
miR-223 Inhibits Lipid Deposition and Inflammation by Suppressing Toll-Like Receptor 4 Signaling in Macrophages

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
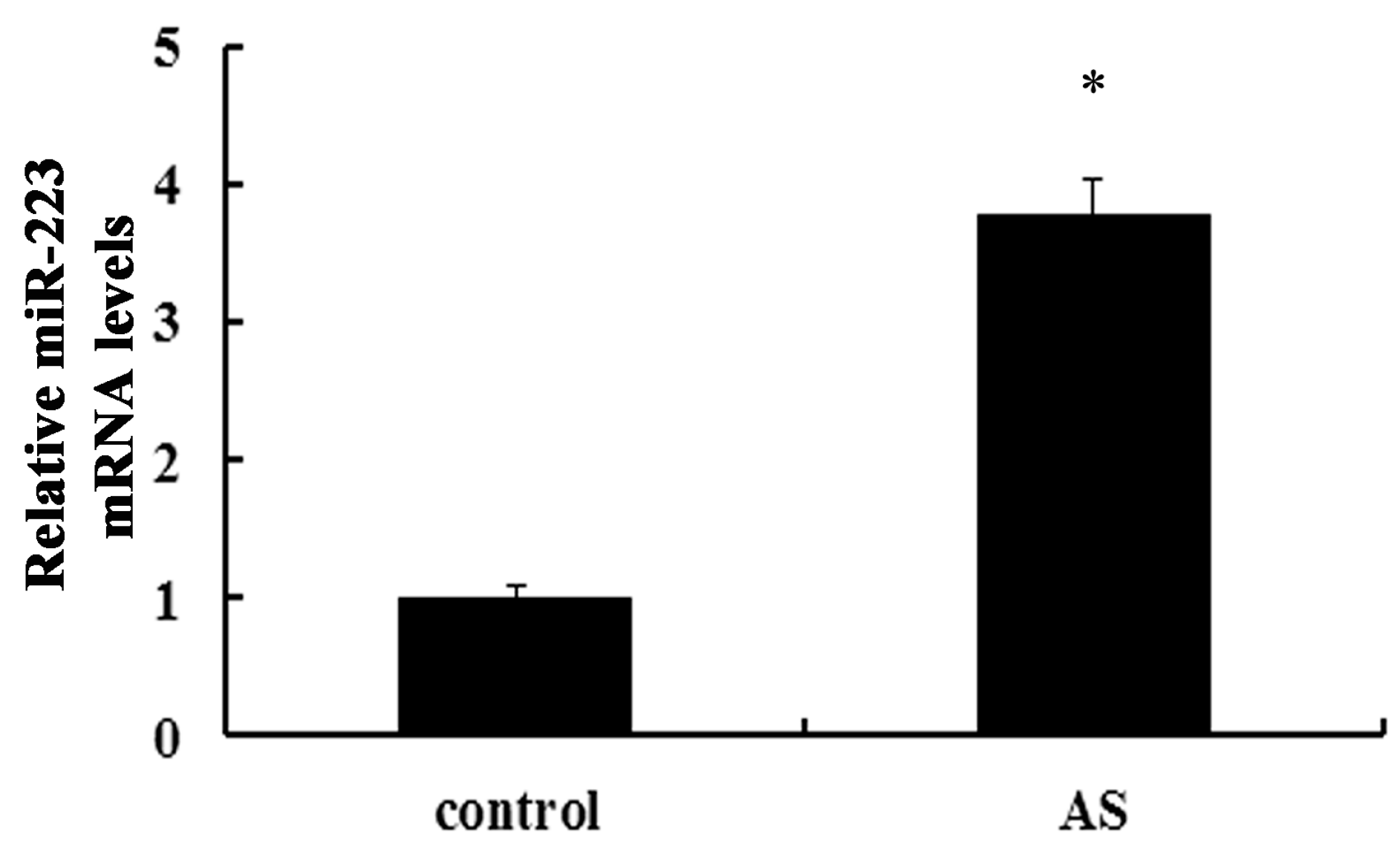
2.1. Expression of miR-223 Is Up-Regulated in Atherosclerotic Lesions in ApoE−/− Mice

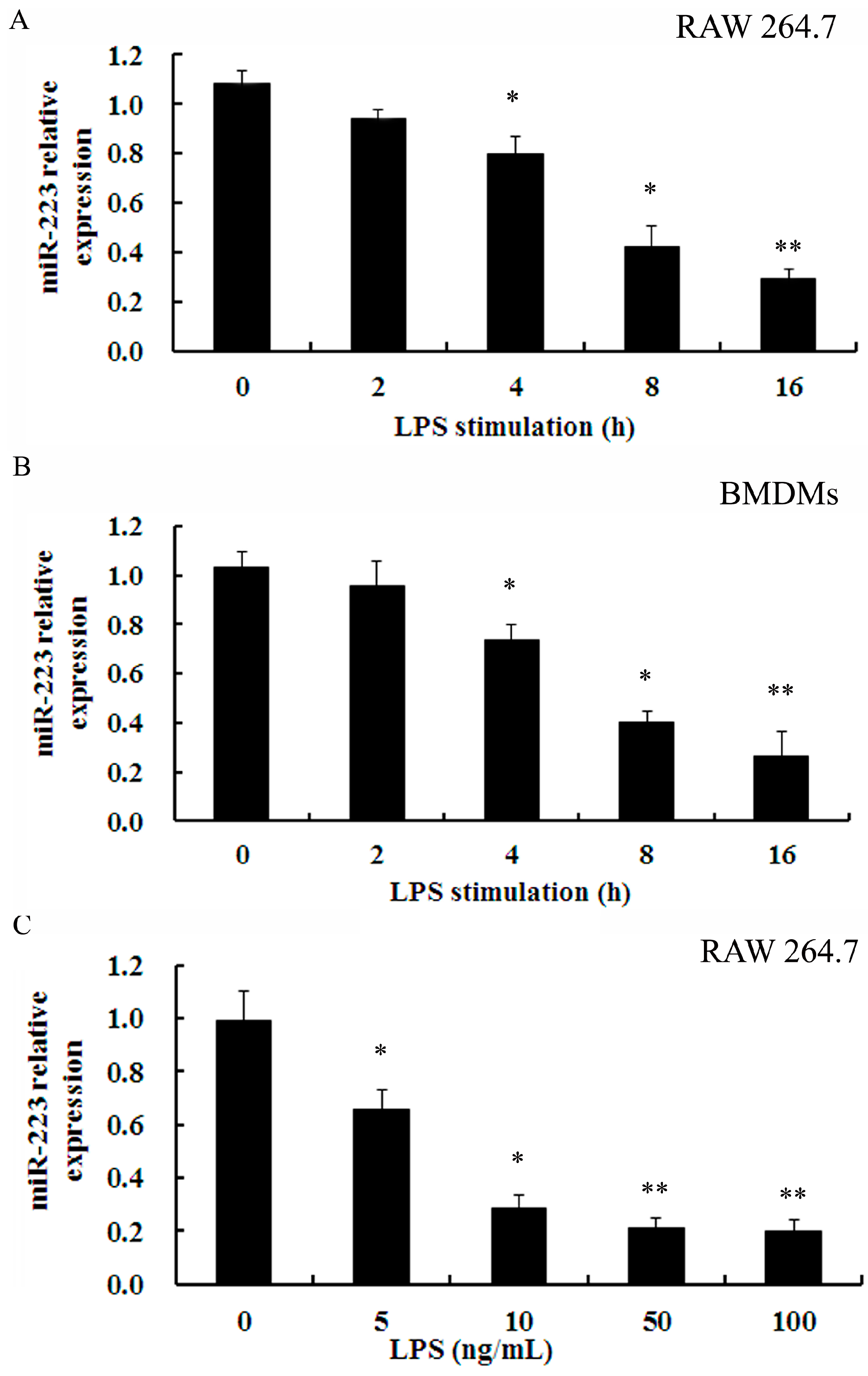
2.2. Down-Regulation of miR-223 Was Confirmed in Macrophages Stimulated with TLR Agonists

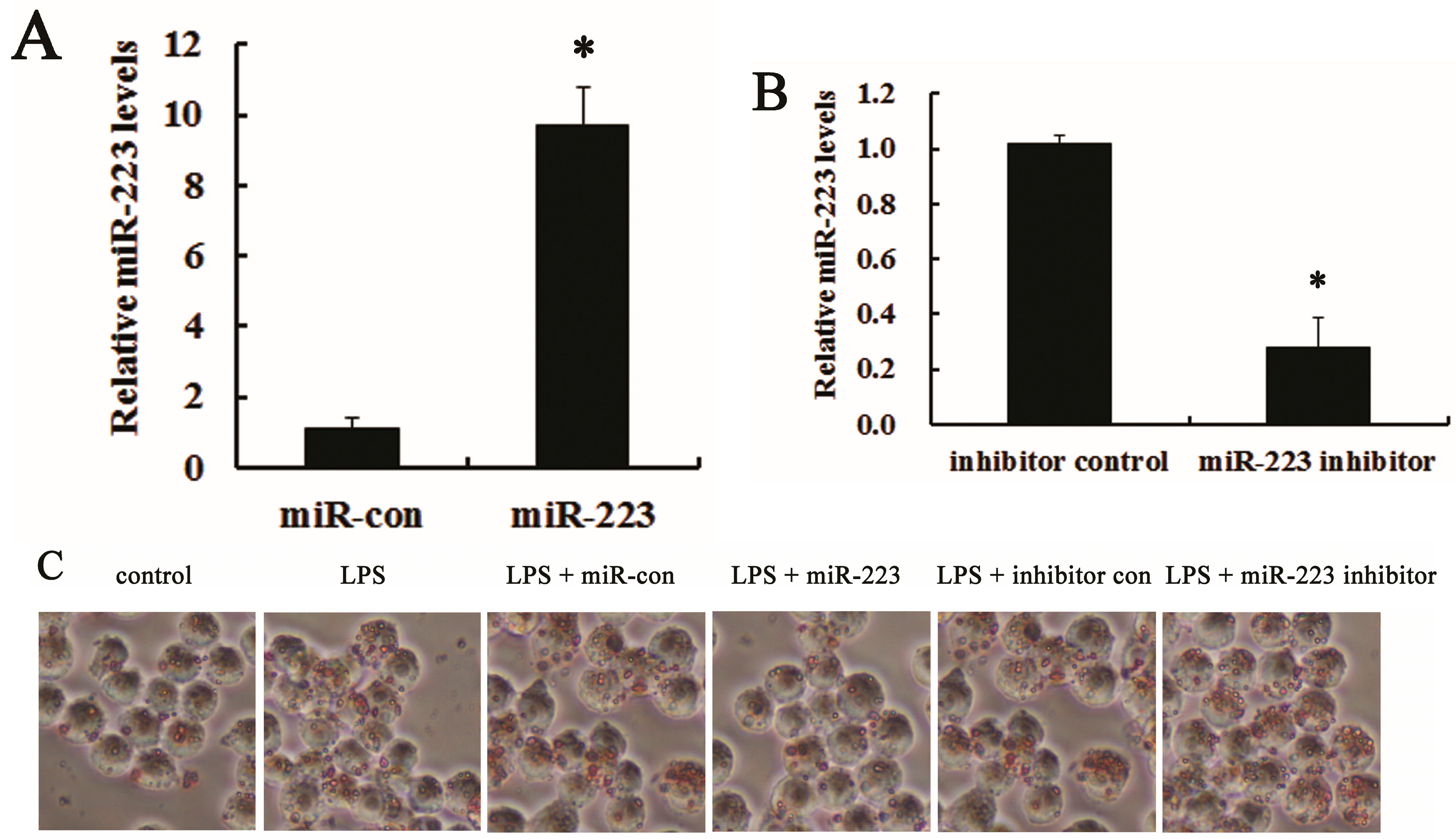
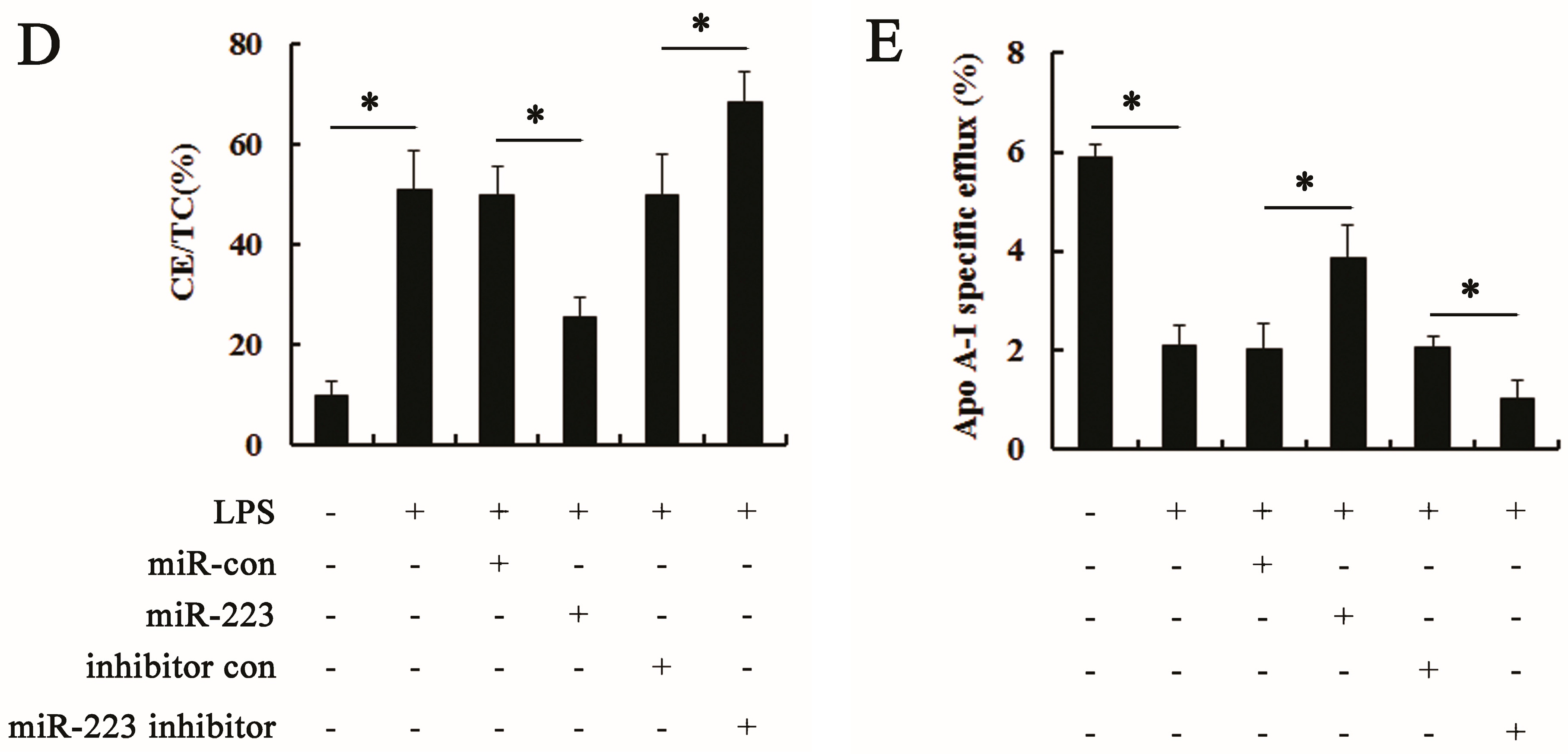
2.3. miR-223 Attenuated LPS-Triggered Foam Cell Formation


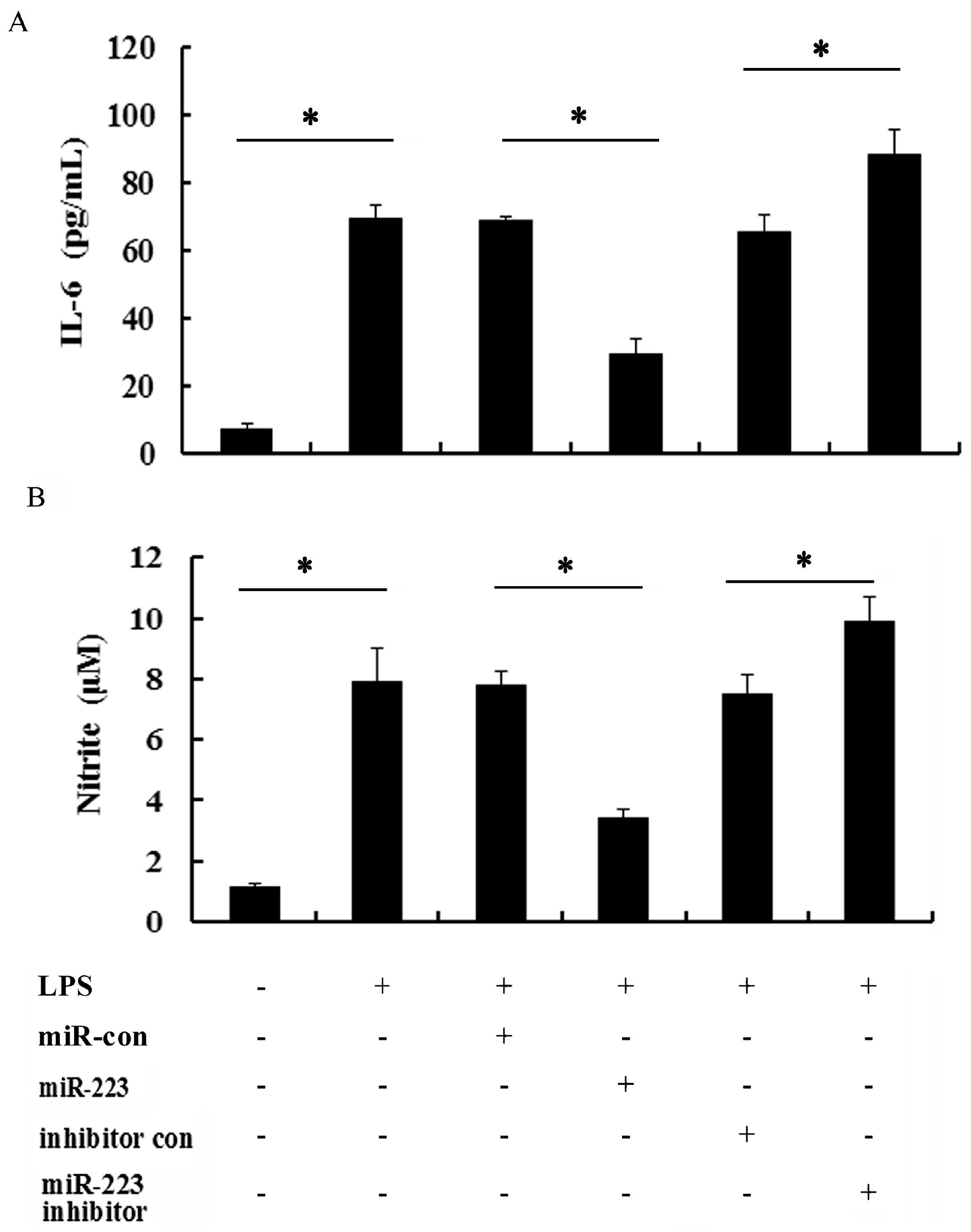
2.4. miR-223 Negatively Regulated Inflammatory Response to LPS in TLR-Triggered Macrophages

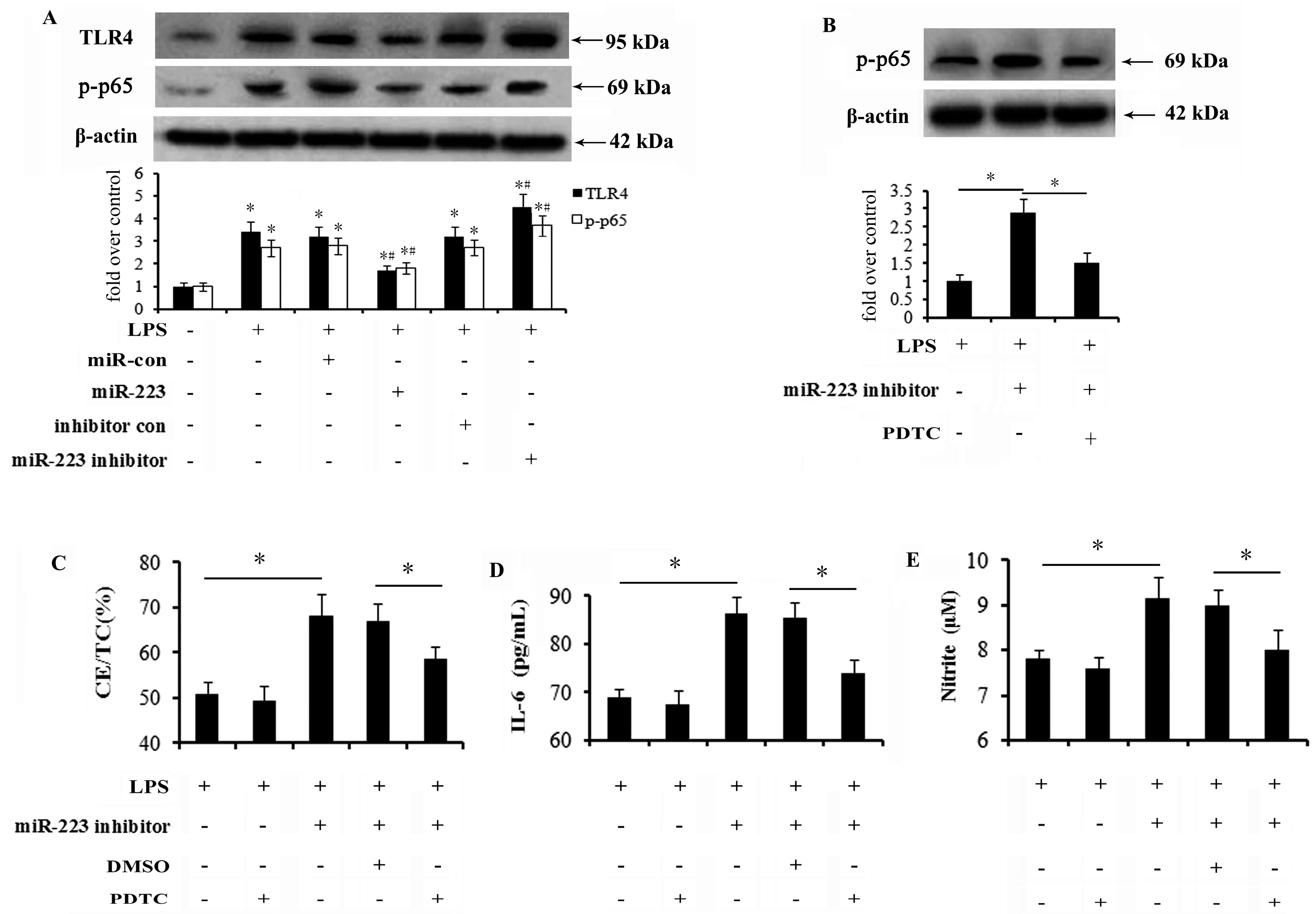
2.5. Activation of the TLR4-NF-κB Pathway Is Abrogated by miR-223 Mimics and Responsible for the Inhibitory Effect of miR-223 on Lipid Accumulation and Inflammatory Response

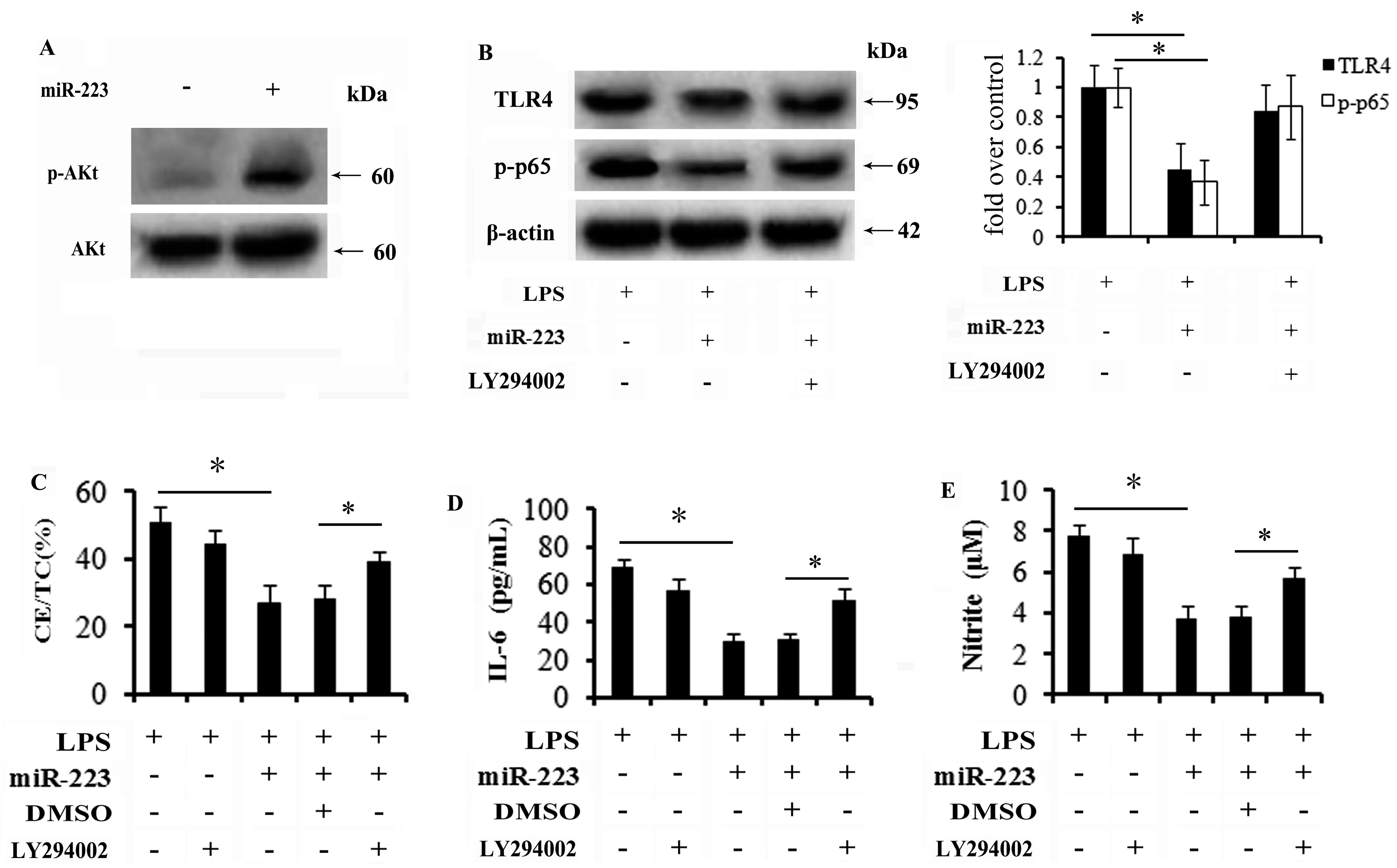
2.6. miR-223 Mitigates TLR4 Signaling Activation through the PI3K/AKT Pathway

3. Discussion
4. Experimental Section
4.1. Reagents and Antibodies
4.2. Animal Model of Atherosclerosis
4.3. Macrophage Cell Culture and Treatment
4.4. Oligonucleotide Transfection
4.5. RNA Extraction and Quantitative RT-PCR
4.6. Oil Red O Staining
4.7. Lipid Assay by High-Performance Liquid Chromatography (HPLC)
4.8. Cholesterol Efflux Analysis
4.9. Measurement of IL-6 Cytokine Production
4.10. Determination of NO Concentration
4.11. Western Blotting Assay
