
Hyperuricemia and Progression of Chronic Kidney Disease: A Review from Physiology and Pathogenesis to the Role of Urate-Lowering Therapy


Abstract
:1. Introduction
2. Physiology of UA
2.1. Source of UA
2.2. UA Clearance
2.3. Genetic Variations Influence the Serum UA Level
3. Hyperuricemia and Renal Disease
4. Mechanism
4.1. Crystalline Effects
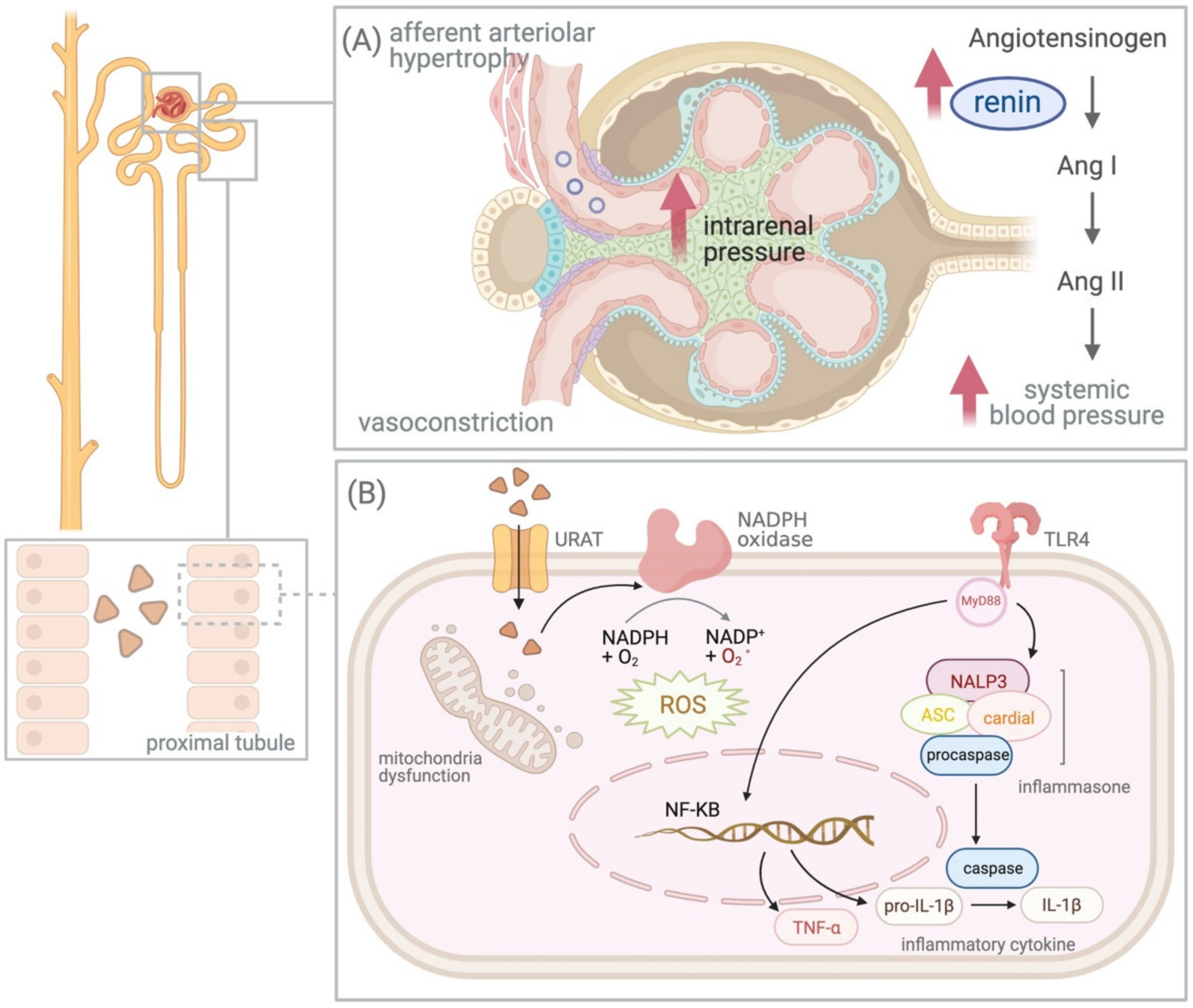
4.2. UA and Renin–Angiotensin–Aldosterone System (RAAS)
4.3. Intracellular Effect of UA
5. Treatment and Evidence of Clinical Trials
5.1. Allopurinol
5.2. Febuxostat
5.3. Combination Therapy: Xanthine Oxidase Inhibitor with Urate Reabsorption Inhibitor
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brook, R.A.; Forsythe, A.; Smeeding, J.E.; Lawrence Edwards, N. Chronic gout: Epidemiology, disease progression, treatment and disease burden. Curr. Med. Res. Opin. 2010, 26, 2813–2821. [Google Scholar] [CrossRef]
- Kim, K.Y.; Ralph Schumacher, H.; Hunsche, E.; Wertheimer, A.I.; Kong, S.X. A literature review of the epidemiology and treatment of acute gout. Clin. Ther. 2003, 25, 1593–1617. [Google Scholar] [CrossRef]
- Harris, M.D.; Siegel, L.B.; Alloway, J.A. Gout and hyperuricemia. Am. Fam. Physician 1999, 59, 925–934. [Google Scholar] [PubMed]
- Van Doornum, S.; Ryan, P.F. Clinical manifestations of gout and their management. Med. J. Aust. 2000, 172, 493–497. [Google Scholar] [CrossRef]
- Agudelo, C.A.; Wise, C.M. Gout: Diagnosis, pathogenesis, and clinical manifestations. Curr. Opin. Rheumatol. 2001, 13, 234–239. [Google Scholar] [CrossRef]
- Shadick, N.A.; Kim, R.; Weiss, S.; Liang, M.H.; Sparrow, D.; Hu, H. Effect of low level lead exposure on hyperuricemia and gout among middle aged and elderly men: The normative aging study. J. Rheumatol. 2000, 27, 1708–1712. [Google Scholar] [PubMed]
- Feig, D.I. Uric acid: A novel mediator and marker of risk in chronic kidney disease? Curr. Opin. Nephrol. Hypertens. 2009, 18, 526–530. [Google Scholar] [CrossRef] [Green Version]
- Chapter 1: Definition and classification of CKD. Kidney Int. Suppl. 2013, 3, 19–62. [CrossRef] [Green Version]
- Talbott, J.H.; Terplan, K.L. The kidney in gout. Medicine 1960, 39, 405–467. [Google Scholar] [CrossRef]
- Clifford, A.J.; Riumallo, J.A.; Young, V.R.; Scrimshaw, N.S. Effect of Oral Purines on Serum and Urinary Uric Acid of Normal, Hyperuricemic and Gouty Humans. J. Nutr. 1976, 106, 428–434. [Google Scholar] [CrossRef]
- Ryu, E.S.; Kim, M.J.; Shin, H.S.; Jang, Y.H.; Choi, H.S.; Jo, I.; Johnson, R.J.; Kang, D.H. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2013, 304, F471–F480. [Google Scholar] [CrossRef]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffler, W.; Grobner, W.; Medina, R.; Zollner, N. Influence of dietary purines on pool size, turnover, and excretion of uric acid during balance conditions. Isotope studies using 15N-uric acid. Res. Exp. Med. 1982, 181, 113–123. [Google Scholar] [CrossRef]
- Merriman, T.R.; Dalbeth, N.; Johnson, R.J. Sugar-sweetened beverages, urate, gout and genetic interaction. Pac. Health Dialog. 2014, 20, 31–38. [Google Scholar]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J.; et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, K.; Malhotra, K.; Sowers, J.; Aroor, A. Uric Acid—Key ingredient in the recipe for cardiorenal metabolic syndrome. Cardiorenal Med. 2013, 3, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Nivorozhkin, A.; Szabo, C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharm. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, J.T.; Lanaspa, M.A.; Murphy, M.N.; Cicerchi, C.; Graves, C.L.; Tipton, P.A.; Ortlund, E.A.; Johnson, R.J.; Gaucher, E.A. Evolutionary history and metabolic insights of ancient mammalian uricases. Proc. Natl. Acad. Sci. USA 2014, 111, 3763–3768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyndman, D.; Liu, S.; Miner, J.N. Urate Handling in the Human Body. Curr. Rheumatol. Rep. 2016, 18, 34. