Cardiotonic Steroids and the Sodium Trade Balance: New Insights into Trade-Off Mechanisms Mediated by the Na+/K+-ATPase

 , , and
, , and
Abstract
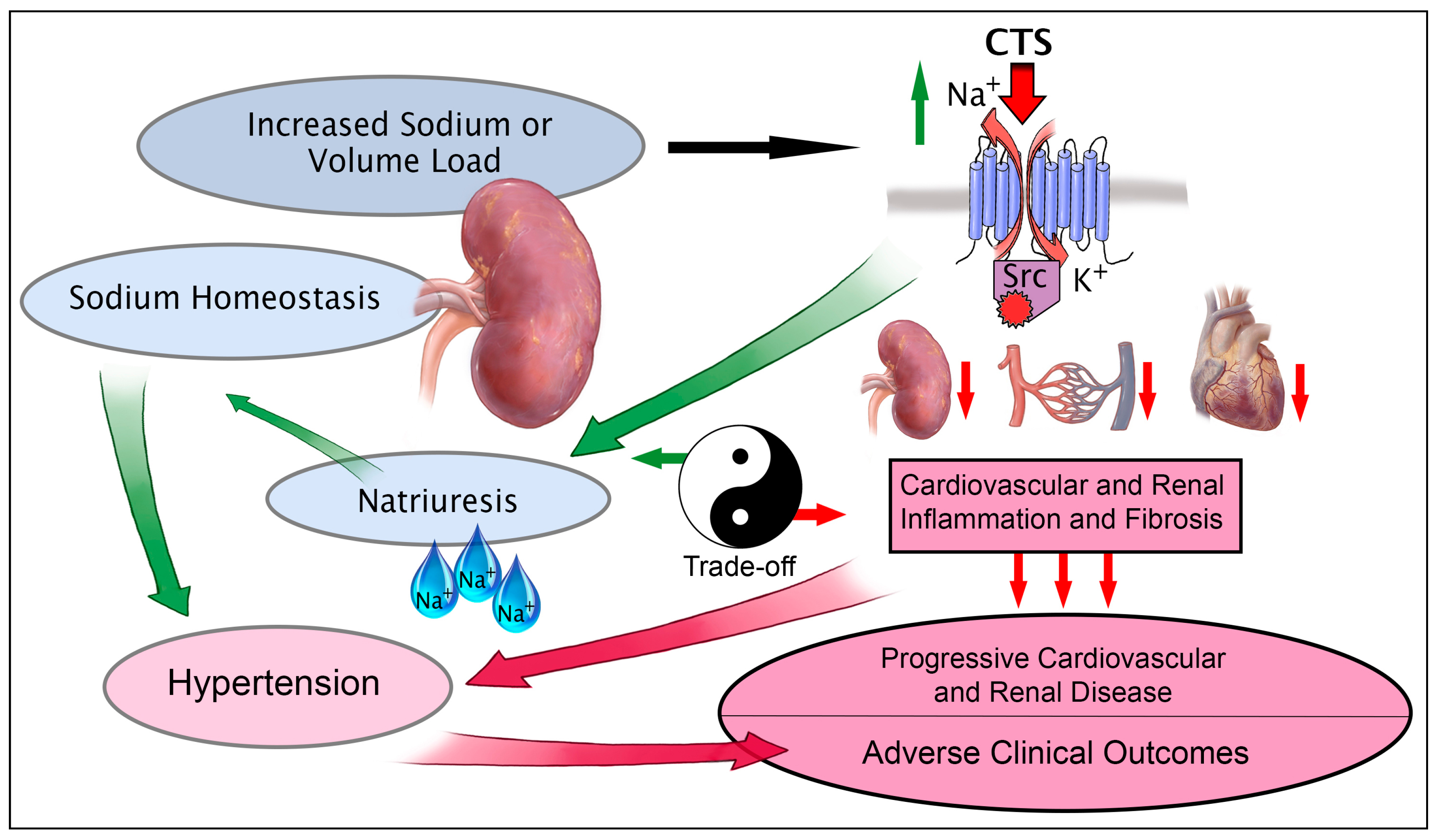
:1. Introduction
2. Structure and Function of the Na+/K+-ATPase (NKA)
3. Cardiotonic Steroids: NKA Ligands Brokering the Sodium Trade Balance
4. CTS (Cardiotonic Steroids), NKA, and Fibrosis
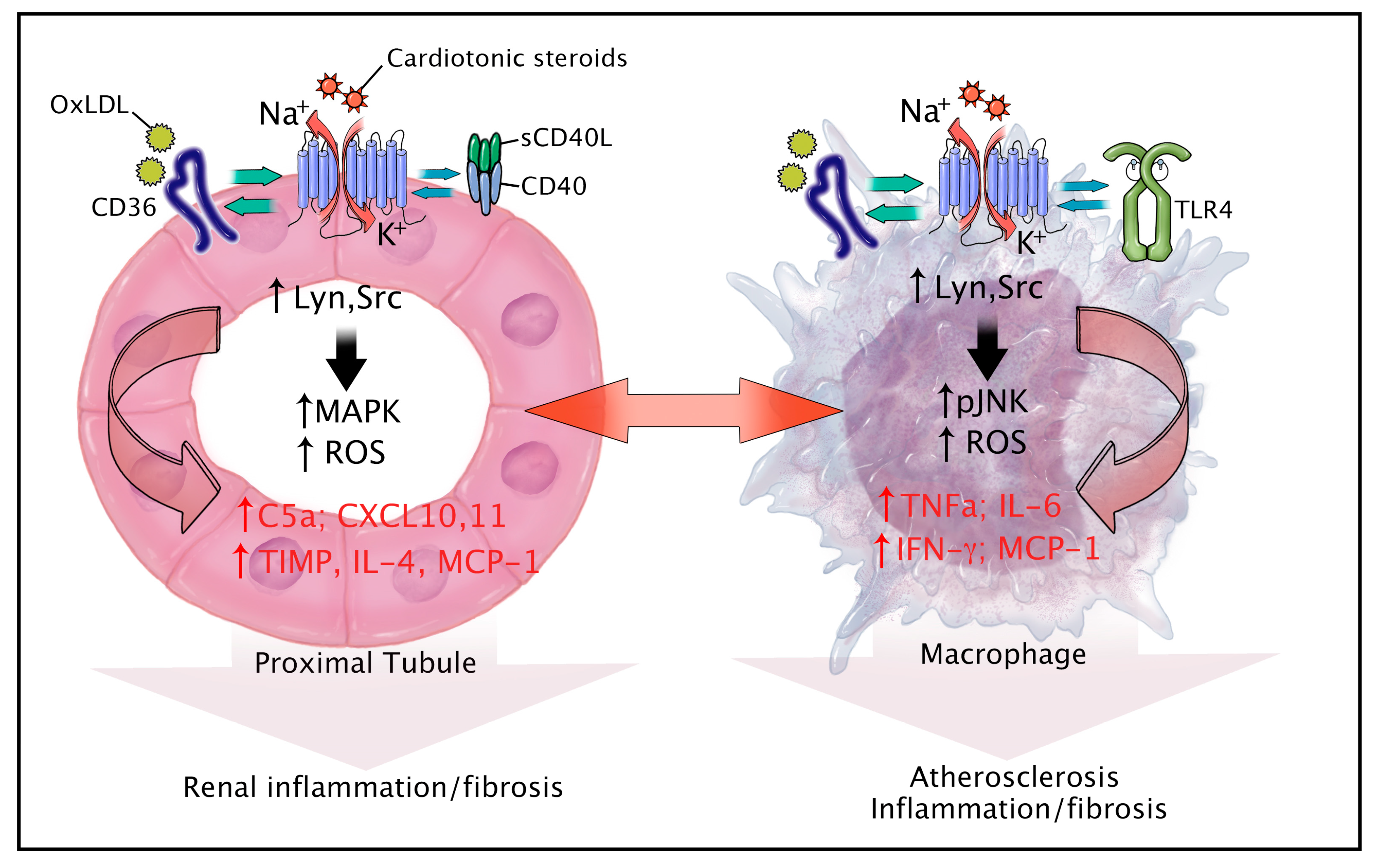
5. CTS, NKA, and Inflammation
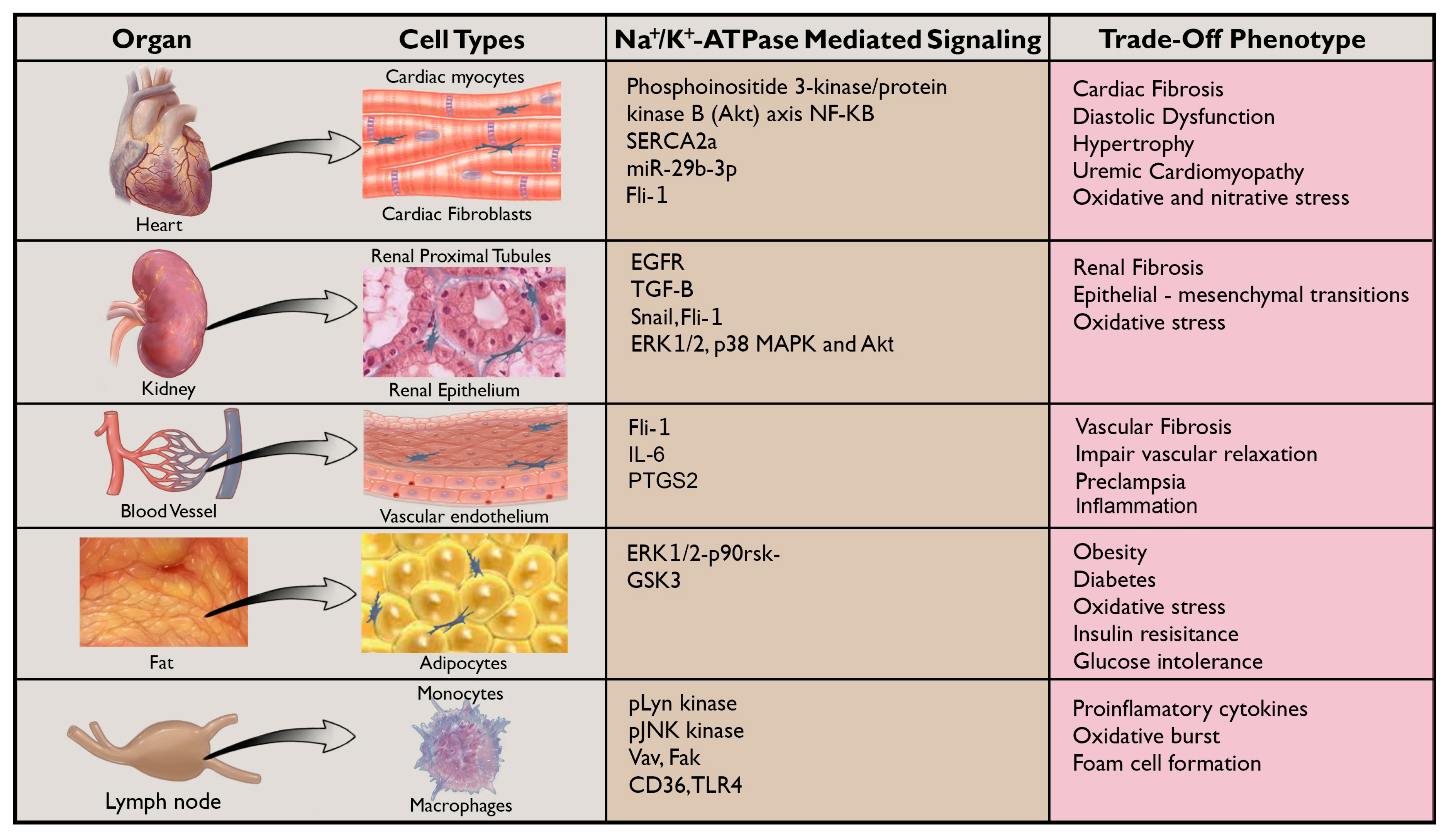
6. New Horizons for NKA Signaling: Aging, Obesity, Diabetes, and Atherosclerosis
7. Novel Signaling Partners of the NKA in Inflammation and Fibrosis
8. Summary
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BP | Blood pressure |
| CKD | Chronic kidney disease |
| CTGF | Connective tissue growth factor |
| CTS | Cardiotonic steroid |
| HDL | High density lipoprotein |
| HF | Heart Failure |
| MBG | Marinobufagenin |
| NKA | Na+/K+-ATPase |
| PNx | Partial (5/6th) Nephrectomy |
| ROS | Reactive oxygen species |
| Src | Sarcoma viral oncogene kinase |
| TCB | Telocinobufagin |
| UUO | Unilateral ureteral obstruction |
References
- Xie, Z. Molecular mechanisms of Na/K-ATPase-mediated signal transduction. Ann. N. Y. Acad. Sci. 2003, 986, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Kometiani, P.; Li, J.; Gnudi, L.; Kahn, B.B.; Askari, A.; Xie, Z. Multiple Signal Transduction Pathways Link Na+/K+-ATPase to growth-related genes in cardiac myocytes the roles of Ras and mitogen-ACTIVATED protein kinases. J. Biol. Chem. 1998, 273, 15249–15256. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Askari, A. Na/K-ATPase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Xie, J.; Tian, J. Reducing Cardiac Fibrosis: Na/K-ATPase Signaling Complex as a Novel Target. Cardiovasc. Pharmacol. Open Access 2017, 6. [Google Scholar] [CrossRef]
- Liu, J.; Lilly, M.N.; Shapiro, J.I. Targeting Na/K-ATPase Signaling: A New Approach to Control Oxidative Stress. Curr. Pharm. Des. 2018, 24, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, J.; Cui, X.; Wang, J.; Cai, L.; Pierre, S.V.; Xie, Z. Na/K-ATPase α1 Isoform as a Critical Signal Integrator in Embryonic Development. FASEB J. 2017, 31, 1007.52. [Google Scholar]
- Buckalew, V.M. Endogenous digitalis-like factors: An overview of the history. Front. Endocrinol. 2015, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Srikanthan, K.; Shapiro, J.I.; Sodhi, K. The role of Na/K-ATPase signaling in oxidative stress related to obesity and cardiovascular disease. Molecules 2016, 21, 1172. [Google Scholar] [CrossRef]
- Sweadner, K.J.; Arystarkhova, E.; Penniston, J.; Cook, J.; Swoboda, K.; Brashear, A.; Ozelius, L. Structure-Phenotype Relationships in ATP1A3 (Na, K-ATPase) Diseases (S17. 007). 2017. Available online: http://n.neurology.org/content/90/15_Supplement/P1.311 (accessed on 24 August 2018).
