Targeting ROS and cPLA2/COX2 Expressions Ameliorated Renal Damage in Obese Mice with Endotoxemia

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
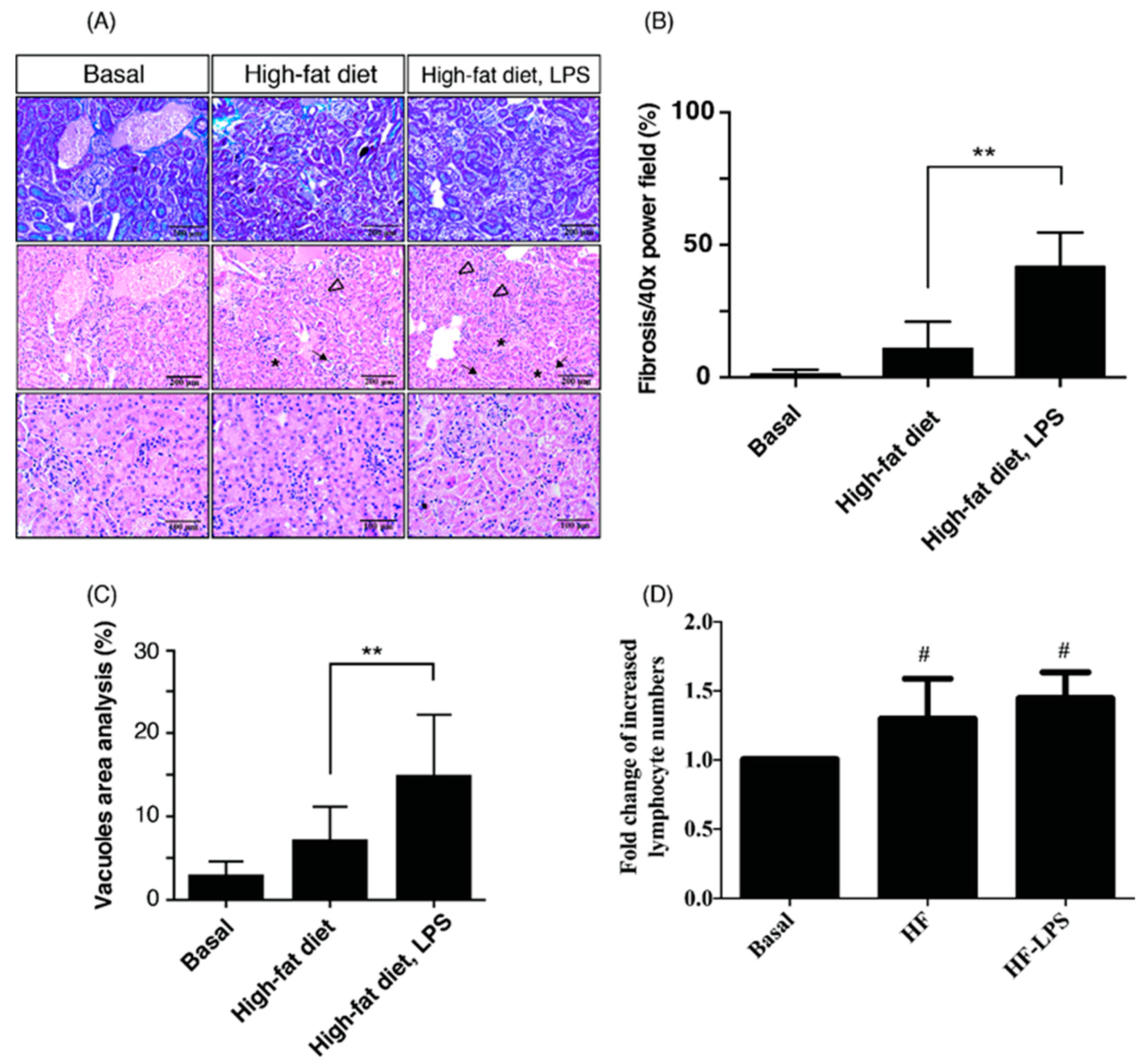
2.1. HF Mice with ME Exhibit the Most Prominent Renal Fibrosis, Depositions of Lipid Vacuoles in Tubular Epithelium, and Lymphocyte Infiltration in Tubulointerstitium
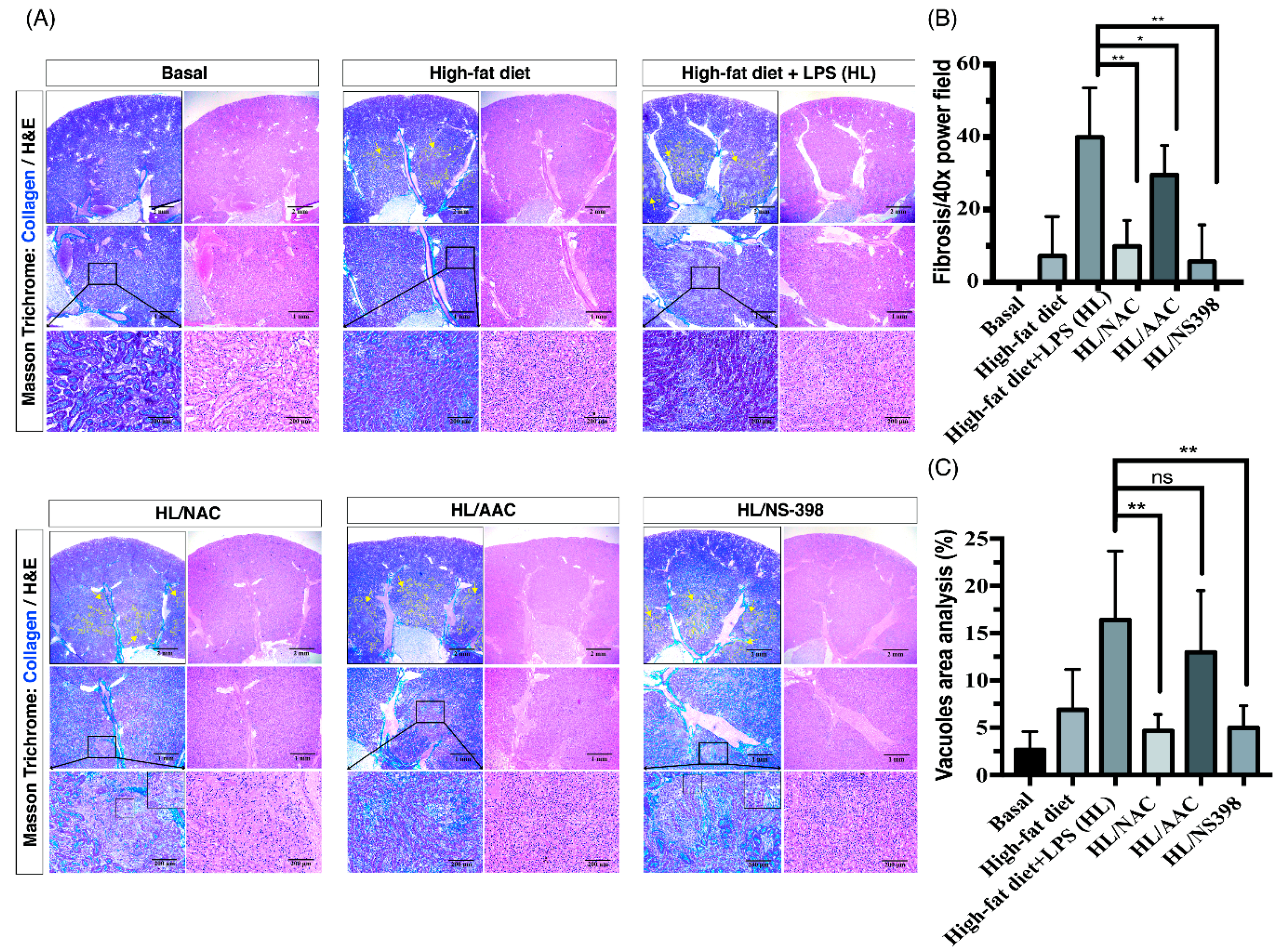
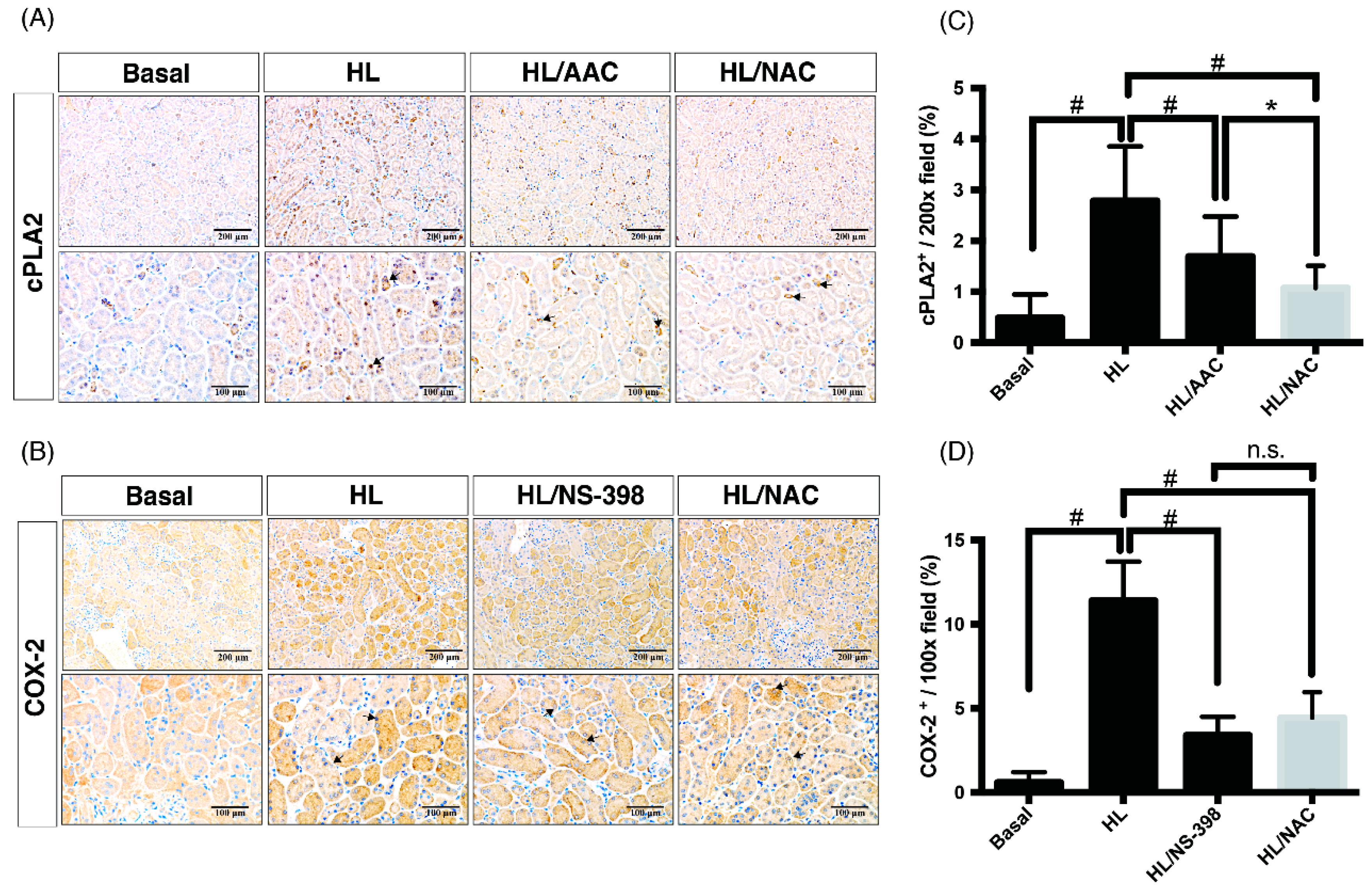
2.2. Inhibitors of ROS, cPLA2, and COX-2 Attenuate the Tubulointerstitial Fibrosis in HF Mice with ME
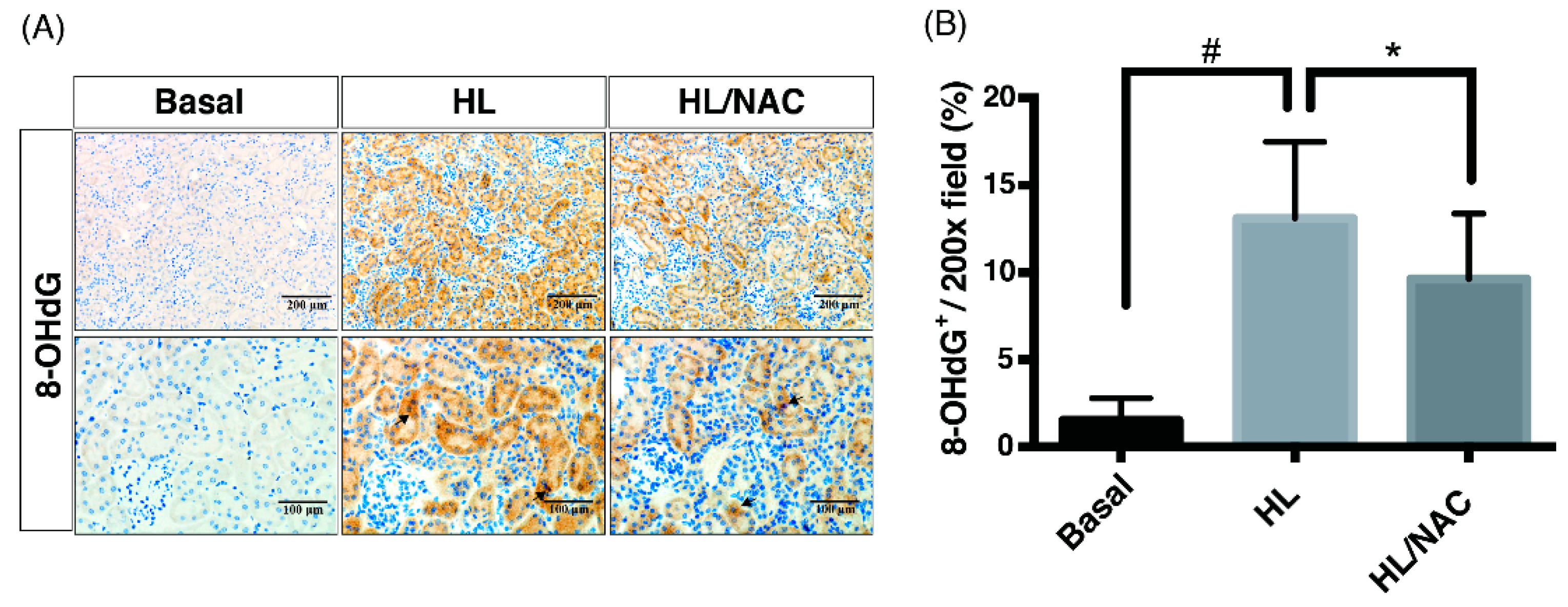
2.3. Scavengers of Non-Specific ROS Attenuate Not Only Oxidative Injury But Also Downstream Pathways of cPLA2 and COX-2 in Obese Kidney Fibrosis with Metabolic Endotoxemia
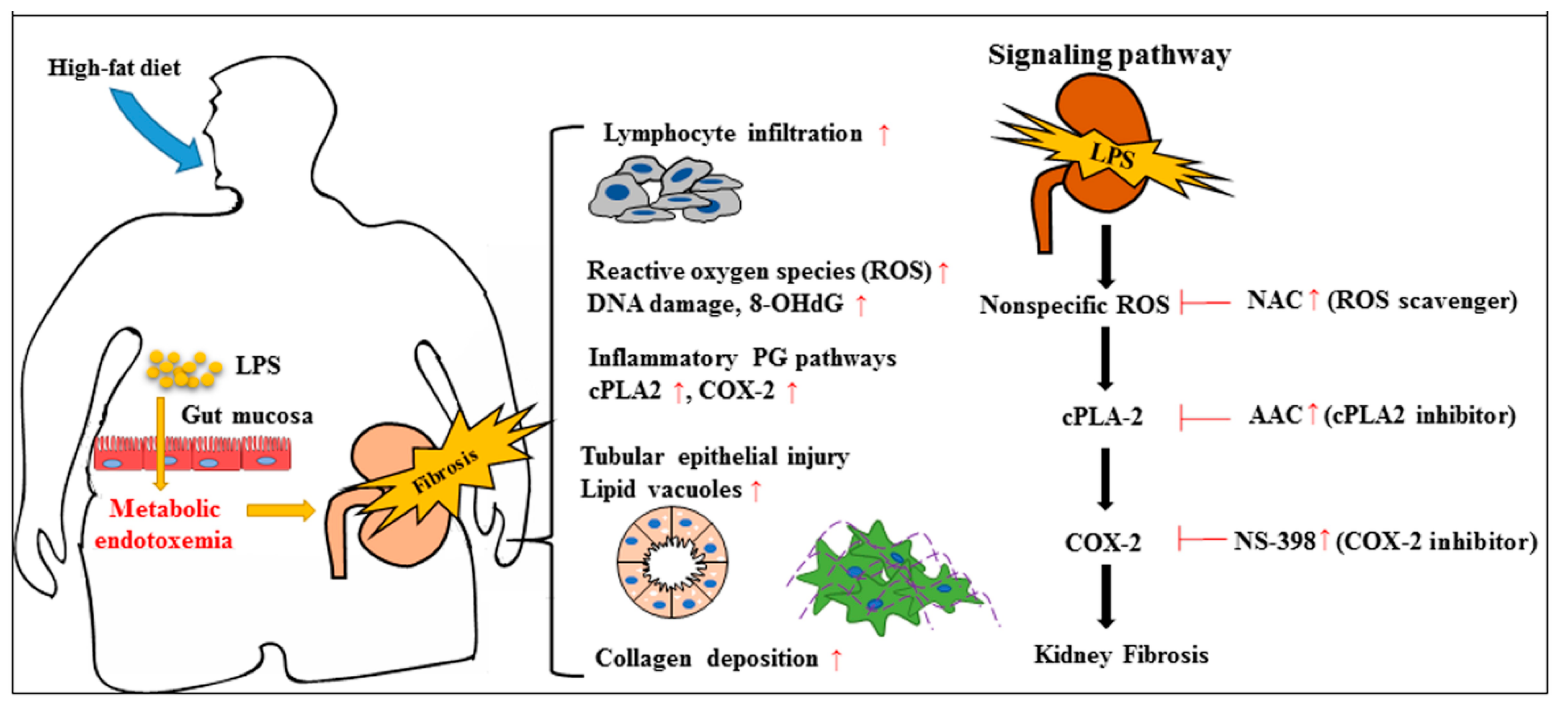
3. Discussion
3.1. The Fat–Intestine–Kidney Axis
3.2. Therapeutic Targets of ROS, cPLA2 and COX-2 in Kidney Diseases
4. Materials and Methods
4.1. Materials
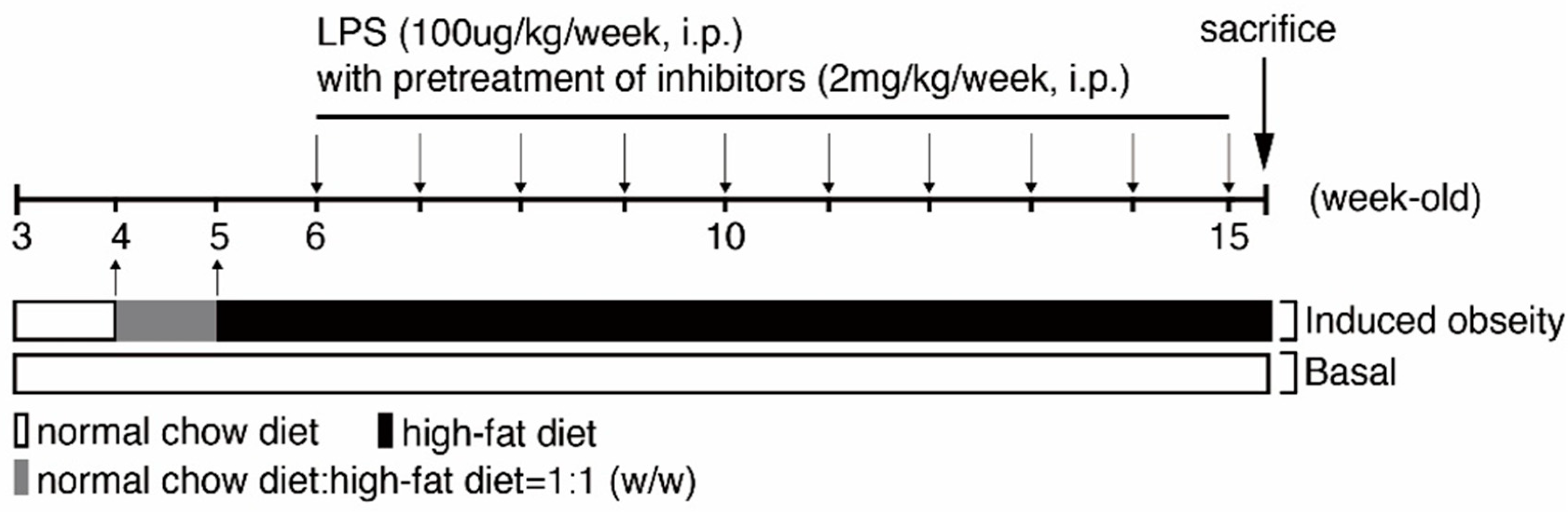
4.2. Creating Animal Models to Mimic Obese Kidney Fibrosis (OKF) in Humans
4.3. Tissue Preparation for Histopathological Evaluation of H&E Stain
4.4. Masson’s Trichrome Staining Method
4.5. Immunohistochemistry Staining Method
4.6. Statistical Analysis of Data
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CKD | chronic kidney disease |
| COX-2 | cyclooxygenase-2 |
| cPLA2 | cytosolic phospholipases A2 |
| HF group | high-fat diet-fed group injected with lipopolysaccharide |
| IHC | immunohistochemistry |
| LPS | lipopolysaccharide |
