Pitavastatin Exerts Potent Anti-Inflammatory and Immunomodulatory Effects via the Suppression of AP-1 Signal Transduction in Human T Cells

 , , , and
, , , and 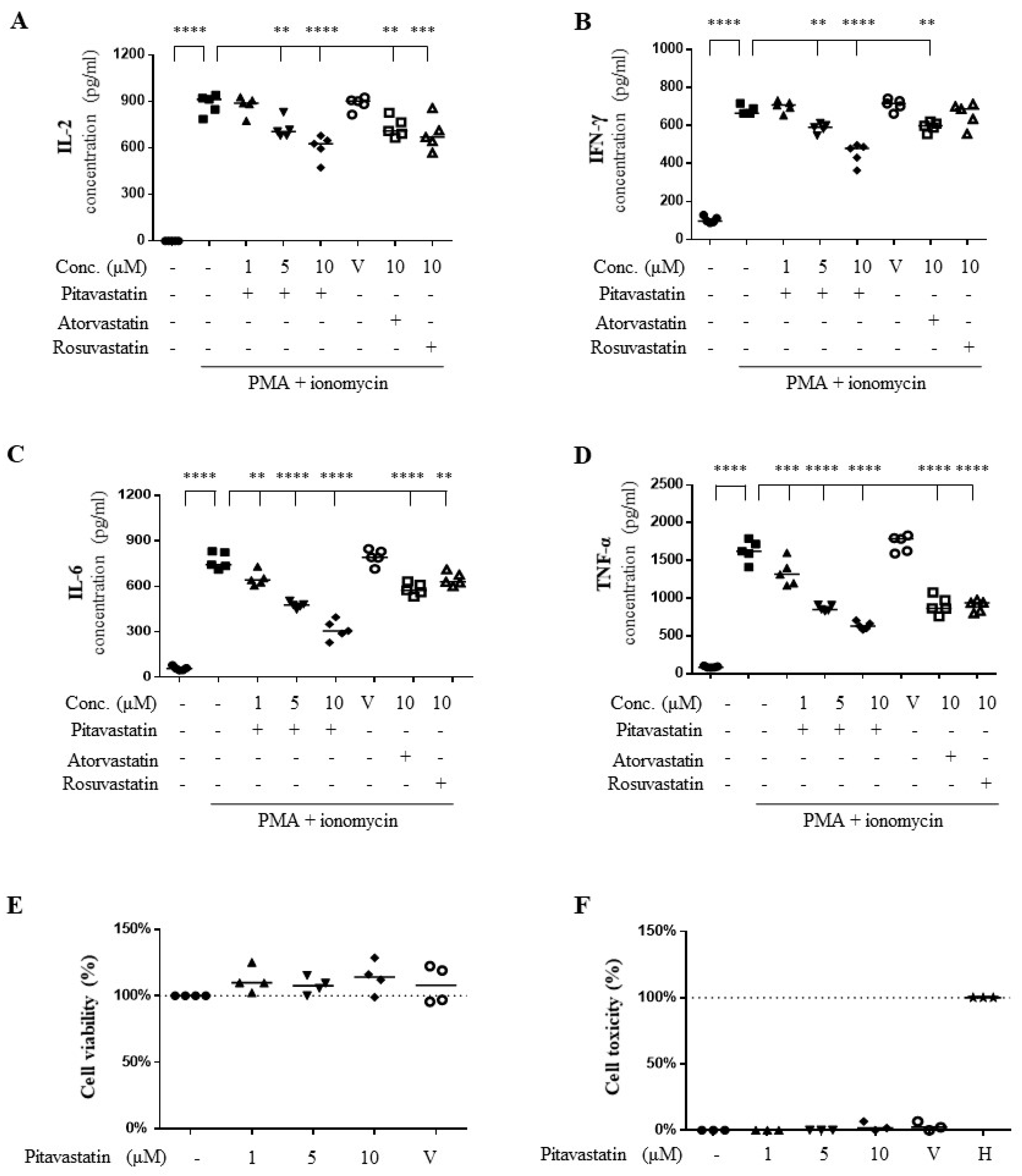{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Pitavastatin Suppressed Phorbol 12-Myristate 13-Acetate (PMA) Plus Ionomycin-Induced Cytokine Production
2.2. Pitavastatin Dose-Dependently Downregulated the mRNA Synthesis of PMA Plus Ionomycin-Induced Pro-Inflammatory Cytokines
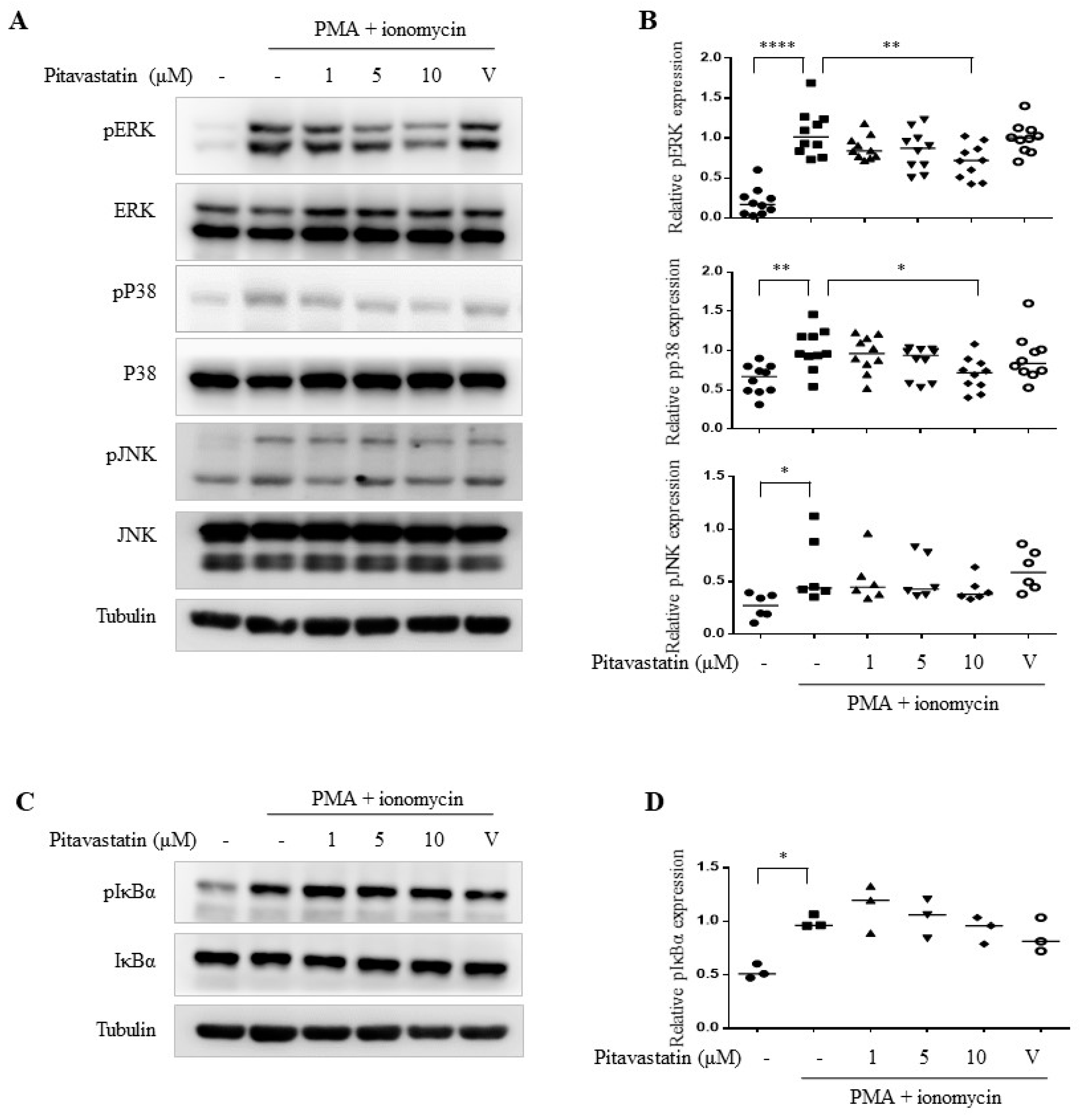
2.3. Pitavastatin Significantly Inhibited PMA Plus Ionomycin-Induced Mitogen-Activated Protein Kinase (MAPK) Pathways Including ERK and p38
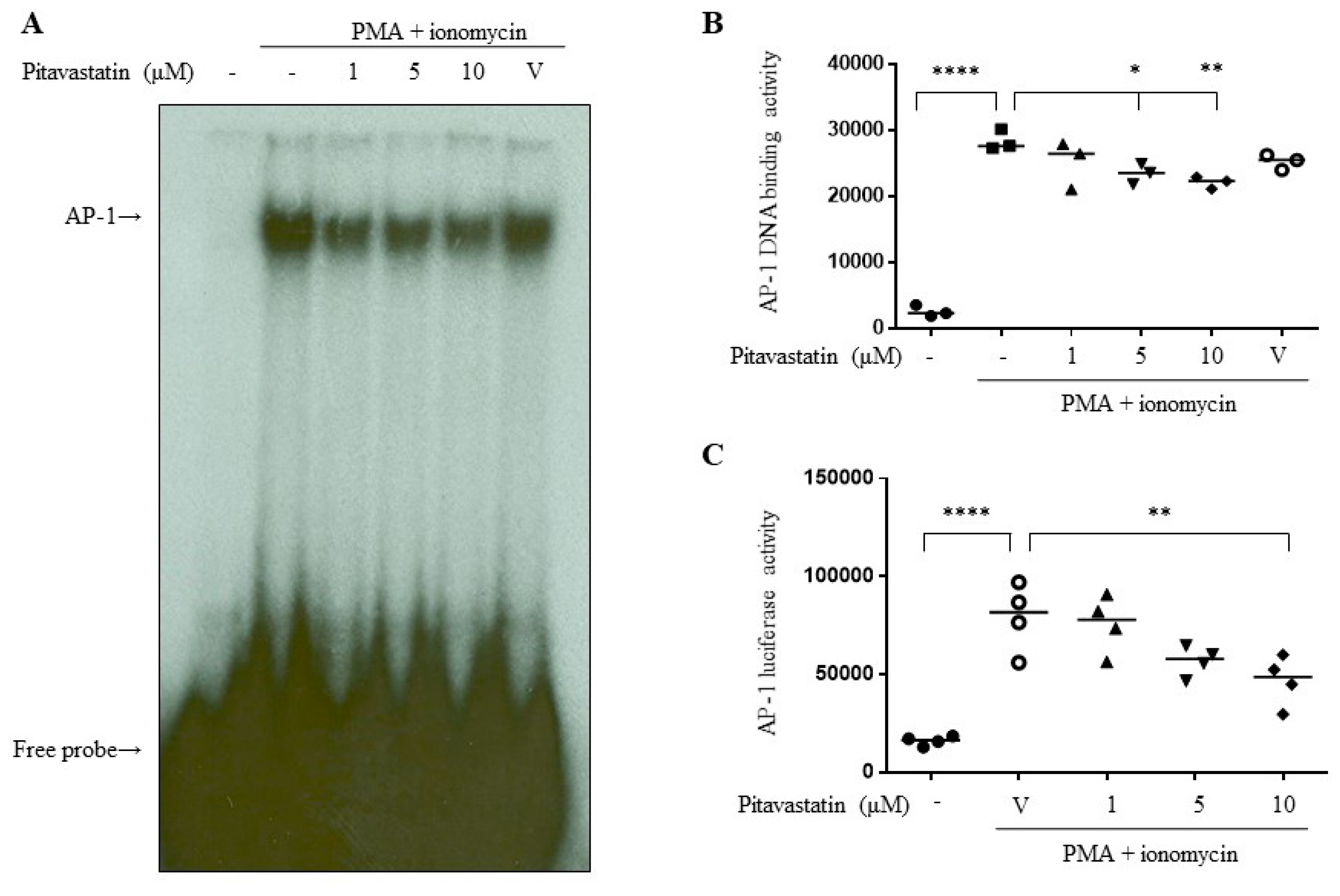
2.4. Pitavastatin Downregulated the Transcriptional Activity of AP-1
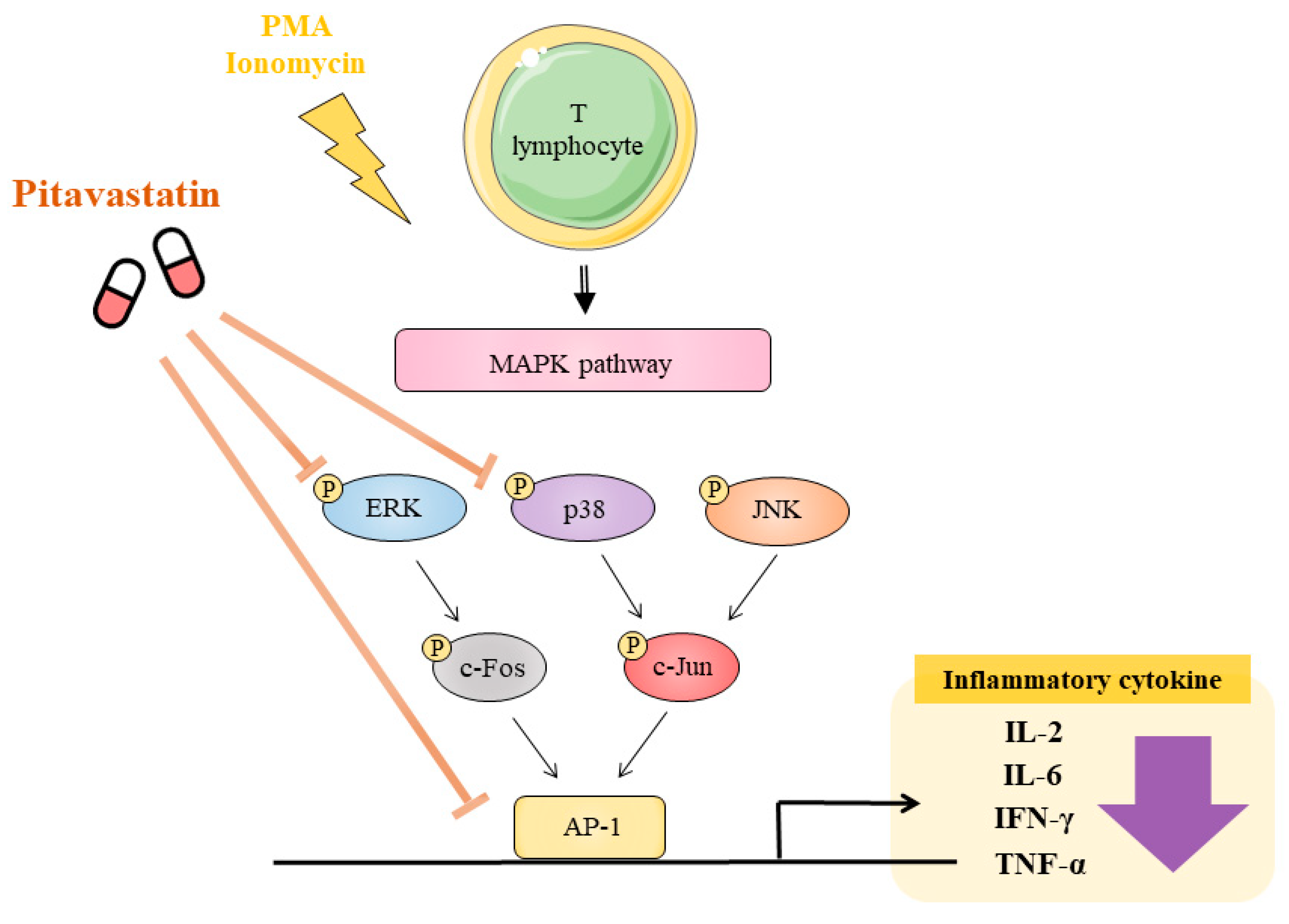
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Isolation of Primary Human T Cells and Cell Culture
4.3. Measurement of Cytotoxicity
4.4. Enzyme-linked Immunosorbent Assay (ELISA)
4.5. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.6. Western Blots
4.7. Nuclear Extract Preparation
4.8. Electrophoresis Mobility Shift Assay (EMSA)
4.9. Transient Transfection and Luciferase Activity Assay
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AP-1 | Activating protein-1 |
| ASCVD | Atherosclerotic cardiovascular disease |
| eNOS | Endothelial NO synthase |
| ECs | Endothelial cells |
| ERK | Extracellular signal-regulated kinase |
| FOX | Forkhead box |
| IFN | Interferon |
| IL | Interleukin |
| JNK | c-Jun N-terminal kinase |
| LDH | Lactate dehydrogenase |
| LDL-C | Low-density lipoprotein cholesterol |
| Ldlr−/− | Low-density lipoprotein receptors knockout |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemotactic protein-1 |
| MTT | 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide |
| NFκB | Nuclear factor-κB |
| PDGF | Platelet-derived growth factor |
| PMA | Phorbol 12-myristate 13-acetate |
| ROCK | Rho-associated protein kinase |
| SMC | Smooth muscle cell |
