Calvo, A.C.; Bonham, S.; et al. Lineage-restricted regulation of SCD and fatty acid saturation by MITF controls melanoma phenotypic plasticity. Mol. Cell 2020, 77, 120–137. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; De Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [Green Version]
- Dobson, S.M.; García-Prat, L.; Vanner, R.J.; Wintersinger, J.; Waanders, E.; Gu, Z.; McLeod, J.; Gan, O.I.; Grandal, I.; Payne-Turner, D.; et al. Relapse-fated latent diagnosis subclones in acute B lineage leukemia are drug tolerant and possess distinct metabolic programs. Cancer Discov. 2020, 10, 568–587. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; MacLean, N.; et al. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, S.U.; Hurren, R.; Voisin, V.; Bridon, G.; Wang, X.; Xu, C.; MacLean, N.; Siriwardena, T.P.; Gronda, M.; Yehudai, D.; et al. Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML. Blood 2017, 129, 2657–2666. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [Green Version]
- Stuani, L.; Sabatier, M.; Sarry, J.-E. Exploiting metabolic vulnerabilities for personalized therapy in acute myeloid leukemia. BMC Biol. 2019, 17, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Bosc, C.; Gadaud, N.; Bousard, A.; Sabatier, M.; Cognet, G.; Saland, E.; Farge, T.; Boet, E.; Gotanègre, M.; Aroua, N.; et al. Mitochondrial determinants of response and resistance to venetoclax plus cytarabine duplet therapy in acute myeloid leukemia. BioRxiv 2020. [Google Scholar] [CrossRef]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol. Cell 2018, 71, 606–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [Green Version]
- Pereira, F.V.; Melo, A.C.L.; Low, J.S.; De Castro, Í.A.; Braga, T.; Almeida, D.C.; De Lima, A.G.U.B.; Hiyane, M.I.; Correa-Costa, M.; Andrade-Oliveira, V.; et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget 2018, 9, 25808–25825. [Google Scholar] [CrossRef] [Green Version]
- Méneyrol, J. Benzylhydroxyde Derivatives, Preparation Thereof and Therapeutic Use Thereof. U.S. Patent 9,878,990 B2, 30 January 2018. [Google Scholar]
- Luna-Yolba, R.; Visentin, V.; Hervé, C.; Chiche, J.; Ricci, J.-E.; Méneyrol, J.; Paillasse, M.R.; Alet, N. EVT-701 is a novel selective and safe mitochondrial complex 1 inhibitor with potent anti-tumor activity in models of solid cancers. Pharmacol. Res. Perspect. 2021. submitted for publication. [Google Scholar]
- The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzeler, K.; Hummel, M.; Bloomfield, C.D.; Spiekermann, K.; Braess, J.; Sauerland, M.-C.; Heinecke, A.; Radmacher, M.D.; Marcucci, G.; Whitman, S.P.; et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood 2008, 112, 4193–4201. [Google Scholar] [CrossRef] [PubMed]
- Mackay, G.M.; Zheng, L.; van den Broek, N.J.; Gottlieb, E. Analysis of Cell Metabolism Using LC-MS and Isotope Tracers. Methods Enzymol. 2015, 561, 171–196. [Google Scholar] [CrossRef]
- Lv, H.; Lv, G.; Chen, C.; Zong, Q.; Jiang, G.; Ye, D.; Cui, X.; He, Y.; Xiang, W.; Han, Q.; et al. NAD+ metabolism maintains inducible PD-L1 expression to drive tumor immune evasion. Cell Metab. 2021, 33, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Scotland, S.; Saland, E.; Skuli, N.; De Toni, F.; Boutzen, H.; Micklow, E.; Sénégas, I.; Peyraud, R.; Peyriga, L.; Theodoro, F.; et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia 2013, 27, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Diehl, F.F.; Lewis, C.A.; Fiske, B.P.; Heiden, M.G.V. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab. 2019, 1, 861–867. [Google Scholar] [CrossRef]
- Yang, L.; García-Cañaveras, J.C.; Chen, Z.; Wang, L.; Liang, L.; Jang, C.; Mayr, J.A.; Zhang, Z.; Ghergurovich, J.M.; Zhan, L.; et al. Serine catabolism feeds NADH when respiration is impaired. Cell Metab. 2020, 31, 809–821. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nat. Cell Biol. 2011, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.R.; Hu, Z.; Shi, X.; Jiang, L.; Boroughs, L.K.; Kovacs, Z.; Boriack, R.; Rakheja, D.; Sullivan, L.B.; Linehan, W.M.; et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014, 7, 1679–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billington, R.A.; Travelli, C.; Ercolano, E.; Galli, U.; Roman, C.B.; Grolla, A.; Canonico, P.L.; Condorelli, F.; Genazzani, A. Characterization of NAD uptake in mammalian cells. J. Biol. Chem. 2008, 283, 6367–6374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilk, A.; Hayat, F.; Cunningham, R.; Li, J.; Garavaglia, S.; Zamani, L.; Ferraris, D.M.; Sykora, P.; Andrews, J.; Clark, J.; et al. Extracellular NAD+ enhances PARP-dependent DNA repair capacity independently of CD73 activity. Sci. Rep. 2020, 10, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kory, N.; De Bos, J.U.; Van Der Rijt, S.; Jankovic, N.; Güra, M.; Arp, N.; Pena, I.A.; Prakash, G.; Chan, S.H.; Kunchok, T.; et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 2020, 6, eabe5310. [Google Scholar] [CrossRef]
