
Four Novel p.N385K, p.V36A, c.1033–1034insT and c.1417–1418delCT Mutations in the Sphingomyelin Phosphodiesterase 1 (SMPD1) Gene in Patients with Types A and B Niemann-Pick Disease (NPD)

Abstract
:1. Introduction
2. Results

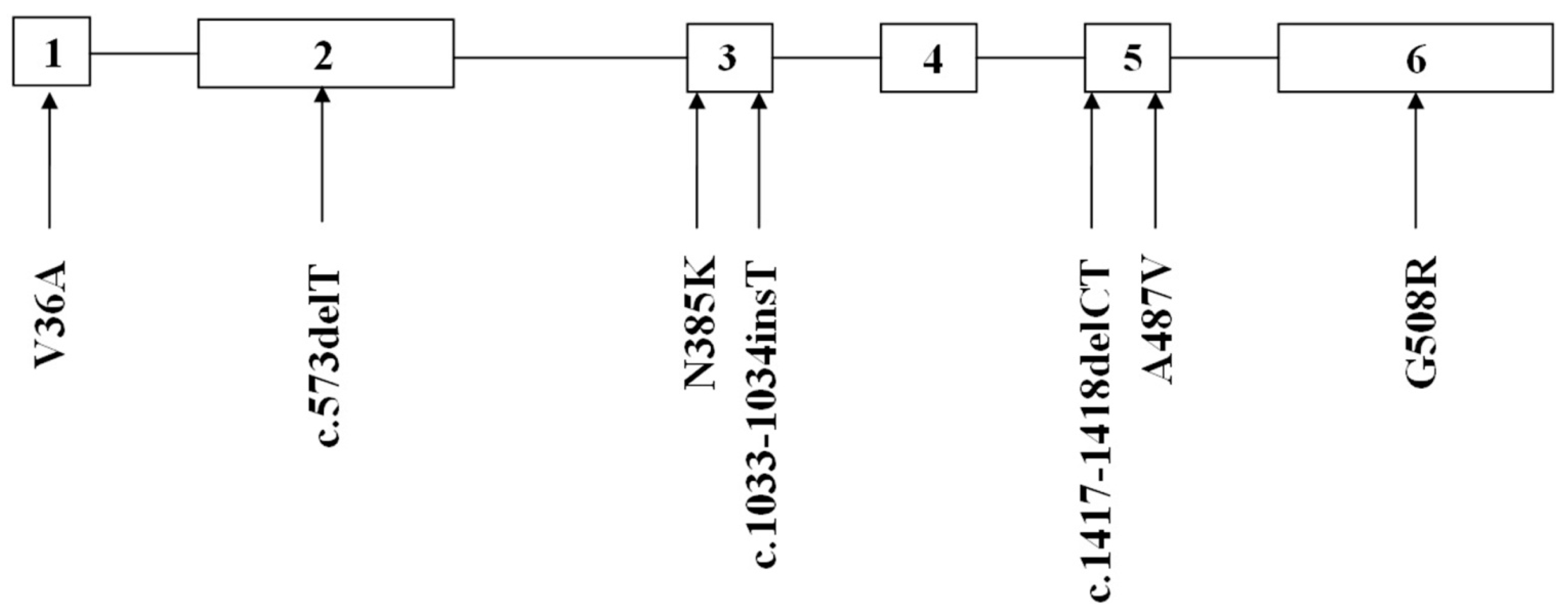{kind=link}
{kind=link}
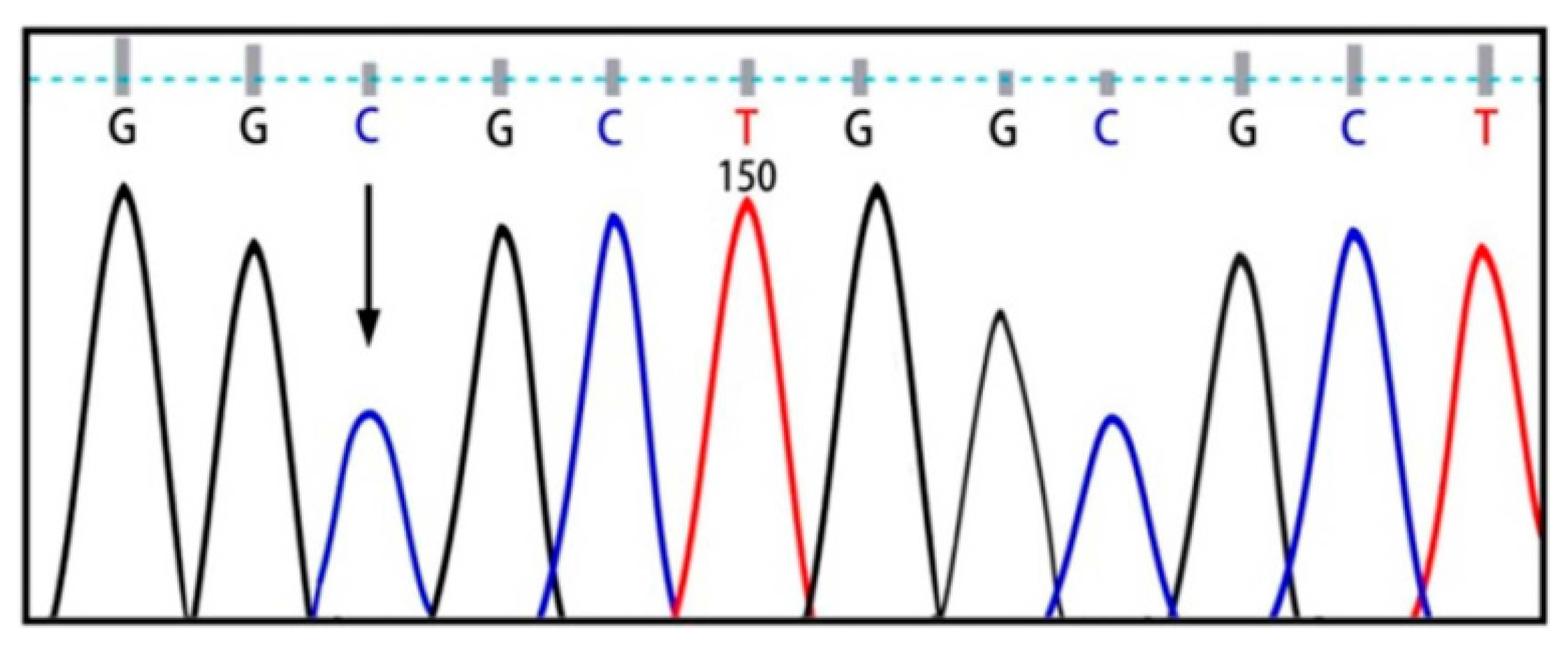{kind=link}
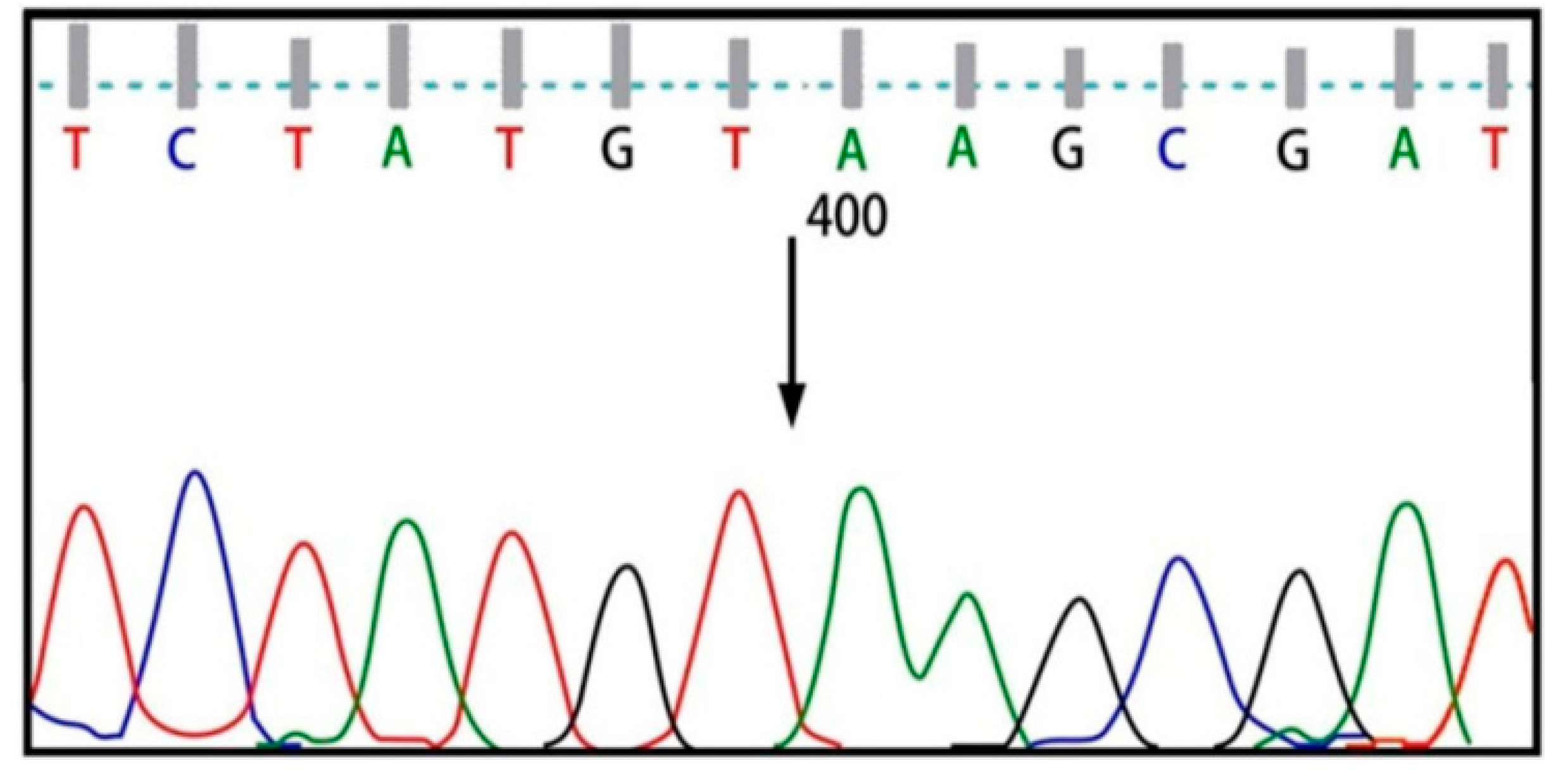{kind=link}
{kind=link}
| No. | Patients No. | Leukocytes-ASM Activity (nmol·17 h−1·mg−1) | Exon | Mutation |
|---|---|---|---|---|
| 1 | P1 | 4.83 | 6 | p.G508R homozygous |
| 2 | P11 | 4.56 | 6 | p.G508R homozygous |
| 3 | P22 | 3.73 | 6 | p.G508R homozygous |
| 4 | P26 | 5.45 | 6 | p.G508R homozygous |
| 5 | P35 | 5.15 | 6 | p.G508R homozygous |
| 6 | P3 | 4.33 | 6 | p.G508R heterozygous |
| 7 | P24 | 3.78 | 6 | p.G508R heterozygous |
| 8 | P28 | 5.08 | 6 | p.G508R heterozygous |
| 9 | P31 | 4.29 | 5, 6 | p.A487V, p.G508R compound heterozygosity |
| 10 | P8 | 4.11 | 3, 6 | p.N385K *, p.G508R compound heterozygosity |
| 11 | P5 | 3.66 | 1 | p.V36A * homozygous |
| 12 | P12 | 3.93 | 1 | p.V36A * heterozygous |
