
MET Amplification in Non-Small Cell Lung Cancer (NSCLC)—A Consecutive Evaluation Using Next-Generation Sequencing (NGS) in a Real-World Setting

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cohort
2.2. MET GCN Detection by NGS
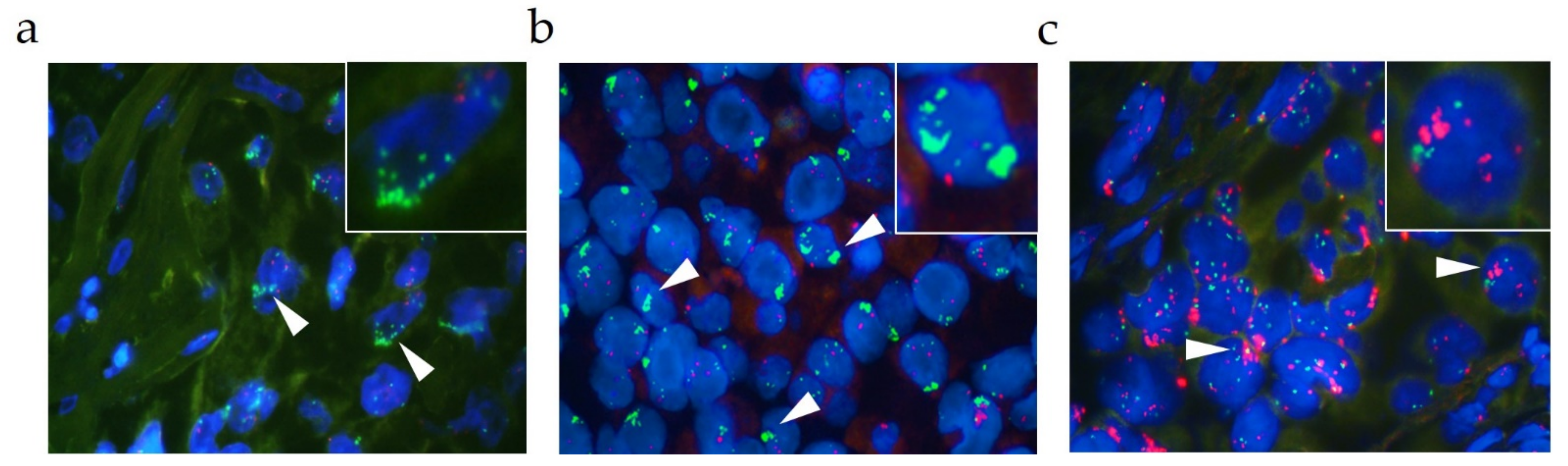
2.3. MET GCN Detection by FISH
2.4. Statistical Analysis
3. Results
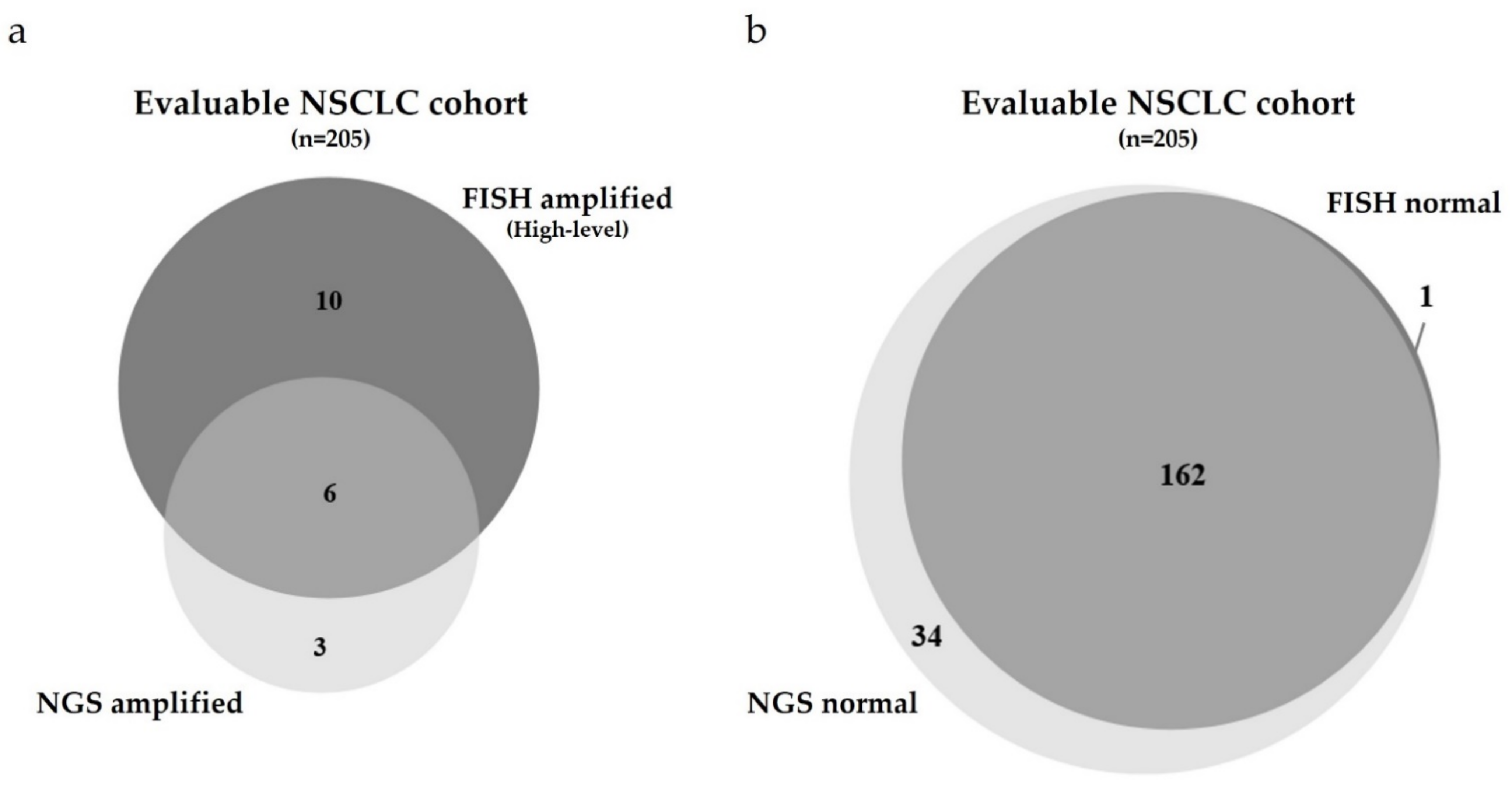
3.1. Comparison of the MET GCN of the NSCLC Cohort Using Two Different Methods: FISH versus NGS
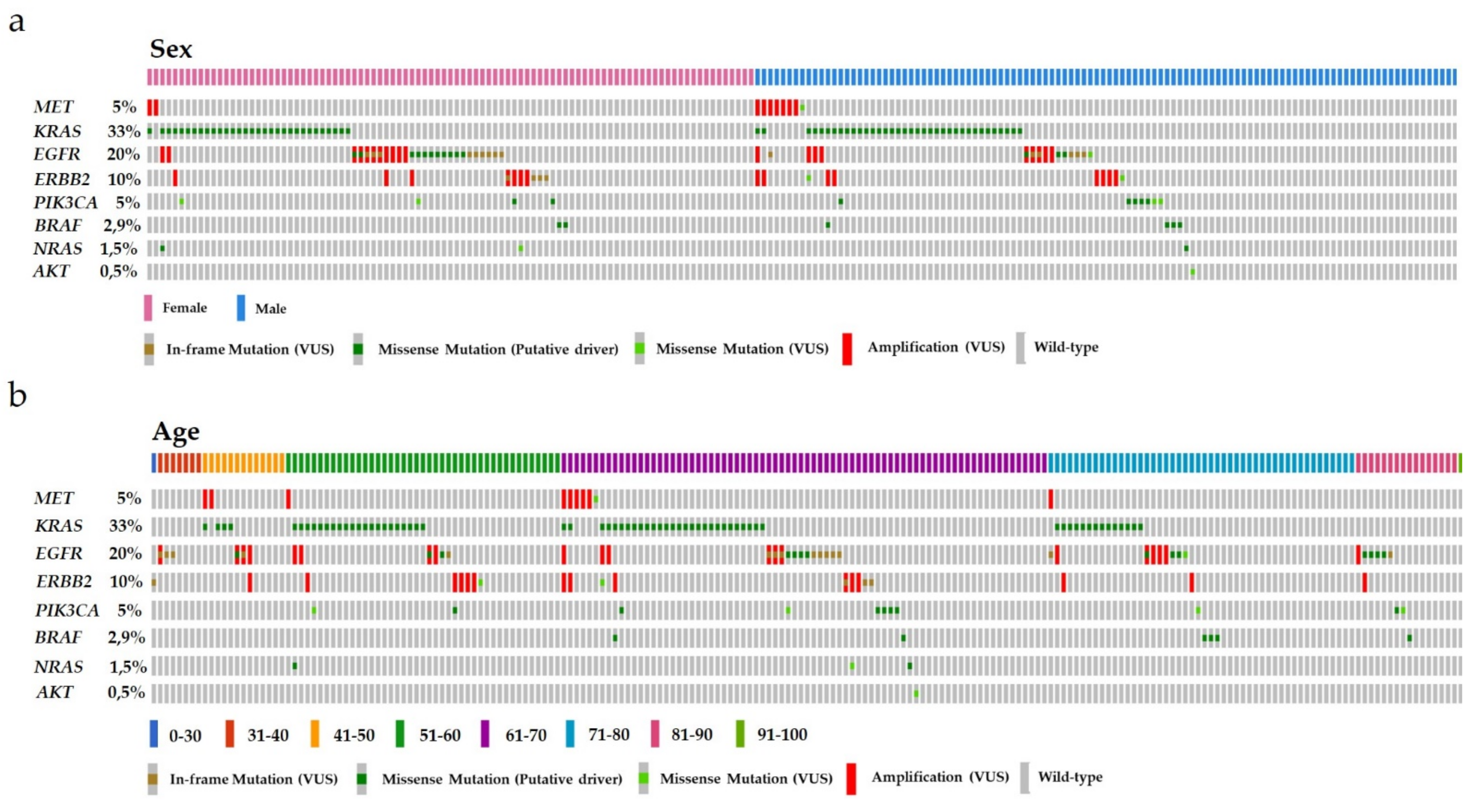
3.2. Correlations of MET GCN with Patient Data
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, N.; Kim, T.; DeMatteo, R.P. Principles of Kinase Inhibitor Therapy for Solid Tumors. Ann. Surg. 2017, 265, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Organ, S.L.; Tsao, M.-S. An overview of the c-MET signaling pathway. Ther. Adv. Med Oncol. 2011, 3 (Suppl. 1), S7–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P.C.; Maulik, G.; Christensen, J.; Salgia, R. c-Met: Structure, functions and potential for therapeutic inhi-bition. J. Cancer Metastasis Rev. 2003, 22, 309–325. [Google Scholar] [CrossRef]
- Tong, J.H.; Yeung, S.F.; Chan, A.; Chung, L.Y.; Chau, S.L.; Lung, R.W.M.; Tong, C.Y.; Chow, C.; Tin, E.K.; Yu, Y.H.; et al. MET Amplification and Exon 14 Splice Site Mutation Define Unique Molecular Subgroups of Non–Small Cell Lung Carcinoma with Poor Prognosis. Clin. Cancer Res. 2016, 22, 3048–3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellman, A.; Zlotorynski, E.; Scherer, S.W.; Cheung, J.; Vincent, J.B.; Smith, D.I.; Trakhtenbrot, L.; Kerem, B. A role for common fragile site induction in amplification ofhuman oncogenes. Cancer Cell 2002, 1, 89–97. [Google Scholar] [CrossRef] [Green Version]
- AACR Project Genie Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consor-tium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; Del Grammastro, M.; Sciar-rotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef] [Green Version]