- Luongo, T.S.; Eller, J.M.; Lu, M.-J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nat. Cell Biol. 2020, 588, 174–179. [Google Scholar] [CrossRef]
- Davila, A.; Liu, L.; Chellappa, K.; Redpath, P.; Nakamaru-Ogiso, E.; Paolella, L.M.; Zhang, Z.; Migaud, M.E.; Rabinowitz, J.D.; Baur, J.A. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. eLife 2018, 7. [Google Scholar] [CrossRef]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between metabolism and epigenetics: A nuclear adaptation to environmental changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Desquiret-Dumas, V.; Gueguen, N.; Leman, G.; Baron, S.; Nivet-Antoine, V.; Chupin, S.; Chevrollier, A.; Vessières, E.; Ayer, A.; Ferre, M.; et al. Resveratrol induces a mitochondrial complex I-dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J. Biol. Chem. 2013, 288, 36662–36675. [Google Scholar] [CrossRef] [Green Version]
- Gowrishankar, K.; Gunatilake, D.; Gallagher, S.J.; Tiffen, J.; Rizos, H.; Hersey, P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-κB. PLoS ONE 2015, 10, e0123410. [Google Scholar] [CrossRef] [Green Version]
- Lucas, J.; Hsieh, T.-C.; Halicka, H.D.; Darzynkiewicz, Z.; Wu, J.M. Upregulation of PD-L1 expression by resveratrol and piceatannol in breast and colorectal cancer cells occurs via HDAC3/p300-mediated NF-κB signaling. Int. J. Oncol. 2018, 53, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Li, Z.; Tao, J.; Hu, H.; Li, Z.; Zhang, Z.; Cheng, F.; Sun, Y.; Zhang, Y.; Yang, J.; et al. Resveratrol induces PD-L1 expression through snail-driven activation of Wnt pathway in lung cancer cells. J. Cancer Res. Clin. Oncol. 2021, 147, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Kiritsy, M.C.; Mott, D.; Behar, S.M.; Sassetti, C.M.; Olive, A.J. Mitochondrial respiration contributes to the interferon gamma response in antigen presenting cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wangpaichitr, M.; Kandemir, H.; Li, Y.; Wu, C.; Nguyen, D.; Feun, L.; Kuo, M.; Savaraj, N. Relationship of metabolic alterations and PD-L1 expression in cisplatin resistant lung cancer. Cell Dev. Biol. 2017, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Valle, S.; Alcalá, S.; Martin-Hijano, L.; Cabezas-Sáinz, P.; Navarro, D.; Muñoz, E.R.; Yuste, L.; Tiwary, K.; Walter, K.; Ruiz-Cañas, L.; et al. Exploiting oxidative phosphorylation to promote the stem and immunoevasive properties of pancreatic cancer stem cells. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.W.; Kong, S.-K.; Kim, B.S.; Kim, H.J.; Lim, H.; Noh, K.; Kim, Y.; Choi, J.-W.; Lee, J.-H.; Kim, Y.-S. IFNγ induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zheng, L.; Du, Q.; Yan, B.; Geller, D.A. Interferon regulatory factor 1 (IRF-1) and IRF-2 regulate PD-L1 expression in hepatocellular carcinoma (HCC) cells. Cancer Immunol. Immunother. 2020, 69, 1891–1903. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Wilhelm, D.; Rajky, O.; Kurscheid, S.; Kresl, P.; Wöhrer, A.; Marosi, C.; Hegi, M.E.; et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro. Oncol. 2017, 19, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Mu, L.; Long, Y.; Yang, C.; Jin, L.; Tao, H.; Ge, H.; Chang, Y.E.; Karachi, A.; Kubilis, P.S.; De Leon, G.; et al. The IDH1 mutation-induced oncometabolite, 2-hydroxyglutarate, may affect DNA methylation and expression of PD-L1 in gliomas. Front. Mol. Neurosci. 2018, 11, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Kadiyala, P.; Carney, S.V.; Gauss, J.C.; Garcia-Fabiani, M.B.; Haase, S.; Alghamri, M.S.; Núñez, F.J.; Liu, Y.; Yu, M.; Taher, A.W.; et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.; Patel, J.; Wise, D.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.; Fantin, V.R.; Hedvat, C.