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.F.; Rudan, I.; Hastie, N.D.; Campbell, H. A ‘complexity’ of urate transporters. Kidney Int. 2010, 78, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Terkeltaub, R.; Bushinsky, D.A.; Becker, M.A. Recent developments in our understanding of the renal basis of hyperuricemia and the development of novel antihyperuricemic therapeutics. Arthritis Res. Ther. 2006, 8 (Suppl. 1), S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieske, J.C.; de la Vega, L.S.; Gettman, M.T.; Slezak, J.M.; Bergstralh, E.J.; Melton, L.J., 3rd; Leibson, C.L. Diabetes mellitus and the risk of urinary tract stones: A population-based case-control study. Am. J. Kidney Dis. 2006, 48, 897–904. [Google Scholar] [CrossRef]
- Bjornstad, P.; Roncal, C.; Milagres, T.; Pyle, L.; Lanaspa, M.A.; Bishop, F.K.; Snell-Bergeon, J.K.; Johnson, R.J.; Wadwa, R.P.; Maahs, D.M. Hyperfiltration and uricosuria in adolescents with type 1 diabetes. Pediatr. Nephrol. 2016, 31, 787–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef] [Green Version]
- Ferns, G.A.; Lanham, J.; Dieppe, P.; Galton, D.J. A DNA polymorphism of an apoprotein gene associates with the hypertriglyceridaemia of primary gout. Hum. Genet. 1988, 78, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.S.; Simmonds, H.A. Hereditary hyperuricemia and renal disease. Semin. Nephrol. 2005, 25, 9–18. [Google Scholar] [CrossRef]
- Ronco, C. Hyperuricemic Syndromes: Pathophysiology and Therapy. Contrib. Nephrol. 2005, 147, 12. [Google Scholar] [CrossRef] [Green Version]
- Wallace, C.; Newhouse, S.J.; Braund, P.; Zhang, F.; Tobin, M.; Falchi, M.; Ahmadi, K.; Dobson, R.J.; Marcano, A.C.; Hajat, C.; et al. Genome-wide association study identifies genes for biomarkers of cardiovascular disease: Serum urate and dyslipidemia. Am. J. Hum. Genet. 2008, 82, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Major, T.J.; Dalbeth, N.; Stahl, E.A.; Merriman, T.R. An update on the genetics of hyperuricaemia and gout. Nat. Rev. Rheumatol. 2018, 14, 341–353. [Google Scholar] [CrossRef]
- Reginato, A.M.; Mount, D.B.; Yang, I.; Choi, H.K. The genetics of hyperuricaemia and gout. Nat. Rev. Rheumatol. 2012, 8, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.Y.; Xiong, J.X.; Mai, H.X.; Lin, J.J.; Jiang, L.N.; Cheng, L.; Ye, Q. Advances on the regulation of telomerase. Yi Chuan 2016, 38, 289–299. [Google Scholar] [CrossRef]
- Dahan, K.; Devuyst, O.; Smaers, M.; Vertommen, D.; Loute, G.; Poux, J.M.; Viron, B.; Jacquot, C.; Gagnadoux, M.F.; Chauveau, D.; et al. A cluster of mutations in the UMOD gene causes familial juvenile hyperuricemic nephropathy with abnormal expression of uromodulin. J. Am. Soc. Nephrol. 2003, 14, 2883–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlig, T.; Zweck, J. Direct observation of switching processes in permalloy rings with lorentz microscopy. Phys. Rev. Lett. 2004, 93, 047203. [Google Scholar] [CrossRef] [PubMed]
- Bleyer, A.J.; Hart, T.C. Genetic factors associated with gout and hyperuricemia. Adv. Chronic Kidney Dis. 2006, 13, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Essex, M.N.; Hopps, M.; Bienen, E.J.; Udall, M.; Mardekian, J.; Makinson, G.T. Evaluation of the Relationship Between Serum Uric Acid Levels and Cardiovascular Events in Patients with Gout: A Retrospective Analysis Using Electronic Medical Record Data. J. Clin. Rheumatol. 2017, 23, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, M.; Niwa, K.; Hisatome, I.; Nakagawa, T.; Roncal-Jimenez, C.A.; Andres-Hernando, A.; Bjornstad, P.; Jensen, T.; Sato, Y.; Milagres, T.; et al. Asymptomatic Hyperuricemia Without Comorbidities Predicts Cardiometabolic Diseases: Five-Year Japanese Cohort Study. Hypertension 2017, 69, 1036–1044. [Google Scholar] [CrossRef]