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.-W.; Seo, J.-W.; Chung, D.H. High MET Gene Copy Number Leads to Shorter Survival in Patients with Non-small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.J.; Mok, T.S.; Chen, Z.H.; Guo, A.L.; Zhang, X.C.; Su, J.; Wu, Y.L. Clinicopathologic and molecu-lar features of epidermal growth factor receptor T790M mutation and c-MET amplification in tyrosine kinase inhibitor-resistant Chinese non-small cell lung cancer. Pathol. Oncol. Res. 2009, 15, 651–658. [Google Scholar] [CrossRef]
- Bean, J.; Brennan, C.; Shih, J.-Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790Mmutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarci-nomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607. [Google Scholar] [CrossRef] [Green Version]
- Rieger, R.; Michaelis, A.; Green, M.M. A Glossary of Genetics and Cytogenetics: Classical and Molecular; Springer: New York, NY, USA, 1968. [Google Scholar]
- Schildhaus, H.-U.; Schultheis, A.M.; Rüschoff, J.; Binot, E.; Merkelbach-Bruse, S.; Fassunke, J.; Schulte, W.; Ko, Y.-D.; Schlesinger, A.; Bos, M.; et al. MET Amplification Status in Therapy-Naïve Adeno- and Squamous Cell Carcinomas of the Lung. Clin. Cancer Res. 2014, 21, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Ther-apy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef]
- Planchard, D.; Loriot, Y.; André, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. EGFR-independent mecha-nisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Yoda, S.; Lennerz, J.K.; Langenbucher, A.; Lin, J.J.; Rooney, M.M.; Prutisto-Chang, K.; Oh, A.; Adams, N.A.; Yeap, B.Y.; et al. MET Alterations Are a Recurring and Ac-tionable Resistance Mechanism in ALK-Positive Lung Cancer. Clin. Cancer Res. 2020, 26, 2535–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.-H.I.; Kwak, E.L.; Siwak-Tapp, C.; Dy, J.; Bergethon, K.; Clark, J.W.; Camidge, D.R.; Solomon, B.J.; Maki, R.G.; Bang, Y.