LOXL2—A New Target in Antifibrogenic Therapy?

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
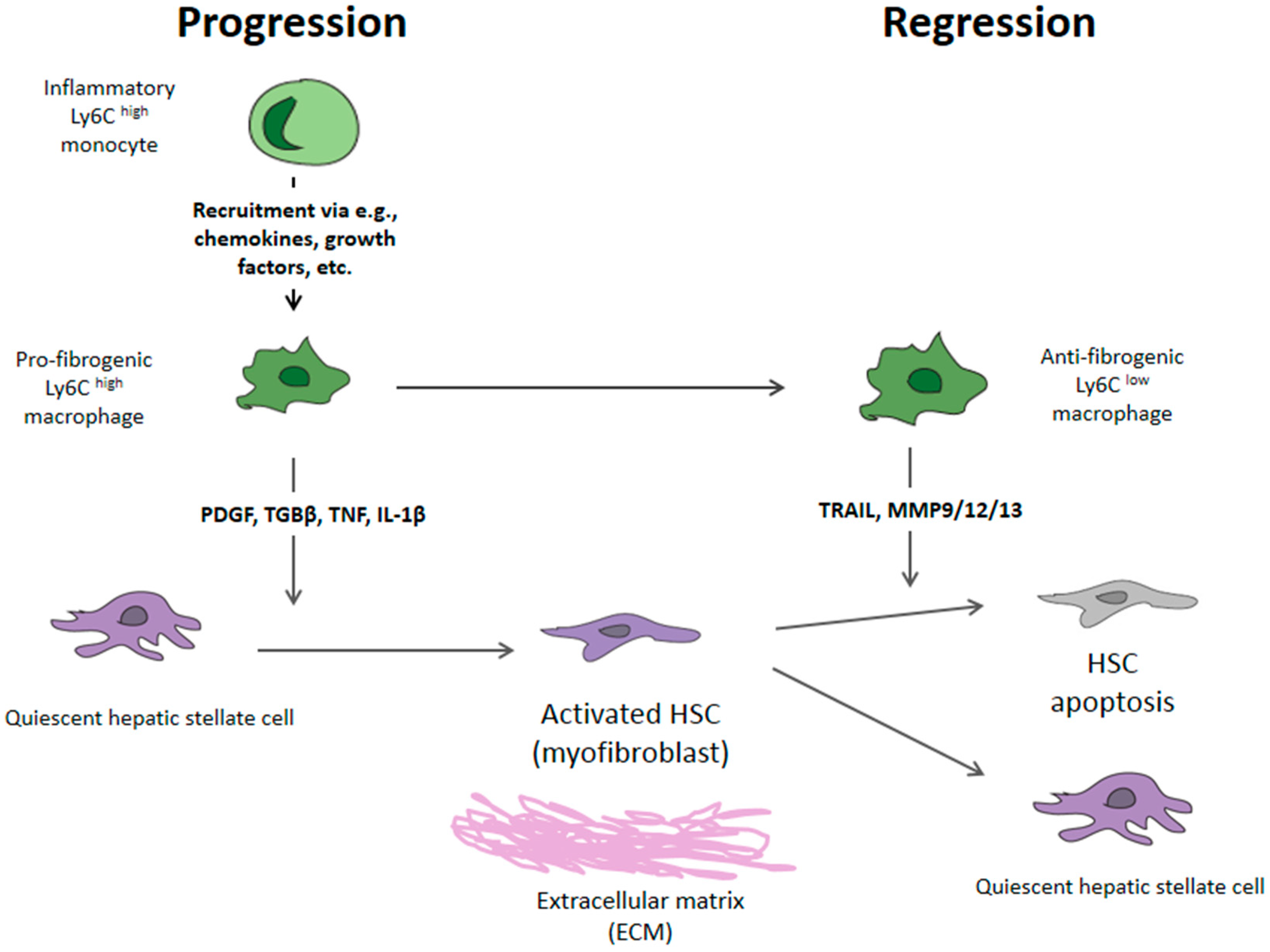
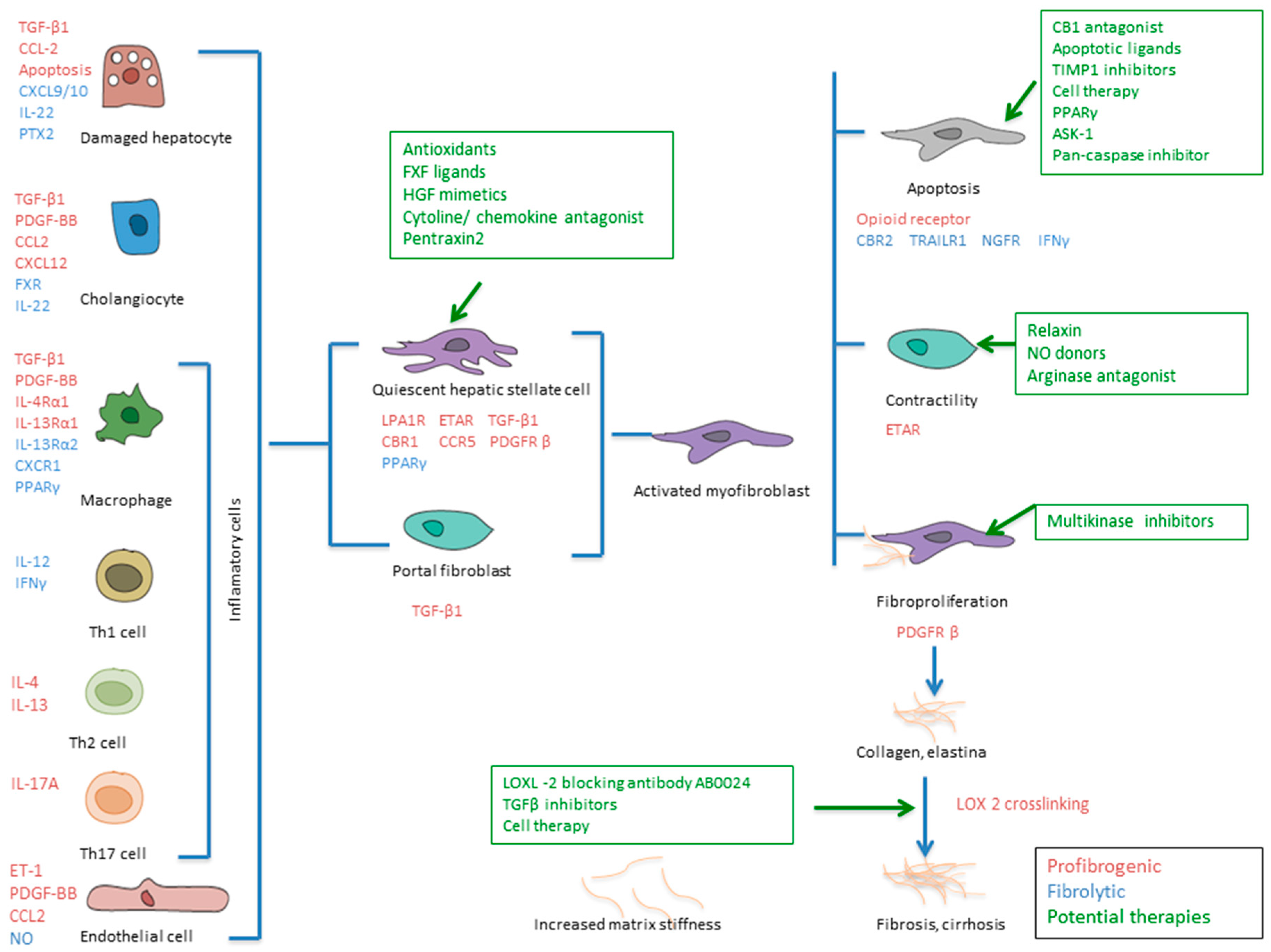
2. Mechanism of Liver Fibrosis
3. Fibrosis Reversion—Lysyl Oxidase 2
4. Non-Invasive Evaluation of Liver Fibrosis
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| HSC | hepatic stellate cell |
| HCV | hepatitis C virus |
| HBV | hepatitis B virus |
| NASH | nonalcoholic steatohepatitis |
| ASH | alcoholic steatohepatitis |
| PBC | primary biliary cirrhosis |
| KC | Kupffer cells |
| EMC | extracellular matrix component |
| CXCR | chemokine receptor |
| CX3CR1 | fractalkine receptor |
| PPARγ | peroxisome proliferator-activated receptors |
| CCL | chemokine ligand |
| MMP | metalloproteinases |
| SVR | sustained virological response |
| LOXL2 | lysyl oxidase-like 2 |
| LSECs | liver sinusoidal endothelial cells |
| LSM | liver stiffness measurement |
| HCC | hepatocelular hepatocarcinoma |
| DAAs | direct antiviral agents |
| NO | nitric oxide |
| PDGF | platelet-derived growth factor |
| TGBβ | transforming growth factor β |
| TNF | tumor necrosis factor |
| IL-1β | interleukin |
| TRAIL | related apoptosis-inducing ligand receptor |
| CBR | cannabinoid receptor 1 |
| ET-1 | endothelin-1 |
| ETAR | endothelin A receptor |
| FXR | farnesoid X receptor |
| Hh(R) | hedgehog (receptor) |
| Int | integrin |
| LPA1R | lyso-phosphatidic acid receptor 1 |
| NGFR | nerve growth factor receptor |
| PTX2 | pentraxin 2 |
| CBR1 | carbonyl reductase 1 |
| HGF | hepatocyte growth factor |
| NGFR | nerve growth factor receptor |
References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef]
- D’Amico, G.; Garcia-Tsao, G.; Pagliaro, L. Natural history and prognostic indicators of survival in cirrhosis: A systematic review of 118 studies. J. Hepatol. 2006, 44, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C. Translating an understanding of the pathogenesis of hepatic fibrosis to novel therapies. Clin. Gastroenterol. Hepatol. 2013, 11, 224–231. [Google Scholar] [CrossRef]
- Hernandez Gea, V.; Friedman, S. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62, S15–S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Novo, E.; Cannito, S.; Zamara, E.; Valfrè di Bonzo, L.; Caligiuri, A.; Cravanzola, C.; Compagnone, A.; Colombatto, S.; Marra, F.; Pinzani, M.; et al. Proangiogenic Cytokines as Hypoxia-Dependent Factors Stimulating Migration of Human Hepatic Stellate Cells. Am. J. Pathol. 