- Bricker, N.S. On the pathogenesis of the uremic state: An exposition of the trade-off hypothesis. N. Engl. J. Med. 1972, 286, 1093–1099. [Google Scholar] [PubMed]
- Liu, C.; Lou, M.; Ding, Y.; Wang, Y.; Huang, Y.; Shao, D.; Chen, W. Ouabain-induced apoptosis and inhibition of viability of tubulointerstitial cells by regulating NKA/pSrc/pERK/pAkt/pS6k/caspase 3 may contribute to lupus nephritis development. Int. J. Clin. Exp. Pathol. 2018, 11, 2305–2313. [Google Scholar]
- Khalaf, F.K.; Mohamed, A.; Kleinhenz, A.; Crawford, E.; Tian, J.; Xie, Z.; Malhotra, D.; Haller, S.; Kennedy, D. Cardiotonic steroid signaling through Na/K-atpase-A-1 and src kinase enhance functional interactions between immune cells and endo/epithelial cells. J. Investig. Med. 2018, 66, 859. [Google Scholar]
- Xie, J.X.; Shapiro, A.P.; Shapiro, J.I. The trade-off between dietary salt and cardiovascular disease; a role for Na/K-ATPase signaling? Front. Endocrinol. 2014, 5, 97. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Dostanic-Larson, I.; Van Huysse, J.W.; Lorenz, J.N.; Lingrel, J.B. The highly conserved cardiac glycoside binding site of Na, K-ATPase plays a role in blood pressure regulation. Proc. Natl. Acad. Sci. USA 2005, 102, 15845–15850. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, W.; Yang, M.; Xin, G.; Cui, W.; Xie, Z.; Silverstein, R.L. Cardiotonic Steroids Stimulate Macrophage Inflammatory Responses Through a Pathway Involving CD36, TLR4, and Na/K-ATPase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.; Hill, M.; Cooper, C.; Shapiro, J.; Tian, J. MicroRNA 29b and Cardiotonic Steroid-Induced Cardiac Fibrosis in Adult Cardiac Fibroblasts. FASEB J. 2015, 29, 814–815. [Google Scholar]
- Cavalcante-Silva, L.H.A.; Lima, É.D.A.; Carvalho, D.M.; Sales-Neto, J.M.; Alves, A.K.D.A.; Galvão, J.G.F.M.; Silva, J.S.d.F.d.; Mascarenhas, S.R. Much More than a Cardiotonic Steroid: Modulation of Inflammation by Ouabain. Front. Physiol. 2018, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Motoyama, K.; Cornelius, F.; Vilsen, B.; Toyoshima, C. X-Ray Crystallographic Study of Na, K-ATPase in Complex with Cardiotonic Steroids. Biophys. J. 2015, 108, 197a. [Google Scholar] [CrossRef]
- Xie, Z. Ouabain interaction with cardiac Na/K-ATPase reveals that the enzyme can act as a pump and as a signal transducer. Cell. Mol. Biol. 2001, 47, 383–390. [Google Scholar] [PubMed]
- Aperia, A.; Akkuratov, E.E.; Fontana, J.M.; Brismar, H. Na+-K+-ATPase, a new class of plasma membrane receptors. Am. J. Physiol.-Cell Physiol. 2016, 310, C491–C495. [Google Scholar] [CrossRef] [PubMed]
- Morth, J.P.; Pedersen, B.P.; Buch-Pedersen, M.J.; Andersen, J.P.; Vilsen, B.; Palmgren, M.G.; Nissen, P. A structural overview of the plasma membrane Na+, K+-ATPase and H+-ATPase ion pumps. Nat. Rev. Mol. Cell Biol. 2011, 12, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, P.L. Structure, function and regulation of Na, K-ATPase in the kidney. Kidney Int. 1986, 29, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.X.; Zhang, S.; Cui, X.; Zhang, J.; Yu, H.; Khalaf, F.K.; Malhotra, D.; Kennedy, D.J.; Shapiro, J.I.; Tian, J. Na/K-ATPase/src complex mediates regulation of CD40 in renal parenchyma. Nephrol. Dial. Transplant. 2018, 33, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol.-Ren. Physiol. 1998, 275, F633–F650. [Google Scholar] [CrossRef]
- Adams, R.J.; Schwartz, A.; Grupp, G.; Grupp, I.; Lee, S.W.; Wallick, E.T.; Powell, T.; Twist, V.W.; Gathiram, P. High-affinity ouabain binding site and low-dose positive inotropic effect in rat myocardium. Nature 1982, 296, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Kutz, L.C.; Mukherji, S.; Marck, P.; Cui, X.; Heiny, J.A.; Blanco, G.; Pierre, S.V.; Xie, Z. Isoform-specific role of Na/K-ATPase α1 in skeletal muscle growth and performance. FASEB J. 2017, 31, 1007.50. [Google Scholar]
- Klimanova, E.A.; Petrushanko, I.Y.; Mitkevich, V.A.; Anashkina, A.A.; Orlov, S.N.; Makarov, A.A.; Lopina, O.D. Binding of ouabain and marinobufagenin leads to different structural changes in Na, K-ATPase and depends on the enzyme conformation. FEBS Lett. 2015, 589, 2668–2674. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Manunta, P. Endogenous cardiotonic steroids in kidney failure: A review and an hypothesis. Adv. Chron. Kidney Dis. 2015, 22, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, B.; Eleanor, E.B. Collection of Toad Venoms and Chemistry of the Toad Venom Steroids; Academic Press: London, UK, 1971. [Google Scholar]
- Bagrov, A.Y.; Fedorova, O.V.; Dmitrieva, R.I.; Howald, W.N.; Hunter, A.P.; Kuznetsova, E.A.; Shpen, V.M. Characterization of a urinary bufodienolide Na+, K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertension 1998, 31, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, L.V.; Raju, V.; El-Okdi, N.; Shidyak, A.; Kennedy, D.J.; Vetteth, S.; Giovannucci, D.R.; Bagrov, A.Y.; Fedorova, O.V.; Shapiro, J.I. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: Implication of epithelial-to-mesenchymal transition. Am. J. Physiol.-Ren. Physiol. 2009, 296, F922–F934. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.; Doris, P.; Bagrov, A. Endogenous marinobufagenin-like factor in acute plasma volume expansion. Clin. Exp. Hypertens. 1998, 20, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Gallice, P.M.; Kovacic, H.N.; Brunet, P.J.; Berland, Y.F.; Crevat, A.D. A non ouabain-like inhibitor of the sodium pump in uremic plasma ultrafiltrates and urine from healthy subjects. Clin. Chim. Acta 1998, 273, 149–160. [Google Scholar] [CrossRef]
- Gonick, H.; Ding, Y.; Vaziri, N.; Bagrov, A.; Fedorova, O. Simultaneous measurement of marinobufagenin, ouabain, and hypertension-associated protein in various disease states. Clin. Exp. Hypertens. 1998, 20, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.; Blaustein, M.; Bova, S.; DuCharme, D.; Harris, D.; Mandel, F.; Mathews, W.; Ludens, J. Identification and characterization of a ouabain-like compound from human plasma. Proc. Natl. Acad. Sci. USA 1991, 88, 6259–6263. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Ringel, R.; Schaeffer, J.; Levinson, P.D.; Hamilton, B.P.; Kowarski, A.A.; Blaustein, M.P. A circulating inhibitor of (Na+ K+) ATPase associated with essential hypertension. Nature 1982, 300, 650. [Google Scholar] [CrossRef] [PubMed]