-J.; et al. Activity of Crizotinib (PF02341066), a Dual Mesenchymal-Epithelial Transition (MET) and Anaplastic Lymphoma Ki-nase (ALK) Inhibitor, in a Non-small Cell Lung Cancer Patient with De Novo MET Amplification. J. Thor. Oncol. 2011, 6, 942–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camidge, D.R.; Ou, S.-H.I.; Shapiro, G.; Otterson, G.A.; Villaruz, L.C.; Villalona-Calero, M.A.; Iafrate, A.J.; Varella-Garcia, M.; Dacic, S.; Cardarella, S.; et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2014, 32, 8001. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.Y.; Reguart, N.; Garon, E.B.; Groen, H.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14–Mutated or MET-Amplified Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and Clonal Selection of MET Amplifi-cation in EGFR Mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Vasudevan, S.; Sharma, S.K.; Panigrahi, M.; Suryavanshi, M.; Saifi, M.; Batra, U. Biomarker testing for advanced lung cancer by next-generation sequencing; a valid method to achieve a comprehensive glimpse at mutational landscape. Appl. Cancer Res. 2020, 40, 1–12. [Google Scholar] [CrossRef]
- Jiang, R.; Zhang, B.; Teng, X.; Hu, P.; Xu, S.; Zheng, Z.; Liu, R.; Tang, T.; Ye, F. Validating a targeted next-generation sequencing assay and profiling somat-ic variants in Chinese non-small cell lung cancer patients. Sci. Rep. 2020, 10, 2070. [Google Scholar] [CrossRef]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profil-ing test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef]
- Viailly, P.-J.; Sater, V.; Viennot, M.; Bohers, E.; Vergne, N.; Berard, C.; Dauchel, H.; Lecroq, T.; Celebi, A.; Ruminy, P.; et al. Improving high-resolution copy number variation analysis from next generation sequencing using unique molecular identifiers. BMC Bioinform. 2021, 22, 120. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, L.E.; Coon, J.S.; Morrison, L.E.; Jacobson, K.K.; Jewell, S.S.; Kaiser, K.A.; Mauer, A.M.; Muzzafar, T.; Polowy, C.; Basu, S.; et al. The Prognostic Value of Chromosome 7 Polysomy in Non-small Cell Lung Cancer Patients Treated with Gefitinib. J. Thorac. Oncol. 