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Di Tullio, M.R.; Russo, C.; Jin, Z.; Sacco, R.L.; Mohr, J.; Homma, S. Aortic arch plaques and risk of recurrent stroke and death. Circulation 2009, 119, 2376–2382. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kim, R.Y.; Imhof, I.; Honbo, N.; Luk, F.S.; Li, K.; Kumar, N.; Zhu, B.-Q.; Eberlé, D.; Ching, D. The immunosuppressant FTY720 prolongs survival in a mouse model of diet-induced coronary atherosclerosis and myocardial infarction. J. Cardiovasc. Pharmacol. 2014, 63, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Graceffa, A. Preclinical atherosclerosis, metabolic syndrome and risk of cardiovascular events. Int. J. Sci. 2014, 3, 33–42. [Google Scholar]
- Yahagi, K.; Davis, H.R.; Arbustini, E.; Virmani, R. Sex differences in coronary artery disease: Pathological observations. Atherosclerosis 2015, 239, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Raul, C.M.; Antonio, C.A.L. Development of anti-atherosclerosis therapy based on the inflammatory and proliferative aspects of the disease. Curr. Pharm. Des. 2015, 21, 1196–1204. [Google Scholar]
- Tian, G.-P.; Tang, Y.-Y.; He, P.-P.; Lv, Y.-C.; Ouyang, X.-P.; Zhao, G.-J.; Tang, S.-L.; Wu, J.-F.; Wang, J.-L.; Peng, J. The effects of miR-467b on lipoprotein lipase (LPL) expression, pro-inflammatory cytokine, lipid levels and atherosclerotic lesions in apolipoprotein E knockout mice. Biochem. Biophys. Res. Commun. 2014, 443, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Yuan, Y.; Wang, C.; Feng, J.; Yuan, Z.; Zhang, X.; Sui, W.; Hu, P.; Zheng, P.; Ye, J. Autophagy involved in lipopolysaccharide-induced foam cell formation is mediated by adipose differentiation-related protein. Lipids Health Dis. 2014, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Murphy, A.J.; Wang, M.; Pagler, T.A.; Vengrenyuk, Y.; Kappus, M.S.; Gorman, D.J.; Nagareddy, P.R.; Zhu, X.; Abramowicz, S. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ. Res. 2013, 112, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Mullick, A.E.; Tobias, P.S.; Curtiss, L.K. Toll-like receptors and atherosclerosis. Immunol. Res. 2006, 34, 193–209. [Google Scholar] [CrossRef]
- Pasterkamp, G.; van Keulen, J.; de Kleijn, D. Role of Toll-like receptor 4 in the initiation and progression of atherosclerotic disease. Eur. J. Clin. Investig. 2004, 34, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Higashimori, M.; Tatro, J.B.; Moore, K.J.; Mendelsohn, M.E.; Galper, J.B.; Beasley, D. Role of Toll-Like receptor 4 in intimal foam cell accumulation in apolipoprotein E-Deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xie, Y.; Zhou, H.; Xu, Y.; Liu, J.; Xie, H.; Yan, J. Involvement of TLR4 in oxidized LDL/β2GPI/Anti-β2GPI-induced transformation of macrophages to foam cells. J. Atheroscler. Thromb. 2014, 21, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Subramanian, S.; Montes, V.N.; Goodspeed, L.; Wang, S.; Han, C.; Teresa, A.S.; Kim, J.; O’Brien, K.D.; Chait, A. Toll-like receptor 4 deficiency decreases atherosclerosis but does not protect against inflammation in obese low-density lipoprotein receptor—Deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Katsargyris, A.; Klonaris, C.; Tsiodras, S.; Bastounis, E.; Giannopoulos, A.; Theocharis, S. Statin treatment is associated with reduced toll-like receptor 4 immunohistochemical expression on carotid atherosclerotic plaques: A novel effect of statins. Vascular 2011, 19, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Ayyappan, J.P.; Abraham, A. Targeting TLR4/MAPKs signaling pathway: A better option for therapeutic inhibition of atherosclerosis. World J. Immunol. 2014, 4, 116–121. [Google Scholar] [CrossRef]