2007, 170, 1942–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, M.; Semela, D.; Bruix, J.; Colle, I.; Pinzani, M.; Bosch, J. Angiogenesis in liver disease. J. Hepatol. 2009, 50, 604–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Dranoff, J.A.; Wells, R.G. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology 2010, 51, 1438–1444. [Google Scholar] [CrossRef]
- Rockey, D. Vascular mediators in the injured liver. Hepatology 2003, 37, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Barron, L.; Wynn, T.A. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am. J. Physiol. Liver Physiol. 2011, 300, G723–G728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Lui, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Madala, S.K.; Ramalingam, T.R.; Gochuico, B.R.; Rosas, I.O.; Cheever, A.W.; Wynn, T.A. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 2010, 207, 535–552. [Google Scholar] [CrossRef]
- Gao, B.; Waisman, A. Th17 Cells Regulate Liver Fibrosis by Targeting Multiple Cell Types: Many Birds With One Stone. Gastroenterology 2012, 143, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Aleffi, S.; Galastri, S.; Provenzano, A. Mononuclear cells in liver fibrosis. Semin. Immunopathol. 2009, 31, 345–358. [Google Scholar] [CrossRef]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Tacke, F. Roles for Chemokines in Liver Disease. Gastroenterology 2014, 147, 577–594.e1. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassiman, D.; Libbrecht, L.; Desmet, V.; Denef, C.; Roskams, T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J. Hepatol. 2002, 36, 200–209. [Google Scholar] [CrossRef]
- Liaw, Y.-F.; Sung, J.J.; Chow, W.C.; Farrell, G.; Lee, C.-Z.; Yuen, H.; Tanwandee, T.; Tao, Q.M.; Shue, K.; Keene, O.N.; et al. Lamivudine for Patients with Chronic Hepatitis B and Advanced Liver Disease. New Engl. J. Med. 2004, 351, 1521–1531. [Google Scholar] [CrossRef]
- Van der Meer, A.J.; Veldt, B.J.; Feld, J.J.; Wedemeyer, H.; Dufour, J.F.; Lammert, F.; Duarte-Rojo, A.; Heathcote, E.J.; Manns, M.P.; Kuske, L.; et al. Association Between Sustained Virological Response and All-Cause Mortality Among Patients With Chronic Hepatitis C and Advanced Hepatic Fibrosis. JAMA 2012, 308, 2584–2593. [Google Scholar] [CrossRef] [Green Version]
- Perelló, C.; Cabezas, J.; Llop, E.; Carrion, J.A.; Ruiz-Antoran, B.; Llerena, S.; Hernandez-Conde, M.; Crespo, J.; Calleja, J.L. Impact of SVR in the development of all complications and fibrosis regression in a cohort of patients treated with interferon-base Triple Therapy and Direct Acting Antiviral. Hepatology 2017, 66, 218A. [Google Scholar]