2007, 2, 414–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsen, T.; Vlieg, J.d.; Alkema, W. BioVenn—A web application for the comparison and visualization of bio-logical lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Heydt, C.; Becher, A.-K.; Wagener-Ryczek, S.; Ball, M.; Schultheis, A.M.; Schallenberg, S.; Rüsseler, V.; Büttner, R.; Merkelbach-Bruse, S. Comparison of in situ and extraction-based methods for the detection of MET amplifications in solid tumors. Comput. Struct. Biotechnol. J. 2019, 17, 1339–1347. [Google Scholar] [CrossRef]
- Guo, R.; Berry, L.D.; Aisner, D.L.; Sheren, J.; Boyle, T.; Bunn, P.A., Jr.; Johnson, B.E.; Kwiatkowski, D.J.; Drilon, A.; Sholl, L.M.; et al. MET IHC is a Poor Screen for MET Amplification or MET exon 14 muta-tions in Lung Adenocarcinomas: Data from a Tri-Institutional Cohort of the Lung Cancer Mutation Consortium. J. Thorac. Oncol. 2019, 14, 1666–1671. [Google Scholar] [CrossRef]
- Clavé, S.; Salido, M.; Rocha, P.; Hardy-Werbin, M.; Gibert, J.; Riera, X.; Weingartner, E.; Cerqueira, C.; Nichol, D.; Simmons, J.; et al. 1991P—Identification of MET gene amplifications using next-generation sequencing in non-small cell lung cancer patients. Ann. Oncol. 2019, 30, v800. [Google Scholar] [CrossRef]
- Noonan, S.A.; Berry, L.; Lu, X.; Gao, D.; Barón, A.E.; Chesnut, P.; Sheren, J.; Aisner, D.L.; Merrick, D.; Doebele, R.C.; et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number–Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J. Thorac. Oncol. 2016, 11, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, C.; Butler, T.; Rhodes, K.; Quist, M.; Neff, T.L.; Moore, S.; Tomlins, S.A.; Reinig, E.; Beadling, C.; Andersen, M.; et al. Assessing Copy Number Alterations in Targeted, Amplicon-Based Next-Generation Sequencing Data. J. Mol. Diagn. 2014, 17, 53–63. [Google Scholar] [CrossRef]