- Bardin, T.; Richette, P. Impact of comorbidities on gout and hyperuricaemia: An update on prevalence and treatment options. BMC Med. 2017, 15, 123. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Ford, E.S.; Li, C.; Curhan, G. Prevalence of the metabolic syndrome in patients with gout: The Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2007, 57, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Hoshiai, H. Detection of numerical chromosome abnormalities in human spermatozoa by three-color fluorescence in situ hybridization. J. Obs. Gynaecol. Res. 1998, 24, 385–392. [Google Scholar] [CrossRef]
- Nakagawa, T.; Cirillo, P.; Sato, W.; Gersch, M.; Sautin, Y.; Roncal, C.; Mu, W.; Sanchez-Lozada, L.G.; Johnson, R.J. The conundrum of hyperuricemia, metabolic syndrome, and renal disease. Intern. Emerg. Med. 2008, 3, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.H.; Nakagawa, T.; Feng, L.; Watanabe, S.; Han, L.; Mazzali, M.; Truong, L.; Harris, R.; Johnson, R.J. A role for uric acid in the progression of renal disease. J. Am. Soc. Nephrol. 2002, 13, 2888–2897. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.R.; Choi, H.S.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Sung, S.A.; Kim, Y.S.; Oh, K.H.; Ahn, C.; Kim, S.W. Hyperuricemia has increased the risk of progression of chronic kidney disease: Propensity score matching analysis from the KNOW-CKD study. Sci. Rep. 2019, 9, 6681. [Google Scholar] [CrossRef]
- Sofue, T.; Nakagawa, N.; Kanda, E.; Nagasu, H.; Matsushita, K.; Nangaku, M.; Maruyama, S.; Wada, T.; Terada, Y.; Yamagata, K.; et al. Prevalences of hyperuricemia and electrolyte abnormalities in patients with chronic kidney disease in Japan: A nationwide, cross-sectional cohort study using data from the Japan Chronic Kidney Disease Database (J-CKD-DB). PLoS ONE 2020, 15, e0240402. [Google Scholar] [CrossRef]
- Section 2: AKI Definition. Kidney Int. Suppl. 2012, 2, 19–36. [CrossRef] [PubMed] [Green Version]
- Sanchez-Lozada, L.G.; Tapia, E.; Avila-Casado, C.; Soto, V.; Franco, M.; Santamaria, J.; Nakagawa, T.; Rodriguez-Iturbe, B.; Johnson, R.J.; Herrera-Acosta, J. Mild hyperuricemia induces glomerular hypertension in normal rats. Am. J. Physiol. Ren. Physiol. 2002, 283, F1105–F1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzali, M.; Hughes, J.; Kim, Y.G.; Jefferson, J.A.; Kang, D.H.; Gordon, K.L.; Lan, H.Y.; Kivlighn, S.; Johnson, R.J. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001, 38, 1101–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Pozo, S.E.; Schold, J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Johnson, R.J.; Lillo, J.L. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: Role of uric acid in the hypertensive response. Int. J. Obes. Lond. 2010, 34, 454–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzali, M.; Kanellis, J.; Han, L.; Feng, L.; Xia, Y.Y.; Chen, Q.; Kang, D.H.; Gordon, K.L.; Watanabe, S.; Nakagawa, T.; et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am. J. Physiol. Ren. Physiol. 2002, 282, F991–F997. [Google Scholar] [CrossRef] [Green Version]
- Bjornstad, P.; Maahs, D.M.; Roncal, C.A.; Snell-Bergeon, J.K.; Shah, V.N.; Milagres, T.; Ellis, S.L.; Hatch, M.; Chung, L.T.; Rewers, M.J.; et al. Role of bicarbonate supplementation on urine uric acid crystals and diabetic tubulopathy in adults with type 1 diabetes. Diabetes Obes. Metab. 2018, 20, 1776–1780. [Google Scholar] [CrossRef]
- Mulay, S.R.; Shi, C.; Ma, X.; Anders, H.J. Novel Insights into Crystal-Induced Kidney Injury. Kidney Dis. 2018, 4, 49–57. [Google Scholar] [CrossRef]