| 13 | P15 | 4.25 | 3 | c.1033–1034insT * homozygous |
| 14 | P32 | 4.29 | 2 | c.573delT homozygous |
| 15 | P29 | 5.88 | 5 | c.1417–1418delCT * homozygous |





| Mutation | Bioinformatic Analysis (I-Mutant 2.0) |
|---|---|
| Prediction of Protein Stability | |
| G508R | decrease stability |
| N385K | decrease stability |
| V36A | decrease stability |
3. Discussion
4. Experimental Section
4.1. Ethics Statement
4.2. DNA Extraction and Primer Design
| Exons | Primer Sequence (5' to 3') | Product Size (bp) | Temperature(°C) |
|---|---|---|---|
| E1 | F: GAGGGCTGGCTAGGGTCCAG | 440 | 68 |
| R: CCAGCCCCAGCAGTCCTT | |||
| E2 | F: TCCTCTGCTCTGCCTCTGATTTCTCACCAT | 900 | 68 |
| R: AATCAGAGACAATGCCCCAGGTTCCCTTCT | |||
| E3 | F: GGAGGACCAGGATTGGAACA | 300 | 62 |
| R: CAGAGGGGCGCCAGCTCAAC | |||
| E4 | F: GATTCAGCTCATGGTCACTG | 300 | 62 |
| R: GGATGGTGAGATGCTCAAGG | |||
| E5, 6 | F: GCATCTCACCATCCCTGTTGTCCCATG | 1000 | 63 |
| R: CTGTTTCACCCTTTCCTACATCAAGAACT |
4.3. Polymerase Chain Reaction (PCR)
4.4. DNA Sequencing
4.5. Bioinformatics Analysis
5. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| NPD | Niemann-Pick disease |
| ASM | acid sphingomyelinase |
| SMC | Special Medical Center |
| SMPD1 | sphingomyelin phosphodiesterase 1 |
Conflicts of Interest
References
- McGovern, M.M.; Pohl-Worgall, T.; Deckelbaum, R.J.; Simpson, W.; Mendelson, D.; Desnick, R.J.; Schuchman, E.H.; Wasserstein, M.P. Lipid abnormalities in children with types A and B Niemann Pick disease. J. Pediatr. 2004, 145, 77–81. [Google Scholar] [CrossRef]
- McGovern, M.; Aron, A.; Brodie, S.; Desnick, R.; Wasserstein, M. Natural history of Type A Niemann-Pick disease Possible endpoints for therapeutic trials. Neurology 2006, 66, 228–232. [Google Scholar] [CrossRef]
- Schuchman, E. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J. Inherit. Metab. Dis 2007, 30, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Desnick, R.J.; Schuchman, E.H.; Hossain, S.; Wallenstein, S.; Lamm, C.; McGovern, M.M. The natural history of type B Niemann-Pick disease: results from a 10-year longitudinal study. Pediatrics 2004, 114, e672–e677. [Google Scholar] [CrossRef] [PubMed]
- Desnick, J.P.; Kim, J.; He, X.; Wasserstein, M.P.; Simonaro, C.M.; Schuchman, E.H. Identification and characterization of eight novel SMPD1 mutations causing types A and B Niemann-Pick disease. Mol. Med. 2010, 16, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Baldi, B.G.; Santana, A.N. C.; Takagaki, T.Y.; Fujita, C.; Kairalla, R.A.; Carvalho, C.R.R. Lung cyst: An unusual manifestation of Niemann-Pick disease. Respirology 2009, 14, 134–136. [Google Scholar] [CrossRef] [PubMed]
- McGovern, M.; Wasserstein, M.; Giugliani, R.; Bembi, B.; Vanier, M.; Mengel, E. A prospective, cross-sectional survey study of the natural history of Niemann-Pick type B disease. Pediatrics 2009, 122, 341–349. [Google Scholar] [CrossRef]
- Hurwitz, R.; Ferlinz, K.; Vielhaber, G.; Moczall, H.; Sandhoff, K. Processing of human acid sphingomyelinase in normal and I-cell fibroblasts. J. Biol. Chem. 1994, 267, 5440–5445. [Google Scholar]
- McGovern, M.M.; Schuchman, E.H. Acid sphingomyelinase deficiency. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1370/ (accessed on 25 January 2009).