| ME | metabolic endotoxemia |
| OKF | obese kidney fibrosis |
| PG | prostaglandin |
| ROS | reactive oxygen species |
References
- Stenvinkel, P.; Zoccali, C.; Ikizler, T.A. Obesity in CKD—What should nephrologists know? J. Am. Soc. Nephrol. 2013, 24, 1727–1736. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Furth, S.L.; Zoccali, C.; on Behalf of the World Kidney Day Steering Committee. Obesity and Kidney Disease: Hidden Consequences of the Epidemic. Am. J. Hypertens. 2017, 30, 328–336. [Google Scholar] [CrossRef]
- Jung, C.H.; Lee, M.J.; Kang, Y.M.; Hwang, J.Y.; Kim, E.H.; Park, J.Y.; Kim, H.K.; Lee, W.J. The risk of chronic kidney disease in a metabolically healthy obese population. Kidney Int. 2015, 88, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Câmara, N.O.S.; Iseki, K.; Kramer, H.; Liu, Z.-H.; Sharma, K. Kidney disease and obesity: Epidemiology, mechanisms and treatment. Nat. Rev. Nephrol. 2017, 13, 181. [Google Scholar] [CrossRef]
- Hobby, G.P.; Karaduta, O.; Dusio, G.F.; Singh, M.; Zybailov, B.L.; Arthur, J.M. Chronic Kidney Disease and the Gut Microbiome. Am. J. Physiol. Renal Physiol. 2019. [Google Scholar] [CrossRef]
- Shah, N.B.; Allegretti, A.S.; Nigwekar, S.U.; Kalim, S.; Zhao, S.; Lelouvier, B.; Servant, F.; Serena, G.; Thadhani, R.I.; Raj, D.S.; et al. Blood Microbiome Profile in CKD: A Pilot Study. Clin. J. Am. Soc. Nephrol. CJASN 2019. [Google Scholar] [CrossRef]
- Lim, P.S.; Chang, Y.K.; Wu, T.K. Serum Lipopolysaccharide-Binding Protein is Associated with Chronic Inflammation and Metabolic Syndrome in Hemodialysis Patients. Blood Purif. 2019, 47, 28–36. [Google Scholar] [CrossRef]