- Lin, P.Y.; Lin, C.C.; Liu, H.C.; Lee, M.D.; Lee, H.C.; Ho, C.S.; Chiu, N.C.; Peng, C.C.; Huang, F.Y.; Tsai, J.D. Rasburicase improves hyperuricemia in patients with acute kidney injury secondary to rhabdomyolysis caused by ecstasy intoxication and exertional heat stroke. Pediatr. Crit. Care Med. 2011, 12, e424–e427. [Google Scholar] [CrossRef]
- Galichon, P.; Hertig, A. Epithelial to mesenchymal transition as a biomarker in renal fibrosis: Are we ready for the bedside? Fibrogenes. Tissue Repair 2011, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grgic, I.; Duffield, J.S.; Humphreys, B.D. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr. Nephrol. 2012, 27, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Noble, N.A.; Miller, D.E.; Border, W.A. Sustained expression of TGF-beta 1 underlies development of progressive kidney fibrosis. Kidney Int. 1994, 45, 916–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellmayr, M.; Hernandez Petzsche, M.R.; Ma, Q.; Kruger, N.; Liapis, H.; Brink, A.; Lenz, B.; Angelotti, M.L.; Gnemmi, V.; Kuppe, C.; et al. Only Hyperuricemia with Crystalluria, but not Asymptomatic Hyperuricemia, Drives Progression of Chronic Kidney Disease. J. Am. Soc. Nephrol. 2020, 31, 2773–2792. [Google Scholar] [CrossRef]
- Gasse, P.; Riteau, N.; Charron, S.; Girre, S.; Fick, L.; Petrilli, V.; Tschopp, J.; Lagente, V.; Quesniaux, V.F.; Ryffel, B.; et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am. J. Respir. Crit. Care. Med. 2009, 179, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, X.L.; Fu, C.; Han, R.; Chen, W.; Lu, Y.; Ye, Z. Soluble uric acid increases NALP3 inflammasome and interleukin-1beta expression in human primary renal proximal tubule epithelial cells through the Toll-like receptor 4-mediated pathway. Int. J. Mol. Med. 2015, 35, 1347–1354. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Fang, L.; Jiang, L.; Wen, P.; Cao, H.; He, W.; Dai, C.; Yang, J. Uric acid induces renal inflammation via activating tubular NF-kappaB signaling pathway. PLoS ONE 2012, 7, e39738. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, G.; Venanzi, S.; Verdura, C.; Saronio, P.; Esposito, A.; Timio, M. Association of uric acid with change in kidney function in healthy normotensive individuals. Am. J. Kidney Dis. 2010, 56, 264–272. [Google Scholar] [CrossRef]
- Saito, I.; Saruta, T.; Kondo, K.; Nakamura, R.; Oguro, T.; Yamagami, K.; Ozawa, Y.; Kato, E. Serum uric acid and the renin-angiotensin system in hypertension. J. Am. Geriatr. Soc. 1978, 26, 241–247. [Google Scholar] [CrossRef]
- Kanabrocki, E.L.; Third, J.L.; Ryan, M.D.; Nemchausky, B.A.; Shirazi, P.; Scheving, L.E.; McCormick, J.B.; Hermida, R.C.; Bremner, W.F.; Hoppensteadt, D.A.; et al. Circadian relationship of serum uric acid and nitric oxide. JAMA 2000, 283, 2240–2241. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, G. Uric acid and chronic kidney disease: A time to act? World J. Nephrol. 2013, 2, 17–25. [Google Scholar] [CrossRef]
- Xu, C.; Lu, A.; Lu, X.; Zhang, L.; Fang, H.; Zhou, L.; Yang, T. Activation of Renal (Pro)Renin Receptor Contributes to High Fructose-Induced Salt Sensitivity. Hypertension 2017, 69, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.A.; Sanchez-Lozada, L.G.; Johnson, R.J.; Kang, D.H. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Feig, D.I.; Soletsky, B.; Johnson, R.J. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: A randomized trial. JAMA 2008, 300, 924–932. [Google Scholar] [CrossRef] [Green Version]
- Talaat, K.M.; El-Sheikh, A.R. The effect of mild hyperuricemia on urinary transforming growth factor beta and the progression of chronic kidney disease. Am. J. Nephrol. 2007, 27, 435–440. [Google Scholar] [CrossRef]