- Puente, Á.; Cabezas, J.; López Arias, M.J.; Fortea, J.I.; Arias, M.T.; Estébanez, Á.; Casafont, F.; Fábrega, E.; Crespo, J. Influence of sustained viral response on the regression of fibrosis and portal hypertension in cirrhotic HCV patients treated with antiviral triple therapy. Rev. Esp. Enferm. Dig. 2017, 109, 17–25. [Google Scholar] [CrossRef]
- Lens, S.; Alvarado-Tapias, E.; Mariño, Z.; Londoño, M.-C.; Llop, E.; Martinez, J.; Fortea, J.I.; Ibañez, L.; Ariza, X.; Baiges, A.; et al. Effects of All-Oral Anti-Viral Therapy on HVPG and Systemic Hemodynamics in Patients With Hepatitis C Virus-Associated Cirrhosis. Gastroenterology 2017, 153, 1273–1283.e1. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, R.; Aghemo, A.; Fraquelli, M.; Rumi, M.G.; Donato, M.F.; Paradis, V.; Bedossa, P.; Colombo, M. The diagnostic accuracy of Fibroscan® for cirrhosis is influenced by liver morphometry in HCV patients with a sustained virological response. J. Hepatol. 2013, 59, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Hjuler, S.T.; Luo, Y.; Rasmussen, D.G.K.; Nielsen, M.J.; Nielsen, S.H.; Leeming, D.J.; Goodman, Z.; Arch, R.H.; Patel, K.; et al. Assessment of liver fibrosis progression and regression by a serological collagen turnover profile. Am. J. Physiol. Liver Physiol. 2019, 316, G25–G31. [Google Scholar] [CrossRef] [PubMed]
- Issa, R.; Zhou, X.; Constandinou, C.M.; Fallowfield, J.; Millward-Sadler, H.; Gaca, M.D.; Sands, E.; Suliman, I.; Trim, N.; Knorr, A. Spontaneous recovery from micronodular cirrhosis: Evidence for incomplete resolution associated with matrix cross-linking☆. Gastroenterology 2004, 126, 1795–1808. [Google Scholar] [CrossRef]
- Grau-Bove, X.; Ruiz-Trillo, I.; Rodriguez-Pascual, F. Origin and evolution of lysyl oxidases. Sci. Rep. 2015, 5, 10568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucero, H.A.; Kagan, H.M. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 2006, 63, 2304–2316. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Baselli, G.A.; Bassani, G.A.; Rametta, R.; Pietrelli, A.; Maggioni, M.; Facciotti, F.; Trunzo, V.; Badiali, S.; et al. Insulin resistance promotes Lysyl Oxidase Like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clin. Sci. 2017, 131, 1301–1315. [Google Scholar] [CrossRef]