| Th1 | T helper-1 |
| TNF-α | Tumor necrosis factor α |
References
- Goldstein, L.J.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar] [PubMed]
- Cholesterol Treatment Trialists, C.; Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [CrossRef]
- Jain, M.; Rosenberg, M. Meta-analysis: Lowering LDL-C levels using statins reduces major vascular events regardless of baseline risk. Ann. Intern. Med. 2012, 157, JC4-2. [Google Scholar] [CrossRef] [PubMed]
- Cholesterol Treatment Trialists’ Collaboration. Efficacy and safety of LDL-lowering therapy among men and women: Meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015, 385, 1397–1405. [Google Scholar]
- Cholesterol Treatment Trialists’ Collaboration. Efficacy and safety of statin therapy in older people: A meta-analysis of individual participant data from 28 randomised controlled trials. Lancet 2019, 393, 407–415. [Google Scholar]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2018, 139, e1082–e1143. [Google Scholar]
- Mohammad, S.; Nguyen, H.; Nguyen, M.; Abdel-Rasoul, M.; Nguyen, V.; Nguyen, C.D.; Nguyen, K.T.; Li, L.; Kitzmiller, J.P. Pleiotropic effects of statins: untapped potential for statin pharmacotherapy. Curr. Vasc. Pharmacol. 2019, 17, 239–261. [Google Scholar] [CrossRef]
- Oesterle, A.; Liao, J.K. The Pleiotropic effects of statins - from coronary artery disease and stroke to atrial fibrillation and ventricular tachyarrhythmia. Curr. Vasc. Pharmacol. 2019, 17, 222–232. [Google Scholar] [CrossRef]
- Takemoto, M.; Sun, J.; Hiroki, J.; Shimokawa, H.; Liao, J.K. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation 2002, 106, 57–62. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Mummidi, S.; Mahimainathan, L.; Patel, D.N.; Bailey, S.R.; Imam, S.Z.; Greene, W.C.; Valente, A.J. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J. Biol. Chem. 2006, 281, 15099–15109. [Google Scholar] [CrossRef] [PubMed]