- Schuchman, E. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. Int. J. Clin. Pharmacol. Ther. 2008, 47, S48–S57. [Google Scholar]
- Brady, R.O.; Kanfer, J.N.; Mock, M.B.; Fredrickson, D.S. The metabolism of sphingomyelin II.Evidence of an enzymatic deficiency in Niemann-Pick diseae. Proc. Natl. Acad. Sci. USA 1966, 55, 366–369. [Google Scholar] [CrossRef]
- Schneider, P.B.; Kennedy, E.P. Sphingomyelinase in normal human spleens and in spleens from subjects with Niemann-Pick disease. J. Lipid Res. 1967, 8, 202–209. [Google Scholar] [PubMed]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.T.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Besley, G.; Hoogeboom, A.; Hoogeveen, A.; Kleijer, W.; Galjaard, H. Somatic cell hybridisation studies showing different gene mutations in Niemann-Pick variants. Hum. Genet. 1980, 54, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Dardis, A.; Zampieri, S.; Filocamo, M.; Burlina, A.; Bembi, B.; Gabriela Pittis, M. Functional in vitro characterization of 14 SMPD1 mutations identified in Italian patients affected by Niemann Pick Type B disease. Hum. Mutat. 2005, 26, 164–164. [Google Scholar] [CrossRef] [PubMed]
- Levran, O.; Desnick, R.; Schuchman, E. Identification and expression of a common missense mutation (L302P) in the acid sphingomyelinase gene of Ashkenazi Jewish type A Niemann-Pick disease patients. Blood 1992, 80, 2081–2087. [Google Scholar] [PubMed]
- Ziegler, R.J.; Brown, C.; Barbon, C.M.; D’Angona, A.M.; Schuchman, E.H.; Andrews, L.; Thurberg, B.L.; McPherson, J.M.; Karey, K.P.; Cheng, S.H. Pulmonary delivery of recombinant acid sphingomyelinase improves clearance of lysosomal sphingomyelin from the lungs of a murine model of Niemann-Pick disease. Mol. Genet. Metab. 2009, 97, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Müssig, K.; Harzer, K.; Mayrhofer, H.; Krägeloh-Mann, I.; Häring, H.U.; Machicao, F. Clinical findings in Niemann-Pick disease type B. Int. Med. J. 2006, 36, 135–136. [Google Scholar] [CrossRef]
- Simonaro, C.M.; Desnick, R.J.; McGovern, M.M.; Wasserstein, M.P.; Schuchman, E.H. The demographics and distribution of type B Niemann-Pick disease: Novel mutations lead to new genotype/phenotype correlations. Am. J. Hum. Genet. 2002, 71, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Gluck, I.; Zeigler, M.; Bargal, R.; Schiff, E.; Bach, G. Niemann Pick disease type A in Israeli Arabs: 677delT, a common novel single mutation. Hum. Mutat. 1999, 12, 136–136. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manshadi, M.D.; Kamalidehghan, B.; Keshavarzi, F.; Aryani, O.; Dadgar, S.; Arastehkani, A.; Tondar, M.; Ahmadipour, F.; Meng, G.Y.; Houshmand, M. Four Novel p.N385K, p.V36A, c.1033–1034insT and c.1417–1418delCT Mutations in the Sphingomyelin Phosphodiesterase 1 (SMPD1) Gene in Patients with Types A and B Niemann-Pick Disease (NPD). Int. J. Mol. Sci. 2015, 16, 6668-6676. https://doi.org/10.3390/ijms16046668
Manshadi MD, Kamalidehghan B, Keshavarzi F, Aryani O, Dadgar S, Arastehkani A, Tondar M, Ahmadipour F, Meng GY, Houshmand M. Four Novel p.N385K, p.V36A, c.1033–1034insT and c.1417–1418delCT Mutations in the Sphingomyelin Phosphodiesterase 1 (SMPD1) Gene in Patients with Types A and B Niemann-Pick Disease (NPD). International Journal of Molecular Sciences. 2015; 16(4):6668-6676. https://doi.org/10.3390/ijms16046668
Chicago/Turabian StyleManshadi, Masoumeh Dehghan, Behnam Kamalidehghan, Fatemeh Keshavarzi, Omid Aryani, Sepideh Dadgar, Ahoora Arastehkani, Mahdi Tondar, Fatemeh Ahmadipour, Goh Yong Meng, and Massoud Houshmand. 2015. "Four Novel p.N385K, p.V36A, c.1033–1034insT and c.1417–1418delCT Mutations in the Sphingomyelin Phosphodiesterase 1 (SMPD1) Gene in Patients with Types A and B Niemann-Pick Disease (NPD)" International Journal of Molecular Sciences 16, no. 4: 6668-6676. https://doi.org/10.3390/ijms16046668