- Wong, C.C.-L.; Tse, A.P.-W.; Huang, Y.-P.; Zhu, Y.-T.; Chiu, D.K.-C.; Lai, R.K.-H.; Au, S.L.-K.; Kai, A.K.-L.; Lee, J.M.-F.; Wei, L.L.; et al. Lysyl oxidase-like 2 is critical to tumor microenvironment and metastatic niche formation in hepatocellular carcinoma. Hepatology 2014, 60, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhöfer, N.; Kong, C.; Le, Q.-T.; Chi, J.-T.A.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Chien, J.W.; Richards, T.J.; Gibson, K.F.; Zhang, Y.; Lindell, K.O.; Lyman, S.K.; Adamkewicz, J.I.; Smith, V.; Kaminski, N.; O’Riordan, T. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur. Respir. J. 2014, 43, 1430–1438. [Google Scholar] [CrossRef]
- Bosch, J.; Ratziu, V.; Rockey, D.; Ghalib, R.; Thuluvath, P.; Schiefke, I.; Flamm, S.; Abdelmalek, M.; Millers, R.; Aguilar, R.; et al. Correlation between noninvasive markers of fibrosis and the hepatic venous pressure gradient (HVPG) in patients with compensated cirrhosis due to nonalcoholic steatohepatitis (NASH). Hepatology 2015, 62, 120A. [Google Scholar]
- Afdha, N.; Everson, G.T.; Calleja, J.L.; McCaughan, G.; Asselah, T.; Myers, R.; Aguilar, R.; Schall, J.; Denning, D.; Brainard, D.; et al. Serum lysyl oxidase-like-2 (sLOXL2) is correlated with the hepatic venous pressure gradient (HVPG) in patients with cirrhosis due to hepatitis C. Hepatology 2015, 62, 121A. [Google Scholar]
- Bourliere, M.; Loustaud-Ratti, V.; Metivier, S.; Leroy, V.; Abergel, A.; Myers, R.; Aguilar, R.; Hyland, R.; Subramanian, M.; McHutchison, J.; et al. Changes in liver stiffness by transient elastography (TE) and serum lysyl oxidase-like-2 (sLOXL2) in patients with cirrhosis treated with ledipasvir/sofosbuvir (LDV/SOF)-based therapy. Hepatology 2015, 62, 123A. [Google Scholar]
- Puente, A.; Fortea, J.I.; Posadas, M.; Garcia, A.; Rasines, L.; Arias Loste, M.T.; Cabezas, J.; Llerena, S.; Iruzubieta, P.; Fabrega, E.; et al. Influence of the metabolic syndrome on fibrosis regression regulated by LOXL2 after sustained virologycal response. Gastroenterol. Hepatol. 2019, 42, 123. [Google Scholar]
- Barry-Hamilton, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.M.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C.; et al. Allosteric inhibition of lysyl oxidase–like-2 impedes the development of a pathologic microenvironment. Nat. Med. 2010, 16, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Ikenaga, N.; Peng, Z.W.; A Vaid, K.; Liu, S.B.; Yoshida, S.; Sverdlov, D.Y.; Mikels-Vigdal, A.; Smith, V.; Schuppan, D.; Popov, Y.V. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 2017, 66, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Brown, K.K.; Collard, H.R.; Cottin, V.; Gibson, K.F.; Kaner, R.J.; Lederer, D.J.; Martinez, F.J.; Noble, P.W.; Song, J.W.; et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: A randomised, double-blind, controlled, phase 2 trial. Respir. Med. 2017, 5, 22–32. [Google Scholar] [CrossRef]