- Rezaie-Majd, A.; Maca, T.; Bucek, R.A.; Valent, P.; Muller, M.R.; Husslein, P.; Kashanipour, A.; Minar, E.; Baghestanian, M. Simvastatin reduces expression of cytokines interleukin-6, interleukin-8, and monocyte chemoattractant protein-1 in circulating monocytes from hypercholesterolemic patients. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Luan, Z.; Chase, A.J.; Newby, A.C. Statins inhibit secretion of metalloproteinases-1, -2, -3, and -9 from vascular smooth muscle cells and macrophages. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Azcutia, V.; Aikawa, E.; Figueiredo, J.L.; Croce, K.; Sonoki, H.; Sacks, F.M.; Luscinskas, F.W.; Aikawa, M. Statins suppress apolipoprotein CIII-induced vascular endothelial cell activation and monocyte adhesion. Eur. Heart J. 2013, 34, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Bekkering, S.; Latz, E.; Riksen, N.P. Long-term activation of the innate immune system in atherosclerosis. Semin. Immunol. 2016, 28, 384–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, K.G.; Jonasson, L. The discovery of cellular immunity in the atherosclerotic plaque. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1714–1717. [Google Scholar] [CrossRef] [PubMed]
- Kishikawa, H.; Shimokama, T.; Watanabe, T. Localization of T lymphocytes and macrophages expressing IL-1, IL-2 receptor, IL-6 and TNF in human aortic intima. Role of cell-mediated immunity in human atherogenesis. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 423, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Frostegard, J.; Ulfgren, A.K.; Nyberg, P.; Hedin, U.; Swedenborg, J.; Andersson, U.; Hansson, G.K. Cytokine expression in advanced human atherosclerotic plaques: Dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 1999, 145, 33–43. [Google Scholar] [CrossRef]
- Tousoulis, D.; Oikonomou, E.; Economou, E.K.; Crea, F.; Kaski, J.C. Inflammatory cytokines in atherosclerosis: Current therapeutic approaches. Eur. Heart J. 2016, 37, 1723–1732. [Google Scholar] [CrossRef]