- Nasrallah, R.; Hassouneh, R.; Hébert, R.L. PGE2, kidney disease, and cardiovascular risk: Beyond hypertension and diabetes. J. Am. Soc. Nephrol. 2016, 27, 666–676. [Google Scholar] [CrossRef]
- Komers, R.; Zdychova, J.; Cahova, M.; Kazdova, L.; Lindsley, J.N.; Anderson, S. Renal cyclooxygenase-2 in obese Zucker (fatty) rats. Kidney Int. 2005, 67, 2151–2158. [Google Scholar] [CrossRef] [Green Version]
- Iyer, A.; Lim, J.; Poudyal, H.; Reid, R.C.; Suen, J.Y.; Webster, J.; Prins, J.B.; Whitehead, J.P.; Fairlie, D.P.; Brown, L. An inhibitor of phospholipase A2 group IIA modulates adipocyte signaling and protects against diet-induced metabolic syndrome in rats. Diabetes 2012, 61, 2320–2329. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, Y.; Ding, G.; Heiney, K.M.; Huang, S.; Zhang, A. Role of COX-2/mPGES-1/prostaglandin E2 cascade in kidney injury. Mediat. Inflamm. 2015, 2015, 147894. [Google Scholar] [CrossRef]
- Câmara, N.O.; Martins, J.O.; Landgraf, R.G. Emerging roles for eicosanoids in renal diseases. Curr. Opin. Nephrol. Hypertens. 2009, 18, 21–27. [Google Scholar] [CrossRef]
- Shen, Y.; Miao, N.-J.; Xu, J.-L.; Gan, X.-X.; Xu, D.; Zhou, L.; Xue, H.; Zhang, W.; Lu, L.-M. N-acetylcysteine alleviates angiotensin II-mediated renal fibrosis in mouse obstructed kidneys. Acta Pharmacol. Sin. 2016, 37, 637. [Google Scholar] [CrossRef]