- Hecht, J.R.; Benson, A.B.; Vyushkov, D.; Yang, Y.; Bendell, J.; Verma, U. A Phase II, Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab in Combination with FOLFIRI for the Second-Line Treatment of Metastatic KRAS Mutant Colorectal Adenocarcinoma. Oncologist 2017, 22, 243-e23. [Google Scholar] [CrossRef]
- Benson, A.B.; Wainberg, Z.A.; Hecht, J.R.; Vyushkov, D.; Dong, H.; Bendell, J.; Kudrik, F. A Phase II Randomized. Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. Oncologist 2017, 22, 241-e15. [Google Scholar] [CrossRef]
- Tatal, A.H.; Feron-Rigodon, M.; Madere, J.; Subramanian, G.M.; Bornstein, J.D. Simtuzumab, an antifibrotic monoclonal antibody against lysyl oxidase like 2, appears safe and well tolerated in patients with liver disease of diverse etiology. J. Hepatol. 2013, 58, S409. [Google Scholar]
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Simtuzumab Is Ineffective for Patients With Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. GS-US-384-1497 Investigators. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.J.; Levy, C.; Janssen, H.L.; Montano-Loza, A.J.; Shiffman, M.L.; Caldwell, S.; Luketic, V.; Ding, D.; Jia, C.; McColgan, B.J.; et al. Simtuzumab for Primary Sclerosing Cholangitis: Phase 2 Study Results With Insights on the Natural History of the Disease. Hepatology 2019, 69, 684–698. [Google Scholar] [CrossRef]
- Garcia-Tsao, G.; Fuchs, M.; Shiffman, M.; Borg, B.B.; Pyrsopoulos, N.; Shetty, K.; Gallegos-Orozco, J.F.; Reddy, K.R.; Feyssa, E.; Chan, J.L.; et al. Emricasan (IDN-6556) Lowers Portal Pressure in Patients With Compensated Cirrhosis and Severe Portal Hypertension. Hepatology 2019, 69, 717–728. [Google Scholar] [CrossRef]
- Castera, L. Noninvasive Methods to Assess Liver Disease in Patients With Hepatitis B or C. Gastroenterology 2012, 142, 1293–1302.e4. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL-ALEH Clinical Practice Guidelines: Non-invasive tests for evaluation of liver disease severity and prognosis. J. Hepatol. 2015, 63, 237–264. [Google Scholar] [CrossRef] [Green Version]