- Frostegard, J.; Zhang, Y.; Sun, J.; Yan, K.; Liu, A. Oxidized low-density lipoprotein (OxLDL)-treated dendritic cells promote activation of T cells in human atherosclerotic plaque and blood, which is repressed by statins: microRNA let-7c is integral to the effect. J. Am. Heart Assoc. 2016, 5, e003976. [Google Scholar] [CrossRef]
- Kim, Y.C.; Kim, K.K.; Shevach, E.M. Simvastatin induces Foxp3+ T regulatory cells by modulation of transforming growth factor-beta signal transduction. Immunology 2010, 130, 484–493. [Google Scholar] [CrossRef]
- Bu, D.X.; Tarrio, M.; Grabie, N.; Zhang, Y.; Yamazaki, H.; Stavrakis, G.; Maganto-Garcia, E.; Pepper-Cunningham, Z.; Jarolim, P.; Aikawa, M.; et al. Statin-induced Kruppel-like factor 2 expression in human and mouse T cells reduces inflammatory and pathogenic responses. J. Clin. Investig. 2010, 120, 1961–1970. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.M.; Lai, J.H.; Yang, S.P.; Tsao, T.P.; Ho, L.J.; Liou, J.T.; Cheng, C.C. Modulation of human T cells signaling transduction by lovastatin. Int. J. Cardiol. 2010, 140, 24–33. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Adachi, K.; Davis, M.M. T-cell receptor ligation induces distinct signaling pathways in naive vs. antigen-experienced T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 1549–1554. [Google Scholar] [CrossRef]
- Taguchi, I.; Iimuro, S.; Iwata, H.; Takashima, H.; Abe, M.; Amiya, E.; Ogawa, T.; Ozaki, Y.; Sakuma, I.; Nakagawa, Y.; et al. High-dose versus low-dose pitavastatin in Japanese patients with stable coronary artery disease (REAL-CAD): A randomized superiority trial. Circulation 2018, 137, 1997–2009. [Google Scholar] [CrossRef]
- Kitahara, M.; Kanaki, T.; Ishii, I.; Saito, Y. Atherosclerosis induced by chronic inhibition of the synthesis of nitric oxide in moderately hypercholesterolaemic rabbits is suppressed by pitavastatin. Br. J. Pharmacol. 2010, 159, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Kohno, M.; Shinomiya, K.; Abe, S.; Noma, T.; Kondo, I.; Oshita, A.; Takeuchi, H.; Takagi, Y.; Yukiiri, K.; Mizushige, K.; et al. Inhibition of migration and proliferation of rat vascular smooth muscle cells by a new HMG-CoA reductase inhibitor, pitavastatin. Hypertens. Res. 2002, 25, 279–285. [Google Scholar]