- Dangwal, S.; Thum, T. microRNA therapeutics in cardiovascular disease models. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.S.A.; Xia, K.; Li, F.; Deng, X.; Salma, U.; Deng, H.; Liu, W.; Yang, T.-L.; Peng, J. Circulating miR-765 and miR-149: Potential noninvasive diagnostic biomarkers for geriatric coronary artery disease patients. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ni, Q.; Tan, L.; Kong, L.; Lu, Y.; Xu, X. The microRNA-146a/b attenuates acute small-for-size liver graft injury in rats. Liver Int. 2015, 35, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Friggeri, A.; Yang, Y.; Park, Y.J.; Tsuruta, Y.; Abraham, E. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc. Natl. Acad. Sci. USA 2009, 106, 15819–15824. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Yin, F.; He, F.; Omran, A.; Li, L.; Wu, T.; Wang, Y.; Peng, J. The effect of miR-132, miR-146a, and miR-155 on MRP8/TLR4-induced astrocyte-related inflammation. J. Mol. Neurosci. 2015, 57, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, J.; Wang, H.; Shi, J.; Wu, K.; Liu, S.; Liu, Y.; Wu, J. HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. PLoS Pathog. 2013, 9, e1003248. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed]
- He, J.; He, Y.; Chen, D.; Yu, B.; Mao, X.; Zheng, P.; Yu, J.; Huang, Z.; Luo, J. miR-628, a microRNA that is induced by Toll-like receptor stimulation, regulates porcine innate immune responses. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Fordham, J.B.; Naqvi, A.R.; Nares, S. Regulation of miR-24, miR-30b, and miR-142-3p during macrophage and dendritic cell differentiation potentiates innate immunity. J. Leukoc. Biol. 2015, 98, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; Gerlic, M.; O’Neill, L.A.; Masters, S.L. miR-223: Infection, inflammation and cancer. J. Intern. Med. 2013, 274, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Ferland-McCollough, D.; Ozanne, S.; Siddle, K.; Willis, A.; Bushell, M. The involvement of microRNAs in Type2 diabetes. Biochem. Soc. Trans. 2010, 38, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Paraskevi, A.; Theodoropoulos, G.; Papaconstantinou, I.; Mantzaris, G.; Nikiteas, N.; Gazouli, M. Circulating microRNA in inflammatory bowel disease. J. Crohn’s Colitis 2012, 6, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Pinatel, E.M.; Orso, F.; Penna, E.; Cimino, D.; Elia, A.R.; Circosta, P.; Dentelli, P.; Brizzi, M.F.; Provero, P.; Taverna, D. miR-223 is a coordinator of breast cancer progression as revealed by bioinformatics predictions. PLoS ONE 2014, 9, e84859. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, H.; Liu, Y.; Song, Y.; Lai, L.; Han, Q.; Cao, X.; Wang, Q. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1β production in macrophages by targeting STAT3. PLoS ONE 2012, 7, e42971. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, R.; Jiang, J.; Yang, B.; Cao, Z.; Cheng, X. miR-223 is upregulated in monocytes from patients with tuberculosis and regulates function of monocyte-derived macrophages. Mol. Immunol. 2015, 67, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Hruska, K.A. MicroRNA-223 is a key factor in osteoclast differentiation. J. Cell. Biochem. 2007, 101, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; de Marchis, M.L.; Nervi, C.; Bozzoni, I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPα regulates human granulopoiesis. Cell 2005, 123, 819–831. [Google Scholar] [PubMed]