- Sanchez-Lozada, L.G. The Pathophysiology of Uric Acid on Renal Diseases. Contrib. Nephrol. 2018, 192, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Feig, D.I.; Stack, A.G.; Kang, D.H.; Lanaspa, M.A.; Ejaz, A.A.; Sanchez-Lozada, L.G.; Kuwabara, M.; Borghi, C.; Johnson, R.J. The case for uric acid-lowering treatment in patients with hyperuricaemia and CKD. Nat. Rev. Nephrol. 2019, 15, 767–775. [Google Scholar] [CrossRef]
- Rodriguez-Iturbe, B.; Pons, H.; Johnson, R.J. Role of the Immune System in Hypertension. Physiol. Rev. 2017, 97, 1127–1164. [Google Scholar] [CrossRef]
- Watanabe, S.; Kang, D.H.; Feng, L.; Nakagawa, T.; Kanellis, J.; Lan, H.; Mazzali, M.; Johnson, R.J. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 2002, 40, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sautin, Y.Y.; Johnson, R.J. Uric acid: The oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.H.; Park, S.K.; Lee, I.K.; Johnson, R.J. Uric acid-induced C-reactive protein expression: Implication on cell proliferation and nitric oxide production of human vascular cells. J. Am. Soc. Nephrol. 2005, 16, 3553–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.M.; Henderson, G.N.; Ouyang, X.; Frye, R.F.; Sautin, Y.Y.; Feig, D.I.; Johnson, R.J. A sensitive and specific liquid chromatography-tandem mass spectrometry method for the determination of intracellular and extracellular uric acid. J. Chromatogr. B 2009, 877, 2032–2038. [Google Scholar] [CrossRef] [Green Version]
- Roncal, C.A.; Reungjui, S.; Sanchez-Lozada, L.G.; Mu, W.; Sautin, Y.Y.; Nakagawa, T.; Johnson, R.J. Combination of captopril and allopurinol retards fructose-induced metabolic syndrome. Am. J. Nephrol. 2009, 30, 399–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittman, J.R.; Bross, M.H. Diagnosis and management of gout. Am. Fam. Physician 1999, 59, 1799–1806, 1810. [Google Scholar]
- Qu, L.H.; Jiang, H.; Chen, J.H. Effect of uric acid-lowering therapy on blood pressure: Systematic review and meta-analysis. Ann. Med. 2017, 49, 142–156. [Google Scholar] [CrossRef]
- Goicoechea, M.; Garcia de Vinuesa, S.; Verdalles, U.; Verde, E.; Macias, N.; Santos, A.; Perez de Jose, A.; Cedeno, S.; Linares, T.; Luno, J. Allopurinol and progression of CKD and cardiovascular events: Long-term follow-up of a randomized clinical trial. Am. J. Kidney Dis. 2015, 65, 543–549. [Google Scholar] [CrossRef]
- Golmohammadi, S.; Almasi, A.; Manouchehri, M.; Omrani, H.R.; Zandkarimi, M.R. Allopurinol Against Progression of Chronic Kidney Disease. Iran. J. Kidney Dis. 2017, 11, 286–293. [Google Scholar] [PubMed]
- Shahid, H.; Singh, J.A. Investigational drugs for hyperuricemia. Expert. Opin. Investig. Drugs 2015, 24, 1013–1030. [Google Scholar] [CrossRef]
- Lee, T.H.; Lee, C.C.; Ng, C.Y.; Chang, M.Y.; Chang, S.W.; Fan, P.C.; Chung, W.H.; Tian, Y.C.; Chen, Y.C.; Chang, C.H. The influence of acute kidney injury on the outcome of Stevens-Johnson syndrome and toxic epidermal necrolysis: The prognostic value of KDIGO staging. PLoS ONE 2018, 13, e0203642. [Google Scholar] [CrossRef]
- Hung, S.I.; Chung, W.H.; Liou, L.B.; Chu, C.C.; Lin, M.; Huang, H.P.; Lin, Y.L.; Lan, J.L.; Yang, L.C.; Hong, H.S.; et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc. Natl. Acad. Sci. USA 2005, 102, 4134–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jick, H.; Porter, J.B. Potentiation of ampicillin skin reactions by allopurinol or hyperuricemia. J. Clin. Pharm. 1981, 21, 456–458. [Google Scholar] [CrossRef]