- Kaneyuki, U.; Ueda, S.; Yamagishi, S.; Kato, S.; Fujimura, T.; Shibata, R.; Hayashida, A.; Yoshimura, J.; Kojiro, M.; Oshima, K.; et al. Pitavastatin inhibits lysophosphatidic acid-induced proliferation and monocyte chemoattractant protein-1 expression in aortic smooth muscle cells by suppressing Rac-1-mediated reactive oxygen species generation. Vasc. Pharmacol. 2007, 46, 286–292. [Google Scholar] [CrossRef]
- Tokoro, T.; Wang, J.; Kitajima, I. The novel HMG-CoA reductase inhibitor, Pitavastatin, induces a protective action in vascular endothelial cells through the production of nitric oxide (NO). Yakugaku Zasshi 2004, 124, 121–126. [Google Scholar] [CrossRef]
- Sato, K.; Nuki, T.; Gomita, K.; Weyand, C.M.; Hagiwara, N. Statins reduce endothelial cell apoptosis via inhibition of TRAIL expression on activated CD4 T cells in acute coronary syndrome. Atherosclerosis 2010, 213, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Kibayashi, E.; Urakaze, M.; Kobashi, C.; Kishida, M.; Takata, M.; Sato, A.; Yamazaki, K.; Kobayashi, M. Inhibitory effect of pitavastatin (NK-104) on the C-reactive-protein-induced interleukin-8 production in human aortic endothelial cells. Clin. Sci. 2005, 108, 515–521. [Google Scholar] [CrossRef] [Green Version]
- Tajiri, K.; Shimojo, N.; Sakai, S.; Machino-Ohtsuka, T.; Imanaka-Yoshida, K.; Hiroe, M.; Tsujimura, Y.; Kimura, T.; Sato, A.; Yasutomi, Y.; et al. Pitavastatin regulates helper T-cell differentiation and ameliorates autoimmune myocarditis in mice. Cardiovasc. Drugs Ther. 2013, 27, 413–424. [Google Scholar] [CrossRef]
- Kitamura, S.; Maeda, K.; Wang, Y.; Sugiyama, Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab. Dispos. 2008, 36, 2014–2023. [Google Scholar] [CrossRef]
- Janneh, O.; Hartkoorn, R.C.; Jones, E.; Owen, A.; Ward, S.A.; Davey, R.; Back, D.J.; Khoo, S.H. Cultured CD4T cells and primary human lymphocytes express hOATPs: Intracellular accumulation of saquinavir and lopinavir. Br. J. Pharmacol. 2008, 155, 875–883. [Google Scholar] [CrossRef]
