Transforming Growth Factor-β1 Selectively Recruits microRNAs to the RNA-Induced Silencing Complex and Degrades CFTR mRNA under Permissive Conditions in Human Bronchial Epithelial Cells

 , and
, and
Abstract
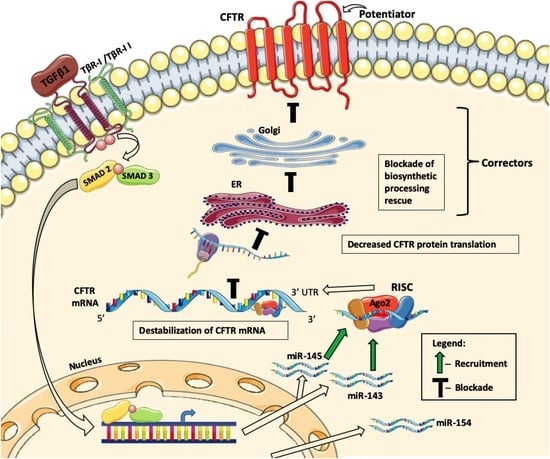
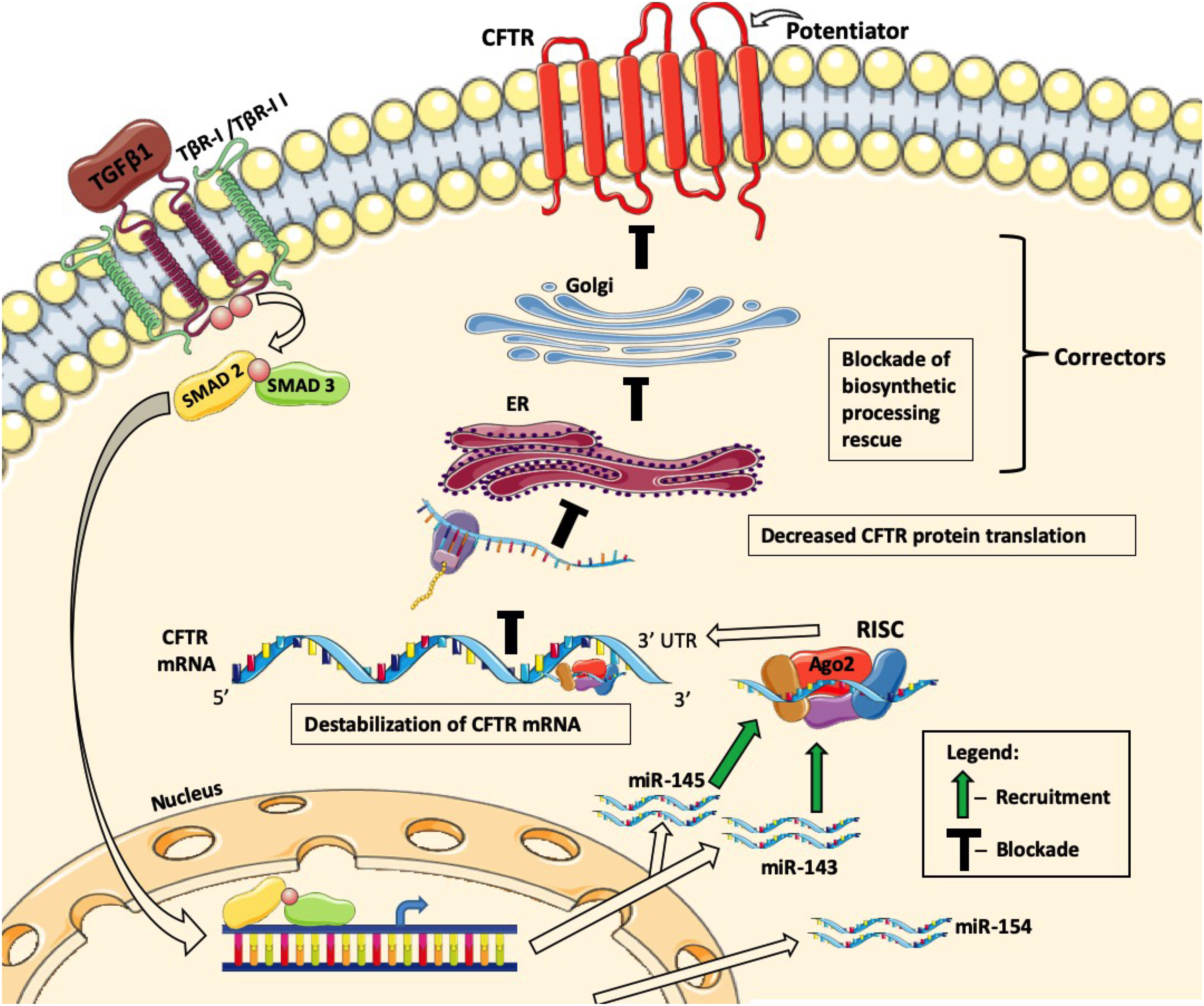
:
1. Introduction
2. Results