- Chen, D.Q.; Cao, G.; Chen, H.; Liu, D.; Su, W.; Yu, X.Y.; Vaziri, N.D.; Liu, X.H.; Bai, X.; Zhang, L.; et al. Gene and protein expressions and metabolomics exhibit activated redox signaling and wnt/beta-catenin pathway are associated with metabolite dysfunction in patients with chronic kidney disease. Redox Biol. 2017, 12, 505–521. [Google Scholar] [CrossRef]
- Khan, N.S.; Song, C.Y.; Thirunavukkarasu, S.; Fang, X.R.; Bonventre, J.V.; Malik, K.U. Cytosolic Phospholipase A2alpha Is Essential for Renal Dysfunction and End-Organ Damage Associated with Angiotensin II-Induced Hypertension. Am. J. Hypertens. 2016, 29, 258–265. [Google Scholar] [CrossRef]
- Montford, J.R.; Lehman, A.M.B.; Bauer, C.D.; Klawitter, J.; Klawitter, J.; Poczobutt, J.M.; Scobey, M.; Weiser-Evans, M.; Nemenoff, R.A.; Furgeson, S.B. Bone marrow-derived cPLA2α contributes to renal fibrosis progression. J. Lipid Res. 2018, 59, 380–390. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Wang, H.L.; Cheng, X.L.; Wei, F.; Bai, X.; Lin, R.C.; Vaziri, N.D. Metabolomics analysis reveals the association between lipid abnormalities and oxidative stress, inflammation, fibrosis, and Nrf2 dysfunction in aristolochic acid-induced nephropathy. Sci. Rep. 2015, 5, 12936. [Google Scholar] [CrossRef]
- Chen, H.M.; Yang, C.M.; Chang, J.F.; Wu, C.S.; Sia, K.C.; Lin, W.N. AdipoR-increased intracellular ROS promotes cPLA2 and COX-2 expressions via activation of PKC and p300 in adiponectin-stimulated human alveolar type II cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L255–L269. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.S.; Lin, C.M.; Chang, J.F.; Wu, C.S.; Sia, K.C.; Lee, I.T.; Huang, K.Y.; Lin, W.N. Participation of NADPH Oxidase-Related Reactive Oxygen Species in Leptin-Promoted Pulmonary Inflammation: Regulation of cPLA2alpha and COX-2 Expression. Int. J. Mol. Sci. 2019, 20, 1078. [Google Scholar] [CrossRef]
- Ji, L.; Wang, Q.; Huang, F.; An, T.; Guo, F.; Zhao, Y.; Liu, Y.; He, Y.; Song, Y.; Qin, G. FOXO1 Overexpression Attenuates Tubulointerstitial Fibrosis and Apoptosis in Diabetic Kidneys by Ameliorating Oxidative Injury via TXNIP-TRX. Oxidative Med. Cell. Longev. 2019, 2019, 3286928. [Google Scholar] [CrossRef]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633. [Google Scholar] [CrossRef]
- De Vries, A.P.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.; Lamb, H.J.; Barlovic, D.P.; Hojs, R. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, Z.; Proctor, G.; Moskowitz, S.; Liebman, S.E.; Rogers, T.; Lucia, M.S.; Li, J.; Levi, M. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J. Biol. Chem. 2005, 280, 32317–32325. [Google Scholar] [CrossRef]
- Zhou, D.; Liu, Y. Renal fibrosis in 2015: Understanding the mechanisms of kidney fibrosis. Nat. Rev. Nephrol. 2016, 12, 68. [Google Scholar] [CrossRef]
- Chang, J.-F.; Liang, S.-S.; Thanasekaran, P.; Chang, H.-W.; Wen, L.-L.; Chen, C.-H.; Liou, J.-C.; Yeh, J.-C.; Liu, S.-H.; Dai, H.-M. Translational Medicine in Pulmonary-Renal Crosstalk: Therapeutic Targeting of p-Cresyl Sulfate Triggered Nonspecific ROS and Chemoattractants in Dyspneic Patients with Uremic Lung Injury. J. Clin. Med. 2018, 7, 266. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species in non-phagocytic cells. J. Leukoc. Biol. 1999, 65, 337–340. [Google Scholar] [CrossRef]