- Nakagomi, A.; Shibui, T.; Kohashi, K.; Kosugi, M.; Kusama, Y.; Atarashi, H.; Shimizu, W. Differential effects of atorvastatin and pitavastatin on inflammation, insulin resistance, and the carotid intima-media thickness in patients with dyslipidemia. J. Atheroscler. Thromb. 2015, 22, 1158–1171. [Google Scholar] [CrossRef]
- Libby, P.; Lichtman, A.H.; Hansson, G.K. Immune effector mechanisms implicated in atherosclerosis: From mice to humans. Immunity 2013, 38, 1092–1104. [Google Scholar] [CrossRef]
- Gupta, S.; Pablo, A.M.; Jiang, X.; Wang, N.; Tall, A.R.; Schindler, C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Investig. 1997, 99, 2752–2761. [Google Scholar] [CrossRef]
- Nagano, H.; Mitchell, R.N.; Taylor, M.K.; Hasegawa, S.; Tilney, N.L.; Libby, P. Interferon-gamma deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. J. Clin. Investig. 1997, 100, 550–557. [Google Scholar] [CrossRef]
- Rocha, V.Z.; Folco, E.J.; Sukhova, G.; Shimizu, K.; Gotsman, I.; Vernon, A.H.; Libby, P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: A role for adaptive immunity in obesity. Circ. Res. 2008, 103, 467–476. [Google Scholar] [CrossRef]
- Schieffer, B.; Schieffer, E.; Hilfiker-Kleiner, D.; Hilfiker, A.; Kovanen, P.T.; Kaartinen, M.; Nussberger, J.; Harringer, W.; Drexler, H. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques, potential implications for inflammation and plaque instability. Circulation 2000, 101, 1372–1378. [Google Scholar] [CrossRef]
- Akita, K.; Isoda, K.; Sato-Okabayashi, Y.; Kadoguchi, T.; Kitamura, K.; Ohtomo, F.; Shimada, K.; Daida, H. An interleukin-6 receptor antibody suppresses atherosclerosis in atherogenic mice. Front. Cardiovasc. Med. 2017, 4, 84. [Google Scholar] [CrossRef]
- Wang, J.; Kitajima, I. Pitavastatin inactivates NF-kappaB and decreases IL-6 production through Rho kinase pathway in MCF-7 cells. Oncol. Rep. 2007, 17, 1149–1154. [Google Scholar]