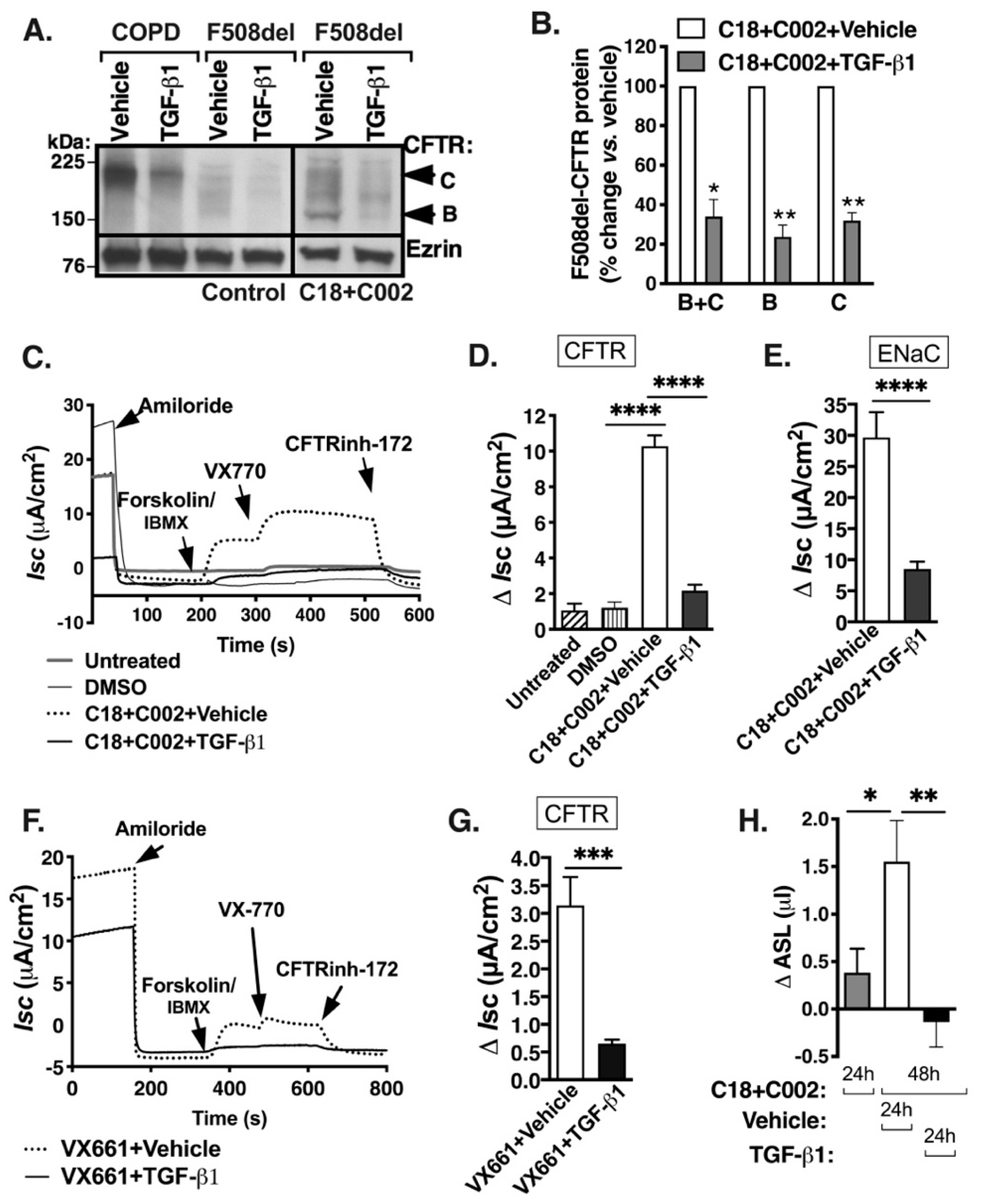
2.1. TGF-β1 Inhibits Rescue of F508del-CFTR and ASL Volume by the New-Generation Correctors