- Harwood, S.; Mullen, A.M.; McMahon, A.C.; Dawnay, A. Plasma OLC is elevated in mild experimental uremia but is not associated with hypertension. Am. J. Hypertens. 2001, 14, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, D.J.; Vetteth, S.; Periyasamy, S.M.; Kanj, M.; Fedorova, L.; Khouri, S.; Kahaleh, M.B.; Xie, Z.; Malhotra, D.; Kolodkin, N.I. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006, 47, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, G.; Jia, J.; Miao, Y.; Gu, S.; Miao, P.; Shi, X.; Wang, Y.; Yu, C. Therapeutic monitoring of serum digoxin for patients with heart failure using a rapid LC-MS/MS method. Clin. Biochem. 2010, 43, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Stella, P.; Rivera, R.; Ciurlino, D.; Cusi, D.; Ferrandi, M.; Hamlyn, J.M.; Bianchi, G. Left ventricular mass, stroke volume, and ouabain-like factor in essential hypertension. Hypertension 1999, 34, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, S.M.; Chen, J.; Cooney, D.; Carter, P.; Omran, E.; Tian, J.; Priyadarshi, S.; Bagrov, A.; Fedorova, O.; Malhotra, D. Effects of uremic serum on isolated cardiac myocyte calcium cycling and contractile function. Kidney Int. 2001, 60, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, Y.; Dong, X.H.; Nishimura, N.; Masaki, H.; Yoshika, M.; Masuda, M.; Takahashi, H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin. Biochem. 2005, 38, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Pierdomenico, S.D.; Bucci, A.; Manunta, P.; Rivera, R.; Ferrandi, M.; Hamlyn, J.M.; Lapenna, D.; Cuccurullo, F.; Mezzetti, A. Endogenous ouabain and hemodynamic and left ventricular geometric patterns in essential hypertension. Am. J. Hypertens. 2001, 14, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, S.S.; Rogowski, A.C.; Weinberg, M.; Krichten, C.M.; Hamilton, B.P.; Hamlyn, J.M. Elevated concentrations of endogenous ouabain in patients with congestive heart failure. Circulation 1992, 86, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Stella, P.; Manunta, P.; Mallamaci, F.; Melandri, M.; Spotti, D.; Tripepi, G.; Hamlyn, J.M.; Malatino, L.S.; Bianchi, G.; Zoccali, C. Endogenous ouabain and cardiomyopathy in dialysis patients. J. Intern. Med. 2008, 263, 274–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Ishii, N.; Nambara, T. Occurrence of bufadienolides in the skin of Bufo viridis Laur. Chem. Pharm. Bull. 1986, 34, 3454–3457. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.K.; Anderson, R.C.; Henderson, F.G. Comparison of cardiac action of bufalin, cinobufotalin, and telocinobufagin with cinobufagin. Proc. Soc. Exp. Biol. Med. 1951, 76, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Touza, N.A.; Pocas, E.S.; Quintas, L.E.; Cunha-Filho, G.; Santos, M.L.; Noel, F. Inhibitory effect of combinations of digoxin and endogenous cardiotonic steroids on Na+/K+-ATPase activity in human kidney membrane preparation. Life Sci. 2011, 88, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Shrestha, K.; Sheehey, B.; Li, X.S.; Guggilam, A.; Wu, Y.; Finucan, M.; Gabi, A.; Medert, C.M.; Westfall, K. Elevated plasma marinobufagenin, an endogenous cardiotonic steroid, is associated with right ventricular dysfunction and nitrative stress in heart failure. Circ. Heart Fail. 2015, 8, 1068. [Google Scholar] [CrossRef] [PubMed]
- Tomaschitz, A.; Piecha, G.; Ritz, E.; Meinitzer, A.; Haas, J.; Pieske, B.; Wiecek, A.; Rus-Machan, J.; Toplak, H.; März, W. Marinobufagenin in essential hypertension and primary aldosteronism: A cardiotonic steroid with clinical and diagnostic implications. Clin. Exp. Hypertens. 2015, 37, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Blaustein, M.P. Endogenous ouabain: Recent advances and controversies. Hypertension 2016, 68, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Weber, M.E.; Guggilam, A.; Westfall, K.M.; Agatisa-Boyle, B.; Bucur, P.; Lingrel, J.B.; Tang, W.W. Telecinobufagin, a novel cardiotonic steroid, promotes myocardial and renal fibrosis via Na/K-ATPase profibrotic signalling pathways. Circulation 2014, 130. [Google Scholar] [CrossRef]
- Sodhi, K.; Nichols, A.; Mallick, A.; Klug, R.L.; Liu, J.; Wang, X.; Srikanthan, K.; Goguet-Rubio, P.; Nawab, A.; Pratt, R.; et al. The Na/K-ATPase Oxidant Amplification Loop Regulates Aging. Sci. Rep. 2018, 8, 9721. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.T.; Drummond, C.A.; Yan, Y.; Liu, J.; Tian, J.; Malhotra, D.; Shapiro, J.I. Passive immunization against marinobufagenin attenuates renal fibrosis and improves renal function in experimental renal disease. Am. J. Hypertens. 2013, 27, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Chen, Y.; Huang, W.; Viterna, J.; Liu, J.; Westfall, K.; Tian, J.; Bartlett, D.J.; Tang, W.W.; Xie, Z. CD36 and Na/K-ATPase-α1 form a proinflammatory signaling loop in kidney. Hypertension 2013, 61, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Fan, F.; Wang, Y.; Gonzalez-Fernandez, E.; Wang, C.; Yang, L.; Booz, G.W.; Roman, R.J. Therapeutic potential of microRNAs for the treatment of renal fibrosis and CKD. Physiol. Genom. 2017, 50, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y.; Nikolic-Paterson, D.J. Advances in Mechanisms of Renal Fibrosis. Front. Physiol. 2018, 9, 284. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.M.K.; Zhang, Y.Y.; Mak, T.S.K.; Tang, P.C.T.; Huang, X.R.; Lan, H.Y. TGF-β signalling in renal fibrosis: From Smads to non-coding RNAs. J. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Lau, W.L.; Jo, H.; Tsujino, K.; Gewin, L.; Reed, N.I.; Atakilit, A.; Nunes, A.C.F.; DeGrado, W.F.; Sheppard, D. Pharmacologic blockade of αvβ1 integrin ameliorates renal failure and fibrosis in vivo. J. Am. Soc. Nephrol. 2017, 28, 1998–2005. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, A.; Florquin, S.; Brok, M.; Claessen, N.; Butter, L.M.; Teske, G.J.; de Boer, O.J.; Vogl, T.; Leemans, J.C.; Dessing, M.C. S100A8/A9 promotes parenchymal damage and renal fibrosis in obstructive nephropathy. Clin. Exp. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Tang, P.M.