- Niu, D.; Li, L.; Yu, Y.; Zang, W.; Li, Z.; Zhou, L.; Jia, L.; Rao, G.; Gao, L.; Cheng, G.; et al. Evaluation of Next Generation Sequencing for Detecting HER2 Copy Number in Breast and Gastric Cancers. Pathol. Oncol. Res. 2020, 26, 2577–2585. [Google Scholar] [CrossRef]
- Okuda, K.; Sasaki, H.; Yukiue, H.; Yano, M.; Fujii, Y. Met gene copy number predicts the prognosis for com-pletely resected non-small cell lung cancer. Cancer Sci. 2008, 99, 2280–2285. [Google Scholar] [CrossRef] [PubMed]
- Tsuta, K.; Kozu, Y.; Mimae, T.; Yoshida, A.; Kohno, T.; Sekine, I.; Tamura, T.; Asamura, H.; Furuta, K.; Tsuda, H. c-MET/Phospho-MET Protein Expression and MET Gene Copy Number in Non-small Cell Lung Carcinomas. J. Thor. Oncol. 2012, 7, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Choi, Y.-L.; Sung, C.O.; An, J.; Seo, J.; Ahn, M.-J.; Ahn, J.S.; Park, K.; Shin, Y.K.; Erkin, O.C.; et al. High MET copy number and MET overexpression: Poor outcome in non-small cell lung cancer patients. Histol. Histopathol. 2012, 27, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Sterlacci, W.; Fiegl, M.; Gugger, M.; Bubendorf, L.; Savic, S.; Tzankov, A. MET overexpression and gene amplification: Prevalence, clinico-pathological characteristics and prognostic significance in a large cohort of patients with surgically resected NSCLC. Virchows Arch. 2017, 471, 49–55. [Google Scholar] [CrossRef]
- Dziadziuszko, R.; Wynes, M.W.; Singh, S.; Asuncion, B.R.; Ranger-Moore, J.; Konopa, K.; Rzyman, W.; Szostakiewicz, B.; Jassem, J.; Hirsch, F.R. Correlation between MET Gene Copy Number by Silver In Situ Hy-bridization and Protein Expression by Immunohistochemistry in Non-small Cell Lung Cancer. J. Thor. Oncol. 2012, 7, 340–347. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| FISH | NGS | |||||
|---|---|---|---|---|---|---|
| Case-ID | MET/CEN7 Ratio | MET GCN | CEN7 GCN | MET Status | Polysomy | NGS Status |
| #1 | >5.00 | >10.00 | 2.00 | High-level amplification | No | Normal |
| #2 | >2.50 | >10.0 | 3.00 | High-level amplification | No | Amplified |
| #3 | 3.30 | >10.0 | 3.00 | High-level amplification | No | Amplified |