- Procopet, B.; Berzigotti, A.; Abraldes, J.G.; Turon, F.; Hernandez-Gea, V.; Garcia-Pagan, J.C.; Bosch, J.; Abraldeṣ, J.G. Real-time shear-wave elastography: Applicability, reliability and accuracy for clinically significant portal hypertension. J. Hepatol. 2015, 62, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Asrani, S.K.; Talwalkar, J.A.; Kamath, P.S.; Shah, V.H.; Saracino, G.; Jennings, L.; Gross, J.; Venkatesh, S.; Ehman, R. Role of magnetic resonance elastography in compensated and decompensated liver disease. J. Hepatol. 2014, 60, 934–939. [Google Scholar] [CrossRef]
- Banerjee, R.; Pavlides, M.; Tunnicliffe, E.M.; Piechnik, S.K.; Sarania, N.; Philips, R.; Collier, J.D.; Booth, J.C.; Schneider, J.E.; Wang, L.M.; et al. Multiparametric magnetic resonance for the non-invasive diagnosis of liver disease. J. Hepatol. 2014, 60, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, B.C.; Wang, H.; Yang, Y.; Wei, L.; Polasek, M.; Schühle, D.T.; Lauwers, G.Y.; Parkar, A.; Sinskey, A.J.; Tanabe, K.K.; et al. Molecular MRI of Collagen to Diagnose and Stage Liver Fibrosis. J. Hepatol. 2013, 59, 992–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalazar, G.; Pappo, O.; Hershcovici, T.; Hadjaj, T.; Shubi, M.; Ohana, H.; Hemed, N.; Ilan, Y. A continuous 13C methacetin breath test for noninvasive assessment of intrahepatic inflammation and fibrosis in patients with chronic HCV infection and normal ALT. J. Hepat. 2008, 15, 716–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stravitz, R.T.; Ilan, Y. Potential use of metabolic breath tests to assess liver disease and prognosis: Has the time arrived for routine use in the clinic? Liver Int. 2016, 37, 328–336. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puente, A.; Fortea, J.I.; Cabezas, J.; Arias Loste, M.T.; Iruzubieta, P.; Llerena, S.; Huelin, P.; Fábrega, E.; Crespo, J. LOXL2—A New Target in Antifibrogenic Therapy? Int. J. Mol. Sci. 2019, 20, 1634. https://doi.org/10.3390/ijms20071634
Puente A, Fortea JI, Cabezas J, Arias Loste MT, Iruzubieta P, Llerena S, Huelin P, Fábrega E, Crespo J. LOXL2—A New Target in Antifibrogenic Therapy? International Journal of Molecular Sciences. 2019; 20(7):1634. https://doi.org/10.3390/ijms20071634
Chicago/Turabian StylePuente, Angela, Jose Ignacio Fortea, Joaquin Cabezas, Maria Teresa Arias Loste, Paula Iruzubieta, Susana Llerena, Patricia Huelin, Emilio Fábrega, and Javier Crespo. 2019. "LOXL2—A New Target in Antifibrogenic Therapy?" International Journal of Molecular Sciences 20, no. 7: 1634. https://doi.org/10.3390/ijms20071634