-K.; Huang, X.-R.; Sun, S.-F.; You, Y.-K.; Xiao, J.; Lv, L.-L.; Xu, A.-P.; Lan, H.-Y. TGF-β Mediates Renal Fibrosis via the Smad3-Erbb4-IR Long Noncoding RNA Axis. Mol. Therapy 2018, 26, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Elkareh, J.; Kennedy, D.J.; Yashaswi, B.; Vetteth, S.; Shidyak, A.; Kim, E.G.; Smaili, S.; Periyasamy, S.M.; Hariri, I.M.; Fedorova, L. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 2007, 49, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Song, Y.; Wang, Y. pNaKtide ameliorates renal interstitial fibrosis through inhibition of sodium-potassium adenosine triphosphatase-mediated signaling pathways in unilateral ureteral obstruction mice. Nephrol. Dial. Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Elkareh, J.; Periyasamy, S.M.; Shidyak, A.; Vetteth, S.; Schroeder, J.; Raju, V.; Hariri, I.M.; El-Okdi, N.; Gupta, S.; Fedorova, L.; et al. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli-1: Implications for uremic cardiomyopathy. Am. J. Physiol. Ren. Physiol. 2009, 296, F1219–F1226. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Zernetkina, V.I.; Shilova, V.Y.; Grigorova, Y.N.; Juhasz, O.; Wei, W.; Marshall, C.A.; Lakatta, E.G.; Bagrov, A.Y. Synthesis of an Endogenous Steroidal Na Pump Inhibitor Marinobufagenin, Implicated in Human Cardiovascular Diseases, Is Initiated by CYP27A1 via Bile Acid Pathway. Circ. Cardiovasc. Genet. 2015, 8, 736–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishkaraeva-Yakovleva, V.V.; Fedorova, O.V.; Solodovnikova, N.G.; Frolova, E.V.; Bzhelyansky, A.M.; Emelianov, I.V.; Adair, C.D.; Zazerskaya, I.E.; Bagrov, A.Y. DigiFab Interacts with Endogenous Cardiotonic Steroids and Reverses Preeclampsia-Induced Na/K.-ATPase Inhibition. Reprod Sci. 2012, 19, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Adair, C.D.; Buckalew, V.M.; Graves, S.W.; Lam, G.K.; Johnson, D.D.; Saade, G.; Lewis, D.F.; Robinson, C.; Danoff, T.M.; Chauhan, N.; et al. Digoxin immune fab treatment for severe preeclampsia. Am. J. Perinatol. 2010, 27, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Tapilskaya, N.I.; Bzhelyansky, A.M.; Frolova, E.V.; Nikitina, E.R.; Reznik, V.A.; Kashkin, V.A.; Bagrov, A.Y. Interaction of Digibind with endogenous cardiotonic steroids from preeclamptic placentae. J. Hypertens. 2010, 28, 361–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitina, E.R.; Mikhailov, A.V.; Nikandrova, E.S.; Frolova, E.V.; Fadeev, A.V.; Shman, V.V.; Shilova, V.Y.; Tapilskaya, N.I.; Shapiro, J.I.; Fedorova, O.V.; et al. In preeclampsia endogenous cardiotonic steroids induce vascular fibrosis and impair relaxation of umbilical arteries. J. Hypertens. 2011, 29, 769–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigorova, Y.N.; Juhasz, O.; Zernetkina, V.; Fishbein, K.W.; Lakatta, E.G.; Fedorova, O.V.; Bagrov, A.Y. Aortic Fibrosis, Induced by High Salt Intake in the Absence of Hypertensive Response, is Reduced by a Monoclonal Antibody to Marinobufagenin. Am. J. Hypertens. 2016, 29, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Emelianov, I.V.; Bagrov, K.A.; Grigorova, Y.N.; Wei, W.; Juhasz, O.; Frolova, E.V.; Marshall, C.A.; Lakatta, E.G.; Konradi, A.O.; et al. Marinobufagenin-induced vascular fibrosis is a likely target for mineralocorticoid antagonists. J. Hypertens. 2015, 33, 1602–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, S.T.; Yan, Y.; Drummond, C.A.; Xie, J.; Tian, J.; Kennedy, D.J.; Shilova, V.Y.; Xie, Z.; Liu, J.; Cooper, C.J. Rapamycin attenuates cardiac fibrosis in experimental uremic cardiomyopathy by reducing marinobufagenin levels and inhibiting downstream pro-fibrotic signaling. J. Am. Heart Assoc. 2016, 5, e004106. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.A.; Hill, M.C.; Shi, H.; Fan, X.; Xie, J.X.; Haller, S.T.; Kennedy, D.J.; Liu, J.; Garrett, M.R.; Xie, Z. Na/K-ATPase signaling regulates collagen synthesis through microRNA-29b-3p in cardiac fibroblasts. Physiol. Genom. 2015, 48, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Dostanic-Larson, I.; Lorenz, J.N.; Van Huysse, J.W.; Neumann, J.C.; Moseley, A.E.; Lingrel, J.B. Physiological role of the α1-and α2-isoforms of the Na+-K+-ATPase and biological significance of their cardiac glycoside binding site. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2006, 290, R524–R528. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.J.; Kent, M.-A.H.; El-Shahat, E.; Wang, H.; Tan, J.; White, R.; Leenen, F.H. Central and peripheral renin-angiotensin systems in ouabain-induced hypertension. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H624–H630. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Xavier, F.E.; Arribas, S.M.; González, M.C.; Rossoni, L.V.; Alonso, M.J.; Salaices, M. Alterations in structure and mechanics of resistance arteries from ouabain-induced hypertensive rats. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H193–H201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrandi, M.; Molinari, I.; Barassi, P.; Minotti, E.; Bianchi, G.; Ferrari, P. Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J. Biol. Chem. 2004, 279, 33306–33314. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.T.; Kennedy, D.J.; Shidyak, A.; Budny, G.V.; Malhotra, D.; Fedorova, O.V.; Shapiro, J.I.; Bagrov, A.Y. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am. J. Hypertens. 2012, 25, 690–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, V.; Kölbel, F.; Štěpán, J.; Gregorova, I.; Přibyl, T. Digoxin-like immunoreactivity in the serum of rats with cardiac overload. J. Mol. Cell. Cardiol. 1981, 13, 107–110. [Google Scholar] [CrossRef]
- Morise, T.; Okamoto, S.; Takasaki, H.; Ikeda, M.; Takeda, R.; Kiuti, F.; Tuda, Y. Biological Activity of Partially Purified Digitalis-like Substance and Na-K-ATPase Inhibitor in Rats: the 17th Conference on the Pathogenesis of Hypertension. Jpn. Circ. J. 1988, 52, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Leenen, F.H.; Yuan, B.; Huang, B.S. Brain “ouabain” and angiotensin II contribute to cardiac dysfunction after myocardial infarction. Am. J. Physiol.-Heart Circ. Physiol. 1999, 277, H1786–H1792. [Google Scholar] [CrossRef]