- Iwata, A.; Shirai, R.; Ishii, H.; Kushima, H.; Otani, S.; Hashinaga, K.; Umeki, K.; Kishi, K.; Tokimatsu, I.; Hiramatsu, K.; et al. Inhibitory effect of statins on inflammatory cytokine production from human bronchial epithelial cells. Clin. Exp. Immunol. 2012, 168, 234–240. [Google Scholar] [Green Version]
- Khalaf, H.; Jass, J.; Olsson, P.E. Differential cytokine regulation by NF-kappaB and AP-1 in Jurkat T-cells. BMC Immunol. 2010, 11, 26. [Google Scholar] [CrossRef]
- Schonthaler, H.B.; Guinea-Viniegra, J.; Wagner, E.F. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann. Rheum. Dis. 2011, 70, i109–i112. [Google Scholar] [CrossRef]
- Yamakawa, T.; Tanaka, S.; Kamei, J.; Kadonosono, K.; Okuda, K. Pitavastatin inhibits vascular smooth muscle cell proliferation by inactivating extracellular signal-regulated kinases 1/2. J. Atheroscler. Thromb. 2003, 10, 37–42. [Google Scholar] [CrossRef]
- Checker, R.; Sandur, S.K.; Sharma, D.; Patwardhan, R.S.; Jayakumar, S.; Kohli, V.; Sethi, G.; Aggarwal, B.B.; Sainis, K.B. Potent anti-inflammatory activity of ursolic acid, a triterpenoid antioxidant, is mediated through suppression of NF-kappaB, AP-1 and NF-AT. PLoS ONE 2012, 7, e31318. [Google Scholar] [CrossRef]
- Cheng, B.F.; Gao, Y.X.; Lian, J.J.; Guo, D.D.; Liu, T.T.; Xie, Y.F.; Wang, L.; Yang, H.J.; Wang, M.; Feng, Z.W. Anti-inflammatory effects of pitavastatin in interleukin-1beta-induced SW982 human synovial cells. Int. Immunopharmacol. 2017, 50, 224–229. [Google Scholar] [CrossRef]
- Ha, Y.M.; Nam, J.O.; Kang, Y.J. Pitavastatin regulates Ang II induced proliferation and migration via IGFBP-5 in VSMC. Korean J. Physiol. Pharmacol. 2015, 19, 499–506. [Google Scholar] [CrossRef]
- Habara, K.; Hamada, Y.; Yamada, M.; Tokuhara, K.; Tanaka, H.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Ito, S.; Okumura, T. Pitavastatin up-regulates the induction of iNOS through enhanced stabilization of its mRNA in pro-inflammatory cytokine-stimulated hepatocytes. Nitric Oxide 2008, 18, 19–27. [Google Scholar] [CrossRef]