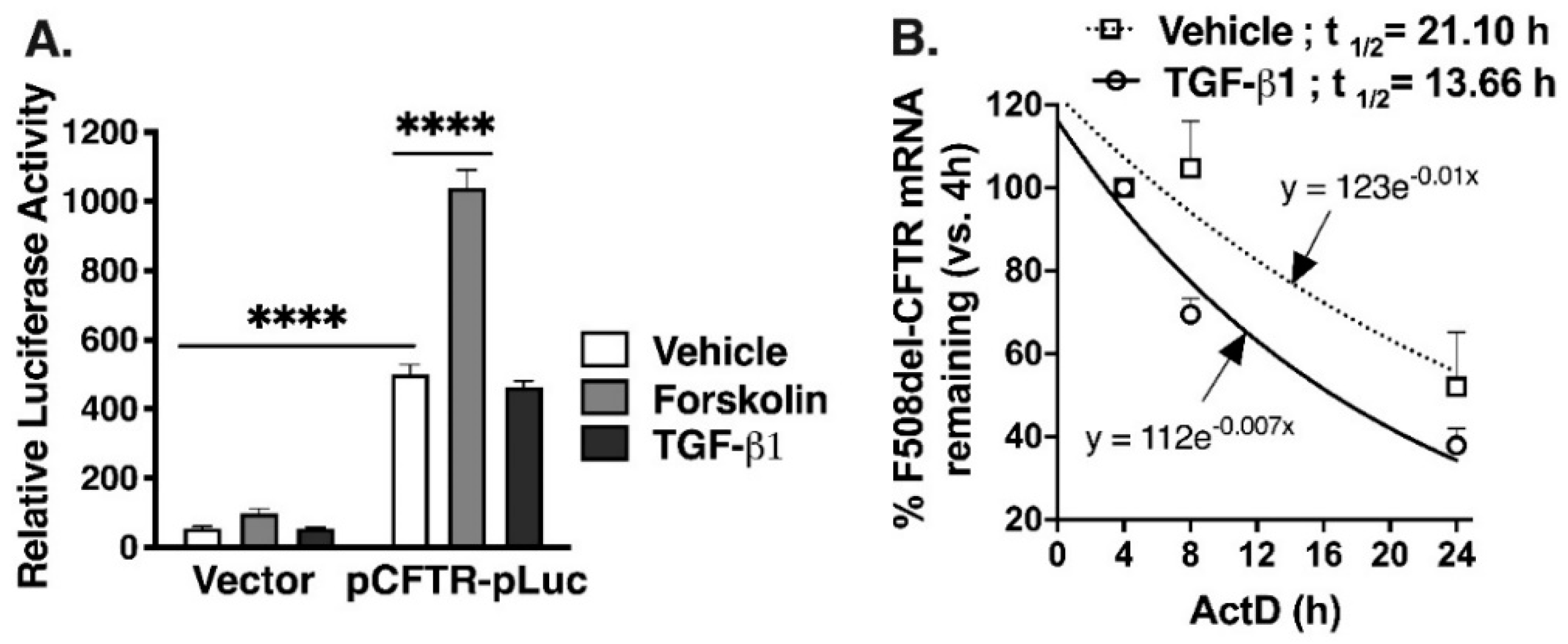
2.2. TGF-β1 Facilitates Degradation of CFTR mRNA
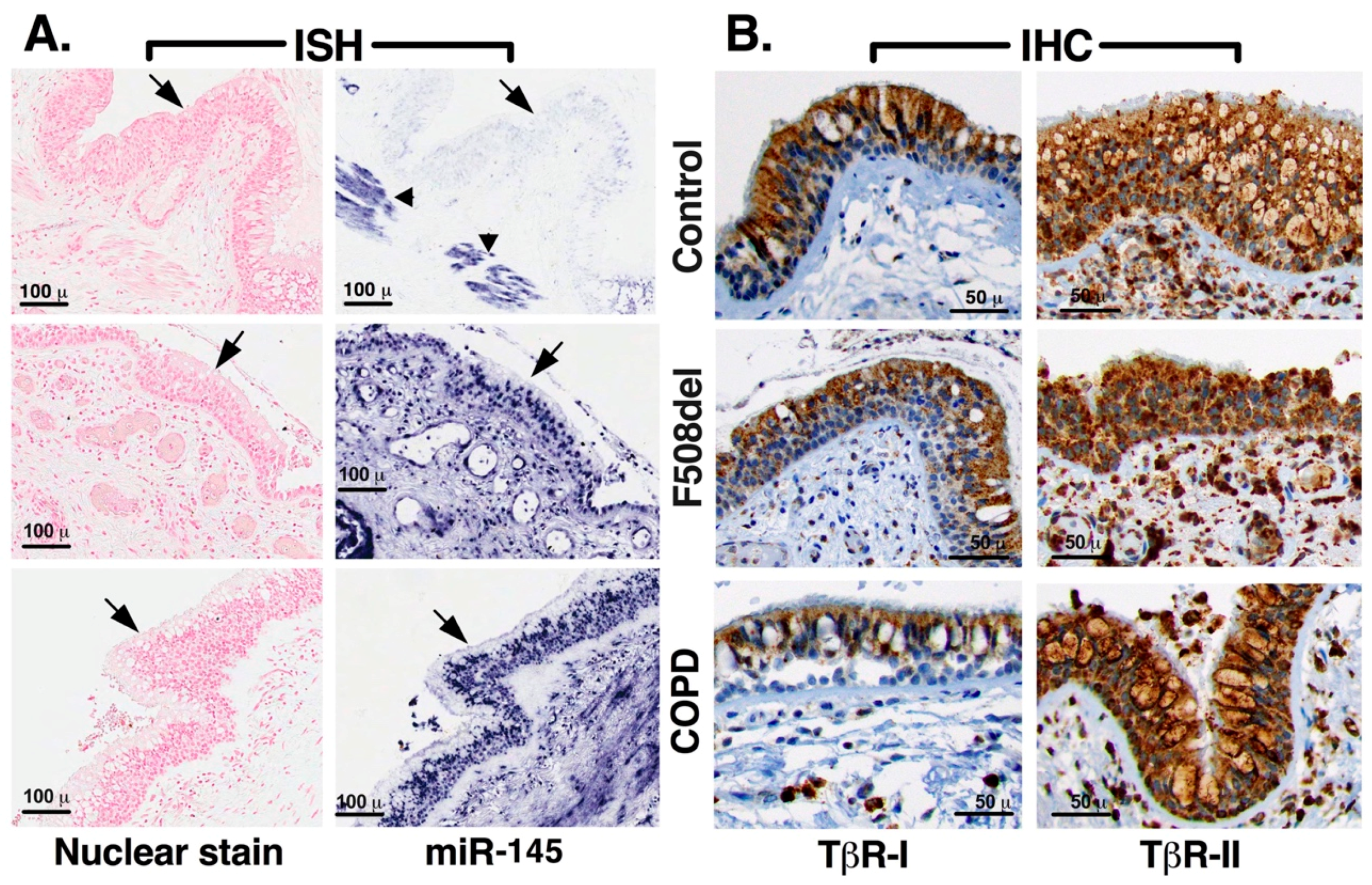
2.3. Native Bronchial Epithelia from Lungs