- Orlov, S.N.; Klimanova, E.A.; Tverskoi, A.M.; Vladychenskaya, E.A.; Smolyaninova, L.V.; Lopina, O.D. Na+ i, K+ i-Dependent and-Independent Signaling Triggered by Cardiotonic Steroids: Facts and Artifacts. Molecules 2017, 22, 635. [Google Scholar] [CrossRef] [PubMed]
- La, J.; Reed, E.B.; Koltsova, S.; Akimova, O.; Hamanaka, R.B.; Mutlu, G.M.; Orlov, S.N.; Dulin, N.O. Regulation of myofibroblast differentiation by cardiac glycosides. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 310, L815–L823. [Google Scholar] [CrossRef] [PubMed]
- La, J.; Reed, E.; Chan, L.; Smolyaninova, L.V.; Akomova, O.A.; Mutlu, G.M.; Orlov, S.N.; Dulin, N.O. Downregulation of TGF-β Receptor-2 Expression and Signaling through Inhibition of Na/K.-ATPase. PLoS ONE 2016, 11, e0168363. [Google Scholar] [CrossRef] [PubMed]
- Agunanne, E.; Horvat, D.; Harrison, R.; Uddin, M.N.; Jones, R.; Kuehl, T.J.; Ghanem, D.A.; Berghman, L.R.; Lai, X.; Li, J.; et al. Marinobufagenin levels in preeclamptic patients: A preliminary report. Am. J. Perinatol. 2011, 28, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Ehrig, J.C.; Afroze, S.H.; Reyes, M.; Allen, S.R.; Drever, N.S.; Pilkinton, K.A.; Kuehl, T.J.; Uddin, M.N. A p38 mitogen-activated protein kinase inhibitor attenuates cardiotonic steroids-induced apoptotic and stress signaling in a Sw-71 cytotrophoblast cell line. Placenta 2015, 36, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Ehrig, J.C.; Horvat, D.; Allen, S.R.; Jones, R.O.; Kuehl, T.J.; Uddin, M.N. Cardiotonic steroids induce anti-angiogenic and anti-proliferative profiles in first trimester extravillous cytotrophoblast cells. Placenta 2014, 35, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.N.; Horvat, D.; Demorrow, S.; Agunanne, E.; Puschett, J.B. Marinobufagenin is an upstream modulator of Gadd45a stress signaling in preeclampsia. Biochim. Biophys. Acta 2011, 1812, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.N.; Horvat, D.; Childs, E.W.; Puschett, J.B. Marinobufagenin causes endothelial cell monolayer hyperpermeability by altering apoptotic signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1726–R1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenaerts, C.; Bond, L.; Tuytten, R.; Blankert, B. Revealing of endogenous Marinobufagin by an ultra-specific and sensitive UHPLC-MS/MS assay in pregnant women. Talanta 2018, 187, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Koltsova, S.V.; Trushina, Y.; Haloui, M.; Akimova, O.A.; Tremblay, J.; Hamet, P.; Orlov, S.N. Ubiquitous [Na+] i/[K+] i-sensitive transcriptome in mammalian cells: Evidence for Ca2+ i-independent excitation-transcription coupling. PLoS ONE 2012, 7, e38032. [Google Scholar] [CrossRef] [PubMed]
- Crujeiras, A.; Díaz-Lagares, A.; Carreira, M.; Amil, M.; Casanueva, F. Oxidative stress associated to dysfunctional adipose tissue: A potential link between obesity, type 2 diabetes mellitus and breast cancer. Free Radic. Res. 2013, 47, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Donath, M.Y.; Pradhan, A.D.; Thuren, T.; Pais, P.; Nicolau, J.C.; Glynn, R.J.; Libby, P.; Ridker, P.M. Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. J. Am. Coll. Cardiol. 2018, 71, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Lafontan, M. Adipose tissue and adipocyte dysregulation. Diabetes Metab. 2014, 40, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kim, D.H.; Tsenovoy, P.L.; Peterson, S.J.; Rezzani, R.; Rodella, L.F.; Aronow, W.S.; Ikehara, S.; Abraham, N.G. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes 2008, 57, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, T.; Tian, J.; Xie, J.X.; Zhao, X.; Liu, L.; Shapiro, J.I.; Xie, Z. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J. Biol. Chem. 2009, 284, 21066–21076. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Brickman, C.; Liu, J.; Sodhi, K.; Shapiro, J.I. Abstract P203: PNaktide Targeted to Adipocytes Inhibits Na/k-atpase Reactive Oxygen Species, Systemic Inflammation, and Obesity Development in Mice Fed a Western Diet (WD). 2017. Available online: https://insights.ovid.com/hypertension/hype/2017/09/001/abstract-p203/249/00004268 (accessed on 24 August 2018).
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, M.; Joseph, I. Na/K-ATPase amplification of oxidant stress; a universal but unrecognized clinical target? Marshall J. Med. 2016, 2, 8. [Google Scholar]
- Sodhi, K.; Maxwell, K.; Yan, Y.; Liu, J.; Chaudhry, M.A.; Getty, M.; Xie, Z.; Abraham, N.G.; Shapiro, J.I. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci. Adv. 2015, 1, e1500781. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Srikanthan, K.; Goguet-Rubio, P.; Nichols, A.; Mallick, A.; Nawab, A.; Martin, R.; Shah, P.T.; Chaudhry, M.; Sigdel, S. pNaKtide Attenuates steatohepatitis and atherosclerosis by blocking Na/K-ATPase/ROS amplification in C57Bl6 and ApoE knockout mice fed a western diet. Sci. Rep. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Turaihi, K.; Baron, D.; Dandona, P. Increased leucocyte Na-K ATPase in obesity: Reversal following weight loss. Metabolism 1987, 36, 851–855. [Google Scholar] [CrossRef]
- Xia, C.; Rao, X.; Zhong, J. Role of T lymphocytes in type 2 diabetes and diabetes-associated inflammation. J. Diabetes Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.Z. Diabetes and its drivers: The largest epidemic in human history? Clin. Diabetes Endocrinol. 2017, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Vannella, K.M.; Wynn, T.A. Mechanisms of organ injury and repair by macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Maillard, M.; Tantardini, C.; Simonini, M.; Lanzani, C.; Citterio, L.; Stella, P.; Casamassima, N.; Burnier, M.; Hamlyn, J.M. Relationships among endogenous ouabain, α-adducin polymorphisms and renal sodium handling in primary hypertension. J. Hypertens. 2008, 26, 914. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Manunta, P.; Hamlyn, J.M.; Pavan, E.; De, R.T.; Semplicini, A.; Pessina, A.C. Immunoreactive endogenous ouabain in primary aldosteronism and essential hypertension: Relationship with plasma renin, aldosterone and blood pressure levels. J. Hypertens. 1995, 13, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Fridman, A.I.; Matveev, S.A.; Agalakova, N.I.; Fedorova, O.V.; Lakatta, E.G.; Bagrov, A.Y. Marinobufagenin, an endogenous ligand of alpha-1 sodium pump, is a marker of congestive heart failure severity. J. Hypertens. 2002, 20, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.; Hall, C.; Krämer, B.; Elbracht, R.; Palitzsch, K.; Lang, B.; Schölmerich, J. Atrial natriuretic factor and digoxin-like immunoreactive factor in diabetic patients: Their interrelation and the influence of the autonomic nervous system. J. Clin. Endocrinol. Metab. 1996, 81, 3385–3389. [Google Scholar] [PubMed]