| #4 | >2.50 | >10.0 | 4.00 | High-level amplification | Yes | Amplified |
| #5 | >2.50 | >10.0 | 4.00 | High-level amplification | Yes | Amplified |
| #6 | >4.00 | >8.00 | 2.00 | High-level amplification | No | Amplified |
| #7 | 1.10 | 7.60 | 7.20 | High-level amplification | Yes | Normal |
| #8 | 1.40 | 7.00 | 5.00 | High-level amplification | Yes | Normal |
| #9 | 2.00 | 6.00 | 3.00 | High-level amplification | No | Normal |
| #10 | 1.32 | 6.00 | 5.00 | High-level amplification | Yes | Normal |
| #11 | 1.50 | 6.00 | 4.00 | High-level amplification | Yes | Normal |
| #12 | 1.30 | 6.00 | 4.50 | High-level amplification | Yes | Normal |
| #13 | 2.50 | 5.00 | 2.00 | High-level amplification | No | Normal |
| #14 | 3.00 | 4.50 | 1.50 | High-level amplification | No | Normal |
| #15 | 2.70 | 4.40 | 1.60 | High-level amplification | No | Amplified |
| #16 | 2.00 | 4.00 | 2.00 | High-level amplification | No | Normal |
| #17 | 1.38 | 5.50 | 4.00 | Intermediate-level amplification | Yes | Normal |
| #18 | 1.70 | 5.00 | 3.00 | Intermediate-level amplification | No | Normal |
| #19 | 1.70 | 5.00 | 3.00 | Intermediate-level amplification | No | Normal |
| #20 | 1.25 | 5.00 | 4.00 | Intermediate-level amplification | Yes | Normal |
| #21 | 1.00 | 5.00 | 5.00 | Intermediate-level amplification | Yes | Normal |
| #22 | 1.25 | 5.00 | 4.00 | Intermediate-level amplification | Yes | Normal |
| #23 | 1.00 | 5.00 | 5.00 | Intermediate-level amplification | Yes | Normal |
| #24 | 1.25 | 5.00 | 4.00 | Intermediate-level amplification | Yes | Normal |
| #25 | 1.00 | 5.00 | 5.00 | Intermediate-level amplification | Yes | Normal |
| #26 | 1.00 | 5.00 | 5.00 | Intermediate-level amplification | Yes | Normal |
| #27 | 1.00 | 5.00 | 5.00 | Intermediate-level amplification | Yes | Normal |
| #28 | 1.25 | 5.00 | 4.00 | Intermediate-level amplification | Yes | Normal |
| #29 | 0.83 | 5.00 | 6.00 | Intermediate-level amplification | Yes | Normal |
| #30 | 1.26 | 5.00 | 4.00 | Intermediate-level amplification | Yes | Amplified |