- Wong, A.C.; Vanhove, A.S.; Watnick, P.I. The interplay between intestinal bacteria and host metabolism in health and disease: Lessons from Drosophila melanogaster. Dis. Models Mech. 2016, 9, 271–281. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care 2010, 33, 2277–2284. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef]
- Rosas-Villegas, A.; Sánchez-Tapia, M.; Avila-Nava, A.; Ramírez, V.; Tovar, A.; Torres, N. Differential effect of sucrose and fructose in combination with a high fat diet on intestinal microbiota and kidney oxidative stress. Nutrients 2017, 9, 393. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet–induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Boutagy, N.E.; McMillan, R.P.; Frisard, M.I.; Hulver, M.W. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie 2016, 124, 11–20. [Google Scholar] [CrossRef]
- Creely, S.J.; McTernan, P.G.; Kusminski, C.M.; Khanolkar, M.; Evans, M.; Louise Harte, A.; Kumar, S. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am. J. Physiol.-Endocrinol. Metabol. 2007, 292, E740–E747. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, P.; Aljada, A.; Ghanim, H.; Hofmeyer, D.; Tripathy, D.; Syed, T.; Al-Haddad, W.; Dhindsa, S.; Dandona, P. Evidence for a potent antiinflammatory effect of rosiglitazone. J. Clin. Endocrinol. Metab. 2004, 89, 2728–2735. [Google Scholar] [CrossRef]