- Gonçalves-de-Albuquerque, C.F.; Burth, P.; Silva, A.R.; de Moraes, I.M.M.; de Jesus Oliveira, F.M.; Santelli, R.E.; Freire, A.S.; de Lima, G.S.; da Silva, E.D.; da Silva, C.I. Murine lung injury caused by Leptospira interrogans glycolipoprotein, a specific Na/K.-ATPase inhibitor. Respir. Res. 2014, 15, 93. [Google Scholar] [CrossRef] [PubMed]
- Quastel, M.; Kaplan, J. Inhibition by ouabain of human lymphocyte transformation induced by phytohaemagglutinin in vitro. Nature 1968, 219, 198. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.; Winger, L.; Rasmussen, H.; Nowell, P. The mitogenic effect of A23187 in human peripheral lymphocytes. Biochim. Biophys. Acta (BBA) Gen. Subj. 1977, 496, 374–383. [Google Scholar] [CrossRef]
- Dornand, J.; Favero, J.; Bonnafous, J.-C.; Mani, J.-C. Mechanism whereby ouabain inhibits human T lymphocyte activation: Effect on the interleukin 2 pathway. Immunobiology 1986, 171, 436–450. [Google Scholar] [PubMed]
- Brodie, C.; Tordai, A.; Saloga, J.; Domenico, J.; Gelfand, E.W. Ouabain induces inhibition of the progression phase in human T-cell proliferation. J. Cell. Physiol. 1995, 165, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, F.K.; Kleinhenz, A.L.; Crawford, E.L.; Tian, J.; Malhotra, D.; Haller, S.T.; Kennedy, D.J. Cardiotonic Steroid Signaling through Na/K-ATPase-alpha-1 and Src Kinase Enhance Immune Cell Pro-Inflammatory Response. Circulation 2017. [Google Scholar] [CrossRef]
- Numazawa, S.; Inoue, N.; Nakura, H.; Sugiyama, T.-I.; Fujino, E.; Shinoki, M.-A.; Yoshida, T.; Kuroiwa, Y. A cardiotonic steroid bufalin-induced differentiation of THP-1 cells: Involvement of Na+, K+-ATPase inhibition in the early changes in proto-oncogene expression. Biochem.Pharmacol. 1996, 52, 321–329. [Google Scholar] [CrossRef]
- Watabe, M.; Masuda, Y.; Nakajo, S.; Yoshida, T.; Kuroiwa, Y.; Nakaya, K. The cooperative interaction of two different signaling pathways in response to bufalin induces apoptosis in human leukemia U937 cells. J. Biol. Chem. 1996, 271, 14067–14073. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, M.; Numazawa, S.; Tani, Y.; Yoshida, T. ERK signaling mediates the induction of inflammatory cytokines by bufalin in human monocytic cells. Am. J. Physiol.-Cell Physiol. 2000, 278, C500–C508. [Google Scholar] [CrossRef] [PubMed]
- Watabe, M.; Ito, K.; Masuda, Y.; Nakajo, S.; Nakaya, K. Activation of AP-1 is required for bufalin-induced apoptosis in human leukemia U937 cells. Oncogene 1998, 16, 779. [Google Scholar] [CrossRef] [PubMed]
- Zhakeer, Z.; Hadeer, M.; Tuerxun, Z.; Tuerxun, K. Bufalin inhibits the inflammatory effects in asthmatic mice through the suppression of nuclear factor-kappa B. activity. Pharmacology 2017, 99, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Bi, Q.-R.; Hou, J.-J.; Qi, P.; Ma, C.-H.; Shen, Y.; Feng, R.-H.; Yan, B.-P.; Wang, J.-W.; Shi, X.-J.; Zheng, Y.-Y. Venenum Bufonis induces rat neuroinflammation by activiating NF-κB pathway and attenuation of BDNF. J. Ethnopharmacol. 2016, 186, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, P.; Wang, F.; Zhou, D.; Wang, R.; Meng, L.; Wang, X.; Zhou, M.; Chen, B.; Ouyang, J. Effects of digoxin on cell cycle, apoptosis and NF-κB pathway in Burkitt’s lymphoma cells and animal model. Leuk. Lymphoma 2017, 58, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Zulian, A.; Linde, C.I.; Pulina, M.V.; Baryshnikov, S.G.; Papparella, I.; Hamlyn, J.M.; Golovina, V.A. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca2+ signaling in arterial smooth muscle cells. Am. J. Physiol. Cell Physiol. 2013, 304, C324–C333. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Ferrandi, M. Cardiac glycosides and cardiomyopathy. Hypertension 2006, 47, 343–344. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Usui-Kawanishi, F.; Karasawa, T.; Kimura, H.; Watanabe, S.; Mise, N.; Kayama, F.; Kasahara, T.; Hasebe, N.; Takahashi, M. The cardiac glycoside ouabain activates NLRP3 inflammasomes and promotes cardiac inflammation and dysfunction. PLoS ONE 2017, 12, e0176676. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kennedy, D.J.; Yan, Y.; Shapiro, J.I. Reactive Oxygen Species Modulation of Na/K-ATPase Regulates Fibrosis and Renal Proximal Tubular Sodium Handling. Int. J. Nephrol. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregg, E.W.; Shaw, J.E. Global health effects of overweight and obesity. N. Engl. J. Med. 2017, 377, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Pozza, C.; Isidori, A.M. What’s Behind the Obesity Epidemic. In Imaging in Bariatric Surgery; Springer: Berlin, Germany, 2018; pp. 1–8. [Google Scholar]
- Burgess, A.; Li, M.; Vanella, L.; Kim, D.H.; Rezzani, R.; Rodella, L.; Sodhi, K.; Canestraro, M.; Martasek, P.; Peterson, S.J. Adipocyte heme oxygenase-1 induction attenuates metabolic syndrome in both male and female obese mice. Hypertension 2010, 56, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Iannello, S.; Milazzo, P.; Belfiore, F. Animal and human tissue Na, K-ATPase in obesity and diabetes: A new proposed enzyme regulation. Am. J. Med. Sci. 2007, 333, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kennedy, D.J.; Ramakrishnan, D.P.; Yang, M.; Huang, W.; Li, Z.; Xie, Z.; Chadwick, A.C.; Sahoo, D.; Silverstein, R.L. Oxidized LDL-bound CD36 recruits an Na+/K+-ATPase-Lyn complex in macrophages that promotes atherosclerosis. Sci. Signal. 2015, 8, ra91. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Xie, Z.-J. The Na-K-ATPase and calcium-signaling microdomains. Physiology 2008, 23, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.X.; Li, X.; Xie, Z. Regulation of renal function and structure by the signaling Na/K-ATPase. IUBMB Life 2013, 65, 991–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endemann, G.; Stanton, L.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar] [PubMed]