WITH Chronic Disease Express Higher mir-145 Levels
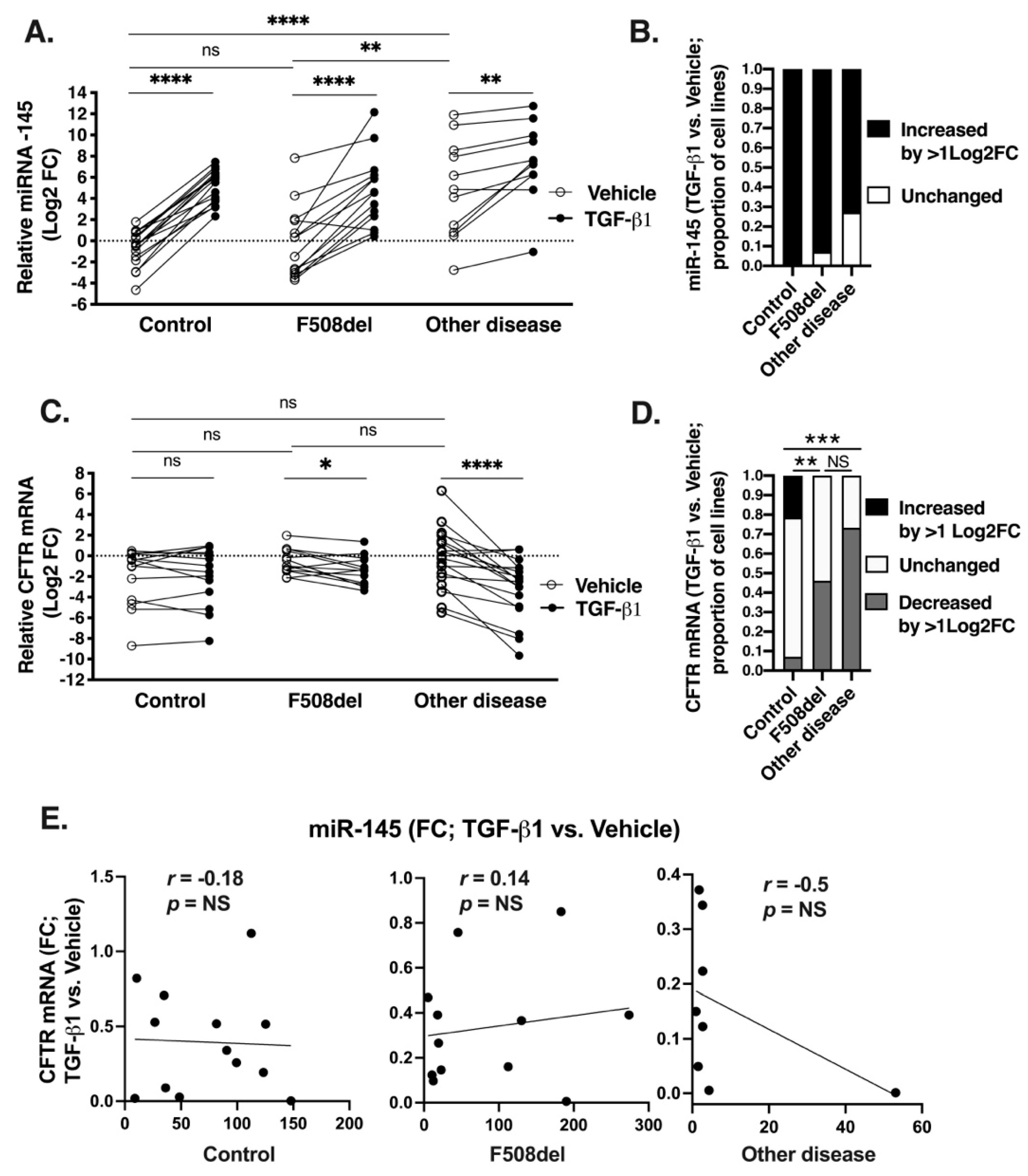
2.4. Upregulation of the Total Cellular miR-145 Levels by TGF-β1 