| #31 | 1.50 | 4.90 | 3.20 | Low-level amplification | No | Amplified |
| #32 | 1.50 | 4.50 | 3.00 | Low-level amplification | No | Normal |
| #33 | 1.50 | 4.50 | 3.00 | Low-level amplification | No | Normal |
| #34 | 1.50 | 4.50 | 3.00 | Low-level amplification | No | Normal |
| #35 | 1.10 | 4.50 | 5.00 | Low-level amplification | Yes | Normal |
| #36 | 1.13 | 4.50 | 4.00 | Low-level amplification | Yes | Normal |
| #37 | 1.13 | 4.50 | 4.00 | Low-level amplification | Yes | Normal |
| #38 | 1.33 | 4.00 | 3.00 | Low-level amplification | No | Normal |
| #39 | 1.30 | 4.00 | 3.00 | Low-level amplification | No | Normal |
| #40 | 1.14 | 4.00 | 3.50 | Low-level amplification | No | Normal |
| #41 | 1.00 | 4.00 | 4.00 | Low-level amplification | Yes | Normal |
| #42 | 1.00 | 4.00 | 4.00 | Low-level amplification | Yes | Normal |
| #43 | 1.00 | 3.50 | 3.50 | Normal | No | Amplified |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schubart, C.; Stöhr, R.; Tögel, L.; Fuchs, F.; Sirbu, H.; Seitz, G.; Seggewiss-Bernhardt, R.; Leistner, R.; Sterlacci, W.; Vieth, M.; et al. MET Amplification in Non-Small Cell Lung Cancer (NSCLC)—A Consecutive Evaluation Using Next-Generation Sequencing (NGS) in a Real-World Setting. Cancers 2021, 13, 5023. https://doi.org/10.3390/cancers13195023
Schubart C, Stöhr R, Tögel L, Fuchs F, Sirbu H, Seitz G, Seggewiss-Bernhardt R, Leistner R, Sterlacci W, Vieth M, et al. MET Amplification in Non-Small Cell Lung Cancer (NSCLC)—A Consecutive Evaluation Using Next-Generation Sequencing (NGS) in a Real-World Setting. Cancers. 2021; 13(19):5023. https://doi.org/10.3390/cancers13195023
Chicago/Turabian StyleSchubart, Christoph, Robert Stöhr, Lars Tögel, Florian Fuchs, Horia Sirbu, Gerhard Seitz, Ruth Seggewiss-Bernhardt, Rumo Leistner, William Sterlacci, Michael Vieth, and et al. 2021. "MET Amplification in Non-Small Cell Lung Cancer (NSCLC)—A Consecutive Evaluation Using Next-Generation Sequencing (NGS) in a Real-World Setting" Cancers 13, no. 19: 5023. https://doi.org/10.3390/cancers13195023