- Okamura, D.M.; López-Guisa, J.M.; Koelsch, K.; Collins, S.; Eddy, A.A. Atherogenic scavenger receptor modulation in the tubulointerstitium in response to chronic renal injury. Am. J. Physiol.-Ren. Physiol. 2007, 293, F575–F585. [Google Scholar] [CrossRef] [PubMed]
- Apostolov, E.O.; Shah, S.V.; Ok, E.; Basnakian, A.G. Quantification of carbamylated LDL in human sera by a new sandwich ELISA. Clin. Chem. 2005, 51, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Ok, E.; Basnakian, A.G.; Apostolov, E.O.; Barri, Y.M.; Shah, S.V. Carbamylated low-density lipoprotein induces death ofendothelial cells: A link to atherosclerosis in patients with kidney disease. Kidney Int. 2005, 68, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Van Kooten, C.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaweco, A.S.; Mitchell, B.L.; Lucas, B.A.; Mcclatchey, K.D.; Van Thiel, D.H. CD40 expression on graft infiltrates and parenchymal CD154 (CD40L) induction in human chronic renal allograft rejection. Kidney Int. 1999, 55, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Bakogiannis, C.; Tousoulis, D.; Antonopoulos, A.S.; Stefanadis, C. The CD40/CD40 ligand system: Linking inflammation with atherothrombosis. J. Am. Coll. Cardiol. 2009, 54, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.T.; Kumarasamy, S.; Folt, D.A.; Wuescher, L.M.; Stepkowski, S.; Karamchandani, M.; Waghulde, H.; Mell, B.; Chaudhry, M.; Maxwell, K. Targeted disruption of Cd40 in a genetically hypertensive rat model attenuates renal fibrosis and proteinuria, independent of blood pressure. Kidney Int. 2017, 91, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Cai, T. Na+-K+–ATPase-mediated signal transduction: From protein interaction to cellular function. Mol. Interv. 2003, 3, 157. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cardiotonic Steroid | Concentration | Biological Matrix | Condition | Species | References |
|---|---|---|---|---|---|
| MBG | 12.3 ± 1.7 nmol | Urine | Acute myocardial ischemia | Human | [31] |
| MBG | 4.2 ± 0.8 nmol | Urine | Angina pectoris | Human | [31] |
| MBG | 1.9 ± 0.38 nmol/L | Plasma | Acute myocardial ischemia | Human | [31] |
| MBG | 0.51 ± 0.07 nmol/L | Plasma | Angina pectoris | Human | [31] |
| MBG | 0.38 ± 0.1 nmol/L | Plasma | Healthy | Human | [31] |
| MBG | 0.49 ± 0.05 nmol/L | Plasma | Volume expansion | Rat | [33] |
| MBG | 0.20 ± 0.06 nmol/L | Plasma | Healthy | Rat | [33] |
| Ouabain | 0.0032 ± 0.0023 nmol/g | Pituitary | Healthy | Rat | [33] |
| Ouabain | 0.0309 ± 0.00312 nmol/g | Pituitary | Volume expansion | Rat | [33] |
| Ouabain | 0.21 ± 0.04 nmol/L | Plasma | Healthy | Rat | [33] |
| Ouabain | 0.09 ± 0.02 nmol/L | Plasma | Volume expansion | Rat | [33] |
| MBG | 0.00007 ± 0.00002 nmol/g | Pituitary | Healthy | Rat | [33] |
| MBG | 0.00005 ± 0.00001 nmol/g | Pituitary | Volume expansion | Rat | [33] |
| Ouabain | 0.138 ± 0.043 nmol/L | Plasma | Healthy | Human | [36] |
| Ouabain | 0.037 ± 0.007 nmol/L | Plasma | Healthy | Dog | [36] |
| Ouabain | 0.0386 nmol/g | Adrenal | Healthy | Rat | [36] |
| Ouabain | 0.0051 nmol/g | Pituitary | Healthy | Rat | [36] |
| Ouabain | 0.0025 nmol/g | Hypothalamus | Healthy | Rat | [36] |
| Ouabain | 0.0025 nmol/g | Atria | Healthy | Rat | [36] |
| Ouabain | 0.0034 nmol/g | Kidney | Healthy | Rat | [36] |
| Ouabain | 0.0021 nmol/g | Liver | Healthy | Rat | [36] |
| Ouabain | 0.08 ± 0.018 nmol/L | Plasma | Healthy | Rat | [36] |
| Ouabain | 0.04 ± 0.012 nmol/L | Plasma | Adrenalectomy | Rat | [36] |
| Ouabain | 0.12 ± 0.043 nmol/L | Plasma | Uninephrectomy + salt | Rat | [36] |
| Ouabain | 0.98 ± 0.079 nmol/L | Plasma | DOCA + salt | Rat | [36] |
| Ouabain | 0.20 ± 0.062 nmol/L | Plasma | 5/6th Nephrectomy | Rat | [38] |
| Ouabain | 0.12 ± 0.062 nmol/L | Plasma | Healthy | Rat | [38] |
| MBG | 0.36 ± 0.016 nmol/L | Plasma | Healthy | Rat | [39] |
| MBG | 0.57 ± 0.036 nmol/L | Plasma | 5/6th Nephrectomy | Rat | [39] |
| MBG | 0.03 ± 0.0023 nmol | Urine | Healthy | Rat | [39] |
| MBG | 0.06 ± 0.0045 nmol | Urine | 5/6th Nephrectomy | Rat | [39] |
| Ouabain | 0.43 ± 0.053 nmol/L | Plasma | Healthy | Rat | [39] |
| Ouabain | 0.44 ± 0.043 nmol/L | Plasma | 5/6th Nephrectomy | Rat | [39] |
| Ouabain | 0.012 ± 0.0015 nmol | Urine | Healthy | Rat | [39] |
| Ouabain | 0.011 ± 0.0019 nmol | Urine | 5/6th Nephrectomy | Rat | [39] |
| Ouabain | 0.25 ± 0.053 nmol/L | Plasma | Healthy | Human | [42] |
| Ouabain | 0.38 ± 0.019 nmol/L | Plasma | Essential Hypertension | Human | [42] |
| Ouabain | 0.04 ± 0.0002 nmol/L | Plasma | Heart Failure | Human | [45] |
| Ouabain | 1.59 ± 2.2 nmol/L | Plasma | Heart Failure | Human | [45] |
| MBG | 2.3 ± 0.7 nmol/L * | Plasma | Healthy | Human | [47] |
| MBG | 9.5 ± 4.8 nmol/L * | Plasma | End Stage Renal Disease | Human | [47] |
| TCB | 4.4 ± 1.4 nmol/L * | Plasma | Healthy | Human | [47] |
| TCB | 17.0 ± 10.7 nmol/L * | Plasma | End Stage Renal Disease | Human | [47] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalaf, F.K.; Dube, P.; Mohamed, A.; Tian, J.; Malhotra, D.; Haller, S.T.; Kennedy, D.J. Cardiotonic Steroids and the Sodium Trade Balance: New Insights into Trade-Off Mechanisms Mediated by the Na+/K+-ATPase. Int. J. Mol. Sci. 2018, 19, 2576. https://doi.org/10.3390/ijms19092576
Khalaf FK, Dube P, Mohamed A, Tian J, Malhotra D, Haller ST, Kennedy DJ. Cardiotonic Steroids and the Sodium Trade Balance: New Insights into Trade-Off Mechanisms Mediated by the Na+/K+-ATPase. International Journal of Molecular Sciences. 2018; 19(9):2576. https://doi.org/10.3390/ijms19092576
Chicago/Turabian StyleKhalaf, Fatimah K., Prabhatchandra Dube, Amal Mohamed, Jiang Tian, Deepak Malhotra, Steven T. Haller, and David J. Kennedy. 2018. "Cardiotonic Steroids and the Sodium Trade Balance: New Insights into Trade-Off Mechanisms Mediated by the Na+/K+-ATPase" International Journal of Molecular Sciences 19, no. 9: 2576. https://doi.org/10.3390/ijms19092576