- Taibi, F.; Metzinger-Le Meuth, V.; M’Baya-Moutoula, E.; Djelouat, M.; Louvet, L.; Bugnicourt, J.M.; Poirot, S.; Bengrine, A.; Chillon, J.M.; Massy, Z.A.; et al. Possible involvement of microRNAs in vascular damage in experimental chronic kidney disease. Biochim. Biophys. Acta 2014, 1842, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Zeller, I.; Srivastava, S. Macrophage functions in atherosclerosis. Circ. Res. 2014, 115, e83–e85. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Calippe, B.; Douin-Echinard, V.; Laffargue, M.; Laurell, H.; Rana-Poussine, V.; Pipy, B.; Guery, J.C.; Bayard, F.; Arnal, J.F.; Gourdy, P. Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: involvement of the phosphatidylinositol 3-kinase pathway. J. Immunol. 2008, 180, 7980–7988. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wei, Z.; Ding, H.; Wang, Q.; Zhou, Z.; Zheng, S.; Zhang, Y.; Hou, D.; Liu, Y.; Zen, K.; et al. MicroRNA-19b/221/222 induces endothelial cell dysfunction via suppression of PGC-1α in the progression of atherosclerosis. Atherosclerosis 2015, 241, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, M.; Yavuzer, S.; Avci, B.K.; Yuruyen, M.; Yavuzer, H.; Dikici, S.A.; Karatas, O.F.; Ozen, M.; Uzun, H.; Ongen, Z. Circulating miR-21 and eNOS in subclinical atherosclerosis in patients with hypertension. Clin. Exp. Hypertens. 2015. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Cybulsky, M.I. ApoE attenuates atherosclerosis via miR-146a. Circ. Res. 2015, 117, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Koga, J.-I.; Aikawa, M. Molecular imaging of macrophages in atherosclerosis. In Cardiovascular Imaging; Springer: Cham, Switzerland, 2015; pp. 65–78. [Google Scholar]
- Ishikawa, Y.; Satoh, M.; Itoh, T.; Minami, Y.; Takahashi, Y.; Akamura, M. Local expression of Toll-like receptor 4 at the site of ruptured plaques in patients with acute myocardial infarction. Clin. Sci. 2008, 115, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Den Dekker, W.K.; Cheng, C.; Pasterkamp, G.; Duckers, H.J. Toll like receptor 4 in atherosclerosis and plaque destabilization. Atherosclerosis 2010, 209, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Serezani, C.H.; Lewis, C.; Jancar, S.; Peters-Golden, M. Leukotriene B4 amplifies NF-κB activation in mouse macrophages by reducing SOCS1 inhibition of MyD88 expression. J. Clin. Investig. 2011, 121, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Wang, Y.; Zhang, M.; Zhang, P.F.; Ding, S.F.; Liu, C.X.; Liu, X.L.; Zhao, Y.X.; Zhang, Y. Atherosclerotic plaque disruption induced by stress and lipopolysaccharide in apolipoprotein E knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1598–H1606. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-Q.; Liu, B.-J.; Wu, J.-F.; Xu, Y.-C.; Duan, X.-H.; Cao, Y.-X.; Dong, J.-C. Icariin attenuates LPS-induced acute inflammatory responses: Involvement of PI3K/Akt and NF-κB signaling pathway. Eur. J. Pharmacol. 