- Bose, B.; Badve, S.V.; Hiremath, S.S.; Boudville, N.; Brown, F.G.; Cass, A.; de Zoysa, J.R.; Fassett, R.G.; Faull, R.; Harris, D.C.; et al. Effects of uric acid-lowering therapy on renal outcomes: A systematic review and meta-analysis. Nephrol. Dial. Transpl. 2014, 29, 406–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanji, T.; Gandhi, M.; Clase, C.M.; Yang, R. Urate lowering therapy to improve renal outcomes in patients with chronic kidney disease: Systematic review and meta-analysis. BMC Nephrol. 2015, 16, 58. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Xu, B.; Yan, B.; Qiao, X.; Wang, L. Effects of uric acid-lowering therapy in patients with chronic kidney disease: A meta-analysis. PLoS ONE 2017, 12, e0187550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badve, S.V.; Pascoe, E.M.; Tiku, A.; Boudville, N.; Brown, F.G.; Cass, A.; Clarke, P.; Dalbeth, N.; Day, R.O.; de Zoysa, J.R.; et al. Effects of Allopurinol on the Progression of Chronic Kidney Disease. N. Engl. J. Med. 2020, 382, 2504–2513. [Google Scholar] [CrossRef]
- Afkarian, M.; Polsky, S.; Parsa, A.; Aronson, R.; Caramori, M.L.; Cherney, D.Z.; Crandall, J.P.; de Boer, I.H.; Elliott, T.G.; Galecki, A.T.; et al. Preventing Early Renal Loss in Diabetes (PERL) Study: A Randomized Double-Blinded Trial of Allopurinol-Rationale, Design, and Baseline Data. Diabetes Care 2019, 42, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martin, G.; Cano, J.; Carriazo, S.; Kanbay, M.; Perez-Gomez, M.V.; Fernandez-Prado, R.; Ortiz, A. The dirty little secret of urate-lowering therapy: Useless to stop chronic kidney disease progression and may increase mortality. Clin. Kidney J. 2020, 13, 936–947. [Google Scholar] [CrossRef]
- Yang, C.Y.; Chen, C.H.; Deng, S.T.; Huang, C.S.; Lin, Y.J.; Chen, Y.J.; Wu, C.Y.; Hung, S.I.; Chung, W.H. Allopurinol Use and Risk of Fatal Hypersensitivity Reactions: A Nationwide Population-Based Study in Taiwan. JAMA Intern. Med. 2015, 175, 1550–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, E.; Viazzi, F.; Pontremoli, R.; Barbagallo, C.M.; Bombelli, M.; Casiglia, E.; Cicero, A.F.G.; Cirillo, M.; Cirillo, P.; Desideri, G.; et al. Association of uric acid with kidney function and albuminuria: The Uric Acid Right for heArt Health (URRAH) Project. J. Nephrol. 2021. [Google Scholar] [CrossRef]
- Ye, P.; Yang, S.; Zhang, W.; Lv, Q.; Cheng, Q.; Mei, M.; Luo, T.; Liu, L.; Chen, S.; Li, Q. Efficacy and tolerability of febuxostat in hyperuricemic patients with or without gout: A systematic review and meta-analysis. Clin. Ther. 2013, 35, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, M.B.; Chavez, B. Febuxostat for the treatment of gout. Expert. Opin. Pharm. 2015, 16, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Hosoya, T.; Uchida, S.; Inaba, M.; Makino, H.; Maruyama, S.; Ito, S.; Yamamoto, T.; Tomino, Y.; Ohno, I.; et al. Febuxostat Therapy for Patients with Stage 3 CKD and Asymptomatic Hyperuricemia: A Randomized Trial. Am. J. Kidney Dis. 2018, 72, 798–810. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.C.; Hung, L.Y.; Chen, Y.C.; Lo, W.C.; Lin, C.H.; Tam, K.W.; Wu, M.Y. Effects of febuxostat on renal function in patients with chronic kidney disease: A systematic review and meta-analysis. Med. Baltim. 2019, 98, e16311. [Google Scholar] [CrossRef]
- Verdoux, H.; Pambrun, E.; Tournier, M.; Cortaredona, S.; Verger, P. Multi-trajectories of antidepressant and antipsychotic use: A 11-year naturalistic study in a community-based sample. Acta Psychiatr. Scand. 2019, 139, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.O.; Wu, I.W.; Chang, S.H.; Lee, C.C.; Tsai, C.Y.; Lin, C.Y.; Lin, W.T.; Huang, Y.T.; Wu, C.Y.; Kuo, G.; et al. Comparative Renoprotective Effect of Febuxostat and Allopurinol in Predialysis Stage 5 Chronic Kidney Disease Patients: A Nationwide Database Analysis. Clin Pharmacol. Ther. 2020, 107, 1159–1169. [Google Scholar] [CrossRef]