- Chen, L.W.; Shih, S.F.; Lin, C.S. The effects of pitavastatin on nuclear factor-κB (NFκB) DNA-binding activity and luciferase activity in activated human T cells and 293T cells, respectively. Unpublished work. 2018. [Google Scholar]
- Cheng, S.M.; Chu, K.M.; Lai, J.H. The modulatory mechanisms of fenofibrate on human primary T cells. Eur. J. Pharm. Sci. 2010, 40, 316–324. [Google Scholar] [CrossRef]
- Hung, L.F.; Huang, K.Y.; Yang, D.H.; Chang, D.M.; Lai, J.H.; Ho, L.J. Advanced glycation end products induce T cell apoptosis: Involvement of oxidative stress, caspase and the mitochondrial pathway. Mech. Ageing Dev. 2010, 131, 682–691. [Google Scholar] [CrossRef]
- Teng, I.J.; Tsai, M.C.; Shih, S.F.; Tsuei, B.F.; Chang, H.; Chuang, Y.P.; Lin, C.S.; Chern, C.Y.; Chen, S.J. Chalcone Derivatives Enhance ATP-Binding Cassette Transporters A1 in Human THP-1 Macrophages. Molecules 2018, 23, 1620. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.W.; Lin, C.-S.; Tsai, M.-C.; Shih, S.-F.; Lim, Z.W.; Chen, S.-J.; Tsui, P.-F.; Ho, L.-J.; Lai, J.-H.; Liou, J.-T. Pitavastatin Exerts Potent Anti-Inflammatory and Immunomodulatory Effects via the Suppression of AP-1 Signal Transduction in Human T Cells. Int. J. Mol. Sci. 2019, 20, 3534. https://doi.org/10.3390/ijms20143534
Chen LW, Lin C-S, Tsai M-C, Shih S-F, Lim ZW, Chen S-J, Tsui P-F, Ho L-J, Lai J-H, Liou J-T. Pitavastatin Exerts Potent Anti-Inflammatory and Immunomodulatory Effects via the Suppression of AP-1 Signal Transduction in Human T Cells. International Journal of Molecular Sciences. 2019; 20(14):3534. https://doi.org/10.3390/ijms20143534
Chicago/Turabian StyleChen, Liv Weichien, Chin-Sheng Lin, Min-Chien Tsai, Shao-Fu Shih, Zhu Wei Lim, Sy-Jou Chen, Pi-Fen Tsui, Ling-Jun Ho, Jenn-Haung Lai, and Jun-Ting Liou. 2019. "Pitavastatin Exerts Potent Anti-Inflammatory and Immunomodulatory Effects via the Suppression of AP-1 Signal Transduction in Human T Cells" International Journal of Molecular Sciences 20, no. 14: 3534. https://doi.org/10.3390/ijms20143534