2010, 642, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kamo, N.; Ke, B.; Busuttil, R.W.; Kupiec-Weglinski, J.W. PTEN-mediated Akt/β-Catenin/foxo1 signaling regulates innate immune responses in mouse liver ischemia/reperfusion injury. Hepatology 2013, 57, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J. IGF-I exerts an anti-inflammatory effect on skeletal muscle cells through down-regulation of TLR4 signaling. Immune Netw. 2011, 11, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.-Y.; Song, Y.-A.; Park, Y.-L.; Myung, E.; Chung, C.-Y.; Park, K.-J.; Cho, S.-B.; Lee, W.-S.; Kim, H.-S.; Rew, J.-S. Epigallocatechin-3-gallate inhibits LPS-induced NF-κB and MAPK signaling pathways in bone marrow-derived macrophages. Gut Liver 2012, 6, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-H.; Xu, C.-F.; Li, Y.-M. Association of MicroRNA-223 expression with hepatic ischemia/reperfusion injury in mice. Dig. Dis. Sci. 2009, 54, 2362–2366. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Bai, X.; Song, Q.; Fan, F.; Hu, Z.; Cheng, G.; Zhang, Y. miR-223 Inhibits Lipid Deposition and Inflammation by Suppressing Toll-Like Receptor 4 Signaling in Macrophages. Int. J. Mol. Sci. 2015, 16, 24965-24982. https://doi.org/10.3390/ijms161024965
Wang J, Bai X, Song Q, Fan F, Hu Z, Cheng G, Zhang Y. miR-223 Inhibits Lipid Deposition and Inflammation by Suppressing Toll-Like Receptor 4 Signaling in Macrophages. International Journal of Molecular Sciences. 2015; 16(10):24965-24982. https://doi.org/10.3390/ijms161024965
Chicago/Turabian StyleWang, Jun, Xiaojun Bai, Qiang Song, Fenling Fan, Zhi Hu, Gesheng Cheng, and Yushun Zhang. 2015. "miR-223 Inhibits Lipid Deposition and Inflammation by Suppressing Toll-Like Receptor 4 Signaling in Macrophages" International Journal of Molecular Sciences 16, no. 10: 24965-24982. https://doi.org/10.3390/ijms161024965