- Chen, H.; Zhu, J.; Liu, Y.; Dong, Z.; Liu, H.; Liu, Y.; Zhou, X.; Liu, F.; Chen, G. Lipopolysaccharide induces chronic kidney injury and fibrosis through activation of mTOR signaling in macrophages. Am. J. Nephrol. 2015, 42, 305–317. [Google Scholar] [CrossRef]
- Magder, S.; Parthenis, D.; Ghouleh, I. Preservation of Renal Blood Flow by the Antioxidant EUK-134 in LPS-Treated Pigs. Int. J. Mol. Sci. 2015, 16, 6801–6817. [Google Scholar] [CrossRef] [Green Version]
- Abbott, M.J.; Tang, T.; Sul, H.S. The role of phospholipase A2-derived mediators in obesity. Drug Discov. Today Disease Mech. 2010, 7, e213–e218. [Google Scholar] [CrossRef]
- Hsieh, P.S.; Jin, J.S.; Chiang, C.F.; Chan, P.C.; Chen, C.H.; Shih, K.C. COX-2-mediated inflammation in fat is crucial for obesity-linked insulin resistance and fatty liver. Obesity 2009, 17, 1150–1157. [Google Scholar] [CrossRef]
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L.; Lan, H.-Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989. [Google Scholar] [CrossRef]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998. [Google Scholar] [CrossRef]
- Montford, J.R.; Lehman, A.M.; Scobey, M.S.; Weiser-Evans, M.C.; Nemenoff, R.A.; Furgeson, S.B. Cytosolic phospholipase A2α increases proliferation and de-differentiation of human renal tubular epithelial cells. Prostaglandins Lipid Mediat. 2016, 126, 1–8. [Google Scholar] [CrossRef]
- Hegen, M.; Sun, L.; Uozumi, N.; Kume, K.; Goad, M.E.; Nickerson-Nutter, C.L.; Shimizu, T.; Clark, J.D. Cytosolic phospholipase A2α–deficient mice are resistant to collagen-induced arthritis. J. Exp. Med. 2003, 197, 1297–1302. [Google Scholar] [CrossRef]