- Chung, T.T.; Yu, K.H.; Kuo, C.F.; Luo, S.F.; Chiou, M.J.; Lan, W.C.; Chen, J.S.; Tseng, W.Y.; Hsieh, A.H.; Wang, L.C. Impact of urate-lowering drugs on the progression and recovery from chronic kidney disease among gout patients. Arthritis Res. Ther. 2019, 21, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, W.B.; Saag, K.G.; Becker, M.A.; Borer, J.S.; Gorelick, P.B.; Whelton, A.; Hunt, B.; Castillo, M.; Gunawardhana, L.; CARES Investigators. Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. N. Engl. J. Med. 2018, 378, 1200–1210. [Google Scholar] [CrossRef]
- Goicoechea, M.; de Vinuesa, S.G.; Verdalles, U.; Ruiz-Caro, C.; Ampuero, J.; Rincon, A.; Arroyo, D.; Luno, J. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin. J. Am. Soc. Nephrol. 2010, 5, 1388–1393. [Google Scholar] [CrossRef]
- Hisatome, I.; Li, P.; Taufiq, F.; Maharani, N.; Kuwabara, M.; Ninomiya, H.; Bahrudin, U. Hyperuricemia as a Risk Factor for Cardiovascular Diseases. J. Biomed. Transl. Res. 2020, 16, e6606. [Google Scholar] [CrossRef]
- Zhang, M.; Solomon, D.H.; Desai, R.J.; Kang, E.H.; Liu, J.; Neogi, T.; Kim, S.C. Assessment of Cardiovascular Risk in Older Patients with Gout Initiating Febuxostat Versus Allopurinol: Population-Based Cohort Study. Circulation 2018, 138, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, Z.; Zhou, J.; Chen, Z.; Li, Y.; Li, S.; Zhao, H.; Badve, S.V.; Lv, J. Effect of Urate-Lowering Therapy on Cardiovascular and Kidney Outcomes: A Systematic Review and Meta-Analysis. Clin. J. Am. Soc. Nephrol. 2020, 15, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, L.; Zhao, T.Y.; Yang, X.; Zhu, X.X.; Zou, H.J.; Wan, W.G.; Xue, Y. Cardiovascular events in hyperuricemia population and a cardiovascular benefit-risk assessment of urate-lowering therapies: A systematic review and meta-analysis. Chin. Med. J. Engl. 2020, 133, 982–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.S.; Ford, I.; Nuki, G.; Hallas, J.; Hawkey, C.J.; Webster, J.; Ralston, S.H.; Walters, M.; Robertson, M.; De Caterina, R.; et al. Long-term cardiovascular safety of febuxostat compared with allopurinol in patients with gout (FAST): A multicentre, prospective, randomised, open-label, non-inferiority trial. Lancet 2020, 396, 1745–1757. [Google Scholar] [CrossRef]
- Feig, D.I. Urate-Lowering Therapy and Chronic Kidney Disease Progression. N. Engl. J. Med. 2020, 382, 2567–2568. [Google Scholar] [CrossRef]
- Fleeman, N.; Pilkington, G.; Dundar, Y.; Dwan, K.; Boland, A.; Dickson, R.; Anijeet, H.; Kennedy, T.; Pyatt, J. Allopurinol for the treatment of chronic kidney disease: A systematic review. Health Technol. Assess. 2014, 18, 1–77. [Google Scholar] [CrossRef] [Green Version]
- Pisano, A.; Cernaro, V.; Gembillo, G.; D’Arrigo, G.; Buemi, M.; Bolignano, D. Xanthine Oxidase Inhibitors for Improving Renal Function in Chronic Kidney Disease Patients: An Updated Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2017, 18, 2283. [Google Scholar] [CrossRef] [Green Version]
- Fitz-Patrick, D.; Roberson, K.; Niwa, K.; Fujimura, T.; Mori, K.; Hall, J.; Yan, X.; Shen, Z.; Liu, S.; Ito, Y.; et al. Safety and efficacy of verinurad, a selective URAT1 inhibitor, for the treatment of patients with gout and/or asymptomatic hyperuricemia in the United States and Japan: Findings from two phase II trials. Mod. Rheumatol. 2019, 29, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Kuriyama, S. Dotinurad: A novel selective urate reabsorption inhibitor as a future therapeutic option for hyperuricemia. Clin. Exp. Nephrol. 2020, 24, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalal, D.I.; Chertow, G.M. Urate Lowering with Combination Therapy in CKD: Reason for Optimism or Einstein’s Definition of Insanity? Am. J. Kidney Dis. 2021, 77, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Stack, A.G.; Dronamraju, N.; Parkinson, J.; Johansson, S.; Johnsson, E.; Erlandsson, F.; Terkeltaub, R. Effect of Intensive Urate Lowering with Combined Verinurad and Febuxostat on Albuminuria in Patients with Type 2 Diabetes: A Randomized Trial. Am. J. Kidney Dis. 2021, 77, 481–489. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stack, A.G.; Terkeltaub, R.; Greene, T.A.; Inker, L.A.; Bjursell, M.; Perl, S.; Rikte, T.; Erlandsson, F.; Perkovic, V.; et al. Rationale, design, demographics, and baseline characteristics of the randomised, controlled, phase 2b SAPPHIRE study of verinurad plus allopurinol in patients with chronic kidney disease and hyperuricaemia. Nephrol. Dial. Transplant. 2021. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study | Year | Sample Size | Population | Intervention Group | Control Group | Outcome | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| eGFR (mL/min/1.73 m2) | Creatinine (mg/dL) | Systolic Pressure (mmHg) | Diastolic Pressure (mmHg) | Other Renal-Associated Outcome | ||||||
| Bose et al. [85] | 2013 | 8 RCTs, 476 participants | Patients with normal or mild decreased eGFR | Allopurinol 100–300 mg/day | Placebo, no treatment, or standard therapy | Favor allopurinol (MD: 3.1, CI: −1.2–7.4) | Favor allopurinol (MD: −0.4, CI: 0.0–0.8) | Favor control (MD: −2.7, CI: −7.3–1.9) | Favor control (MD: −1.9, CI: −4.9–1.2) | Allopurinol group ESRD risk (RR 1.01, CI: 0.15–6.98) |
| Fleeman et al. [108] | 2014 | 4 RCTs, 257 participants | Patients with CKD and hyperuricemia | Allopurinol 100–300 mg/day | Placebo or standard therapy | Favor allopurinol for 6 months (MD: 3.92, CI: −1.33–9.18) | NR | Favor control for 6 months (MD: 1.78, CI: −3.77–7.34) Favor allopurinol in 12 months (MD: −0.32, CI: −5.99–5.35) | Favor allopurinol for 6 months (MD: −0.26, CI: −3.40–2.88) Favor control in 12 months (MD: 0.10, CI: −2.88–3.07) | – |
| Kanji et al. [86] | 2015 | 19 RCTs, 992 participants | Patients with CKD | Urate-lowering therapy | Placebo or no treatment | Favor allopurinol (MD: 3.2; CI: 0.16–6.2) | Favor allopurinol (MD: 0.629; CI: 0.433–0.825) | Favor allopurinol (MD: 6.6, CI: 2.0–11.1) | Favor allopurinol (MD: 2.1, CI: 0.50–3.7) | – |
| Pisano et al. [109] | 2017 | 9 RCTs, 695 participants | Patients with CKD and hyperuricemia | Allopurinol, febuxostat, topiroxostat | Placebo or standard therapy | Favor intervention (MD: 2.33, CI: −0.27–4.92) | Favor intervention (MD: −0.05; CI: −0.12–0.02) | NR | NR | Intervention group ESRD risk (RR 0.42, CI: 0.22–0.80) |
| Lin et al. [96] | 2019 | 11 RCTs, 1317 participants | Patients with CKD and hyperuricemia | Febuxostat 10–80 mg/day | Placebo, allopurinol in two studies benzbromarone in one study | Favor febuxostat (MD: 2.05; CI: −0.24–4.34) | Favor febuxostat (MD: −0.06; CI: −0.21–0.09) | Favor febuxostat (MD: −4.44; CI: −8.08–0.80) | Favor febuxostat (MD: −3.08; CI: −6.25–0.08) | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, T.H.; Chen, J.-J.; Wu, C.-Y.; Yang, C.-W.; Yang, H.-Y. Hyperuricemia and Progression of Chronic Kidney Disease: A Review from Physiology and Pathogenesis to the Role of Urate-Lowering Therapy. Diagnostics 2021, 11, 1674. https://doi.org/10.3390/diagnostics11091674
Lee TH, Chen J-J, Wu C-Y, Yang C-W, Yang H-Y. Hyperuricemia and Progression of Chronic Kidney Disease: A Review from Physiology and Pathogenesis to the Role of Urate-Lowering Therapy. Diagnostics. 2021; 11(9):1674. https://doi.org/10.3390/diagnostics11091674
Chicago/Turabian StyleLee, Tao Han, Jia-Jin Chen, Chao-Yi Wu, Chih-Wei Yang, and Huang-Yu Yang. 2021. "Hyperuricemia and Progression of Chronic Kidney Disease: A Review from Physiology and Pathogenesis to the Role of Urate-Lowering Therapy" Diagnostics 11, no. 9: 1674. https://doi.org/10.3390/diagnostics11091674