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collignon, E.; Canale, A.; Al Wardi, C.; Bizet, M.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Naveaux, C.; Barham, W.; Wilson, A.; et al. Immunity drives TET1 regulation in cancer through NF-κB. Sci. Adv. 2018, 4, eaap7309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [Green Version]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 1–16. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.; Enjyoji, K.; Robson, S.C.; Colgan, S.P. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Köhler, D.; Eckle, T.; Kong, T.; Robson, S.C.; Colgan, S.P. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 2009, 113, 224–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rytelewski, M.; Harutyunyan, K.; Baran, N.; Mallampati, S.; Zal, M.A.; Cavazos, A.; Butler, J.M.; Konoplev, S.; El Khatib, M.; Plunkett, S.; et al. Inhibition of oxidative phosphorylation reverses bone marrow hypoxia visualized in imageable syngeneic B-ALL mouse model. Front. Oncol. 2020, 10, 991. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, J.; Yi, G.; Deng, M.; Liu, H.; Liang, M.; Shi, B.; Fu, X.; Chen, Y.; Chen, L.; et al. Metformin suppresses hypoxia-induced stabilization of HIF-1α through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016, 7, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimarães, T.A.; Farias, L.C.; Santos, E.S.; de Carvalho Fraga, C.A.; Orsini, L.A.; de Freitas Teles, L.; Feltenberger, J.D.; de Jesus, S.F.; de Souza, M.G.; Sousa Santos, S.H.; et al. Metformin increases PDH and suppresses HIF-1α under hypoxic conditions and induces cell death in oral squamous cell carcinoma. Oncotarget 2016, 7, 55057–55068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocemba-Pilarczyk, K.A.; Trojan, S.; Ostrowska, B.; Lasota, M.; Dudzik, P.; Kusior, D.; Kot, M. Influence of metformin on HIF-1 pathway in multiple myeloma. Pharmacol. Rep. 2020, 72, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Malhotra, R.; Woodruff, R.; Guidotti, G. Mammalian plasma membrane ecto-nucleoside triphosphate diphosphohydrolase 1, CD39, is not active intracellularly. J. Biol. Chem. 2001, 276, 41518–41525. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FAB | Karyotype | Sex | FLT3 | NPM1 | IDH1 | Kit | N/K | ||
|---|---|---|---|---|---|---|---|---|---|
| ITD | TKD | IDH2 | Ras | ||||||
| Human | |||||||||
| MOLM13 | M5-AML | ins(11;9)(q23;p22p23) | M | ITD | WT | WT | WT | WT | WT |
| MV4-11 | M5-AML | Complex | M | ITD | WT | WT | WT | WT | WT |
| THP-1 | M5-AML | Complex | M | WT | WT | WT | WT | WT | NRAS p.G12D |
| U937 | M5-AML | t(10;11)(p13;q14) | M | WT | WT | WT | WT | WT | WT |
| Antibody | Dilution | Supplier | Catalogue Number |
|---|---|---|---|
| Anti-mPD-L1-APC | 1:50 | Biolegend, Ozyme | #124312 |
| APC-anti-Rat IgG2b,κ | 1:50 | Biolegend, Ozyme | #400611 |
| Anti-mCD39-PE-Cy7 | 1:50 | Biolegend, Ozyme | #143806 |
| PE/Cyanine7-anti-Rat IgG2a,κ | 1:50 | Biolegend, Ozyme | #400522 |
| Anti-hPD-L1-APC | 1:100 | Biolegend, Ozyme | #329708 |
| APC-anti-Mouse IgG2b,κ | 1:100 | Biolegend, Ozyme | #400320 |
| Anti-hCD39-PE-Cy7 | 1:100 | Biolegend, Ozyme | #328212 |
| PE/Cyanine7-anti-Mouse IgG1,κ | 1:100 | Biolegend, Ozyme | #400126 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luna-Yolba, R.; Marmoiton, J.; Gigo, V.; Marechal, X.; Boet, E.; Sahal, A.; Alet, N.; Abramovich, I.; Gottlieb, E.; Visentin, V.; et al. Disrupting Mitochondrial Electron Transfer Chain Complex I Decreases Immune Checkpoints in Murine and Human Acute Myeloid Leukemic Cells. Cancers 2021, 13, 3499. https://doi.org/10.3390/cancers13143499
Luna-Yolba R, Marmoiton J, Gigo V, Marechal X, Boet E, Sahal A, Alet N, Abramovich I, Gottlieb E, Visentin V, et al. Disrupting Mitochondrial Electron Transfer Chain Complex I Decreases Immune Checkpoints in Murine and Human Acute Myeloid Leukemic Cells. Cancers. 2021; 13(14):3499. https://doi.org/10.3390/cancers13143499
Chicago/Turabian StyleLuna-Yolba, Raquel, Justine Marmoiton, Véronique Gigo, Xavier Marechal, Emeline Boet, Ambrine Sahal, Nathalie Alet, Ifat Abramovich, Eyal Gottlieb, Virgile Visentin, and et al. 2021. "Disrupting Mitochondrial Electron Transfer Chain Complex I Decreases Immune Checkpoints in Murine and Human Acute Myeloid Leukemic Cells" Cancers 13, no. 14: 3499. https://doi.org/10.3390/cancers13143499