- Challis, J.R.; Matthews, S.G.; Gibb, W.; Lye, S.J. Endocrine and paracrine regulation of birth at term and preterm. Endocr. Rev. 2000, 21, 514–550. [Google Scholar] [CrossRef]
- Challis, J.R.; Sloboda, D.M.; Alfaidy, N.; Lye, S.J.; Gibb, W.; Patel, F.A.; Whittle, W.L.; Newnham, J.P. Prostaglandins and mechanisms of preterm birth. Reprod. Camb. 2002, 124, 1–17. [Google Scholar] [CrossRef]
- Belikov, A.V.; Schraven, B.; Simeoni, L. T cells and reactive oxygen species. J. Biomed. Sci. 2015, 22, 85. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.-F.; Yeh, J.-C.; Ho, C.-T.; Liu, S.-H.; Hsieh, C.-Y.; Wang, T.-M.; Chang, S.-W.; Lee, I.-T.; Huang, K.-Y.; Wang, J.-Y.; et al. Targeting ROS and cPLA2/COX2 Expressions Ameliorated Renal Damage in Obese Mice with Endotoxemia. Int. J. Mol. Sci. 2019, 20, 4393. https://doi.org/10.3390/ijms20184393
Chang J-F, Yeh J-C, Ho C-T, Liu S-H, Hsieh C-Y, Wang T-M, Chang S-W, Lee I-T, Huang K-Y, Wang J-Y, et al. Targeting ROS and cPLA2/COX2 Expressions Ameliorated Renal Damage in Obese Mice with Endotoxemia. International Journal of Molecular Sciences. 2019; 20(18):4393. https://doi.org/10.3390/ijms20184393
Chicago/Turabian StyleChang, Jia-Feng, Jih-Chen Yeh, Chun-Ta Ho, Shih-Hao Liu, Chih-Yu Hsieh, Ting-Ming Wang, Shu-Wei Chang, I-Ta Lee, Kuo-Yang Huang, Jen-Yu Wang, and et al. 2019. "Targeting ROS and cPLA2/COX2 Expressions Ameliorated Renal Damage in Obese Mice with Endotoxemia" International Journal of Molecular Sciences 20, no. 18: 4393. https://doi.org/10.3390/ijms20184393