Does Not Correlate with Repression of CFTR mRNA
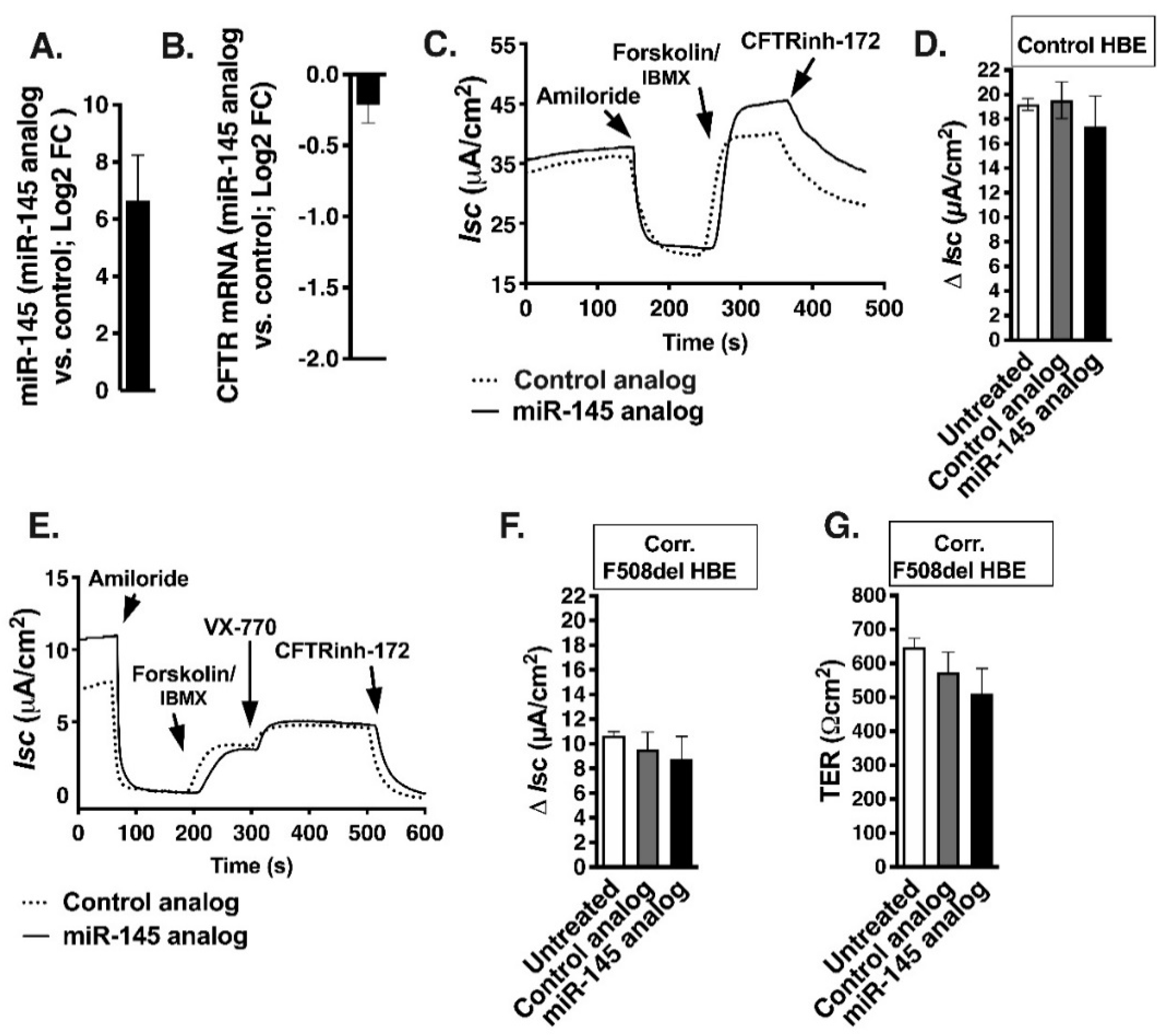
2.5. Decreasing miR-145 Expression Does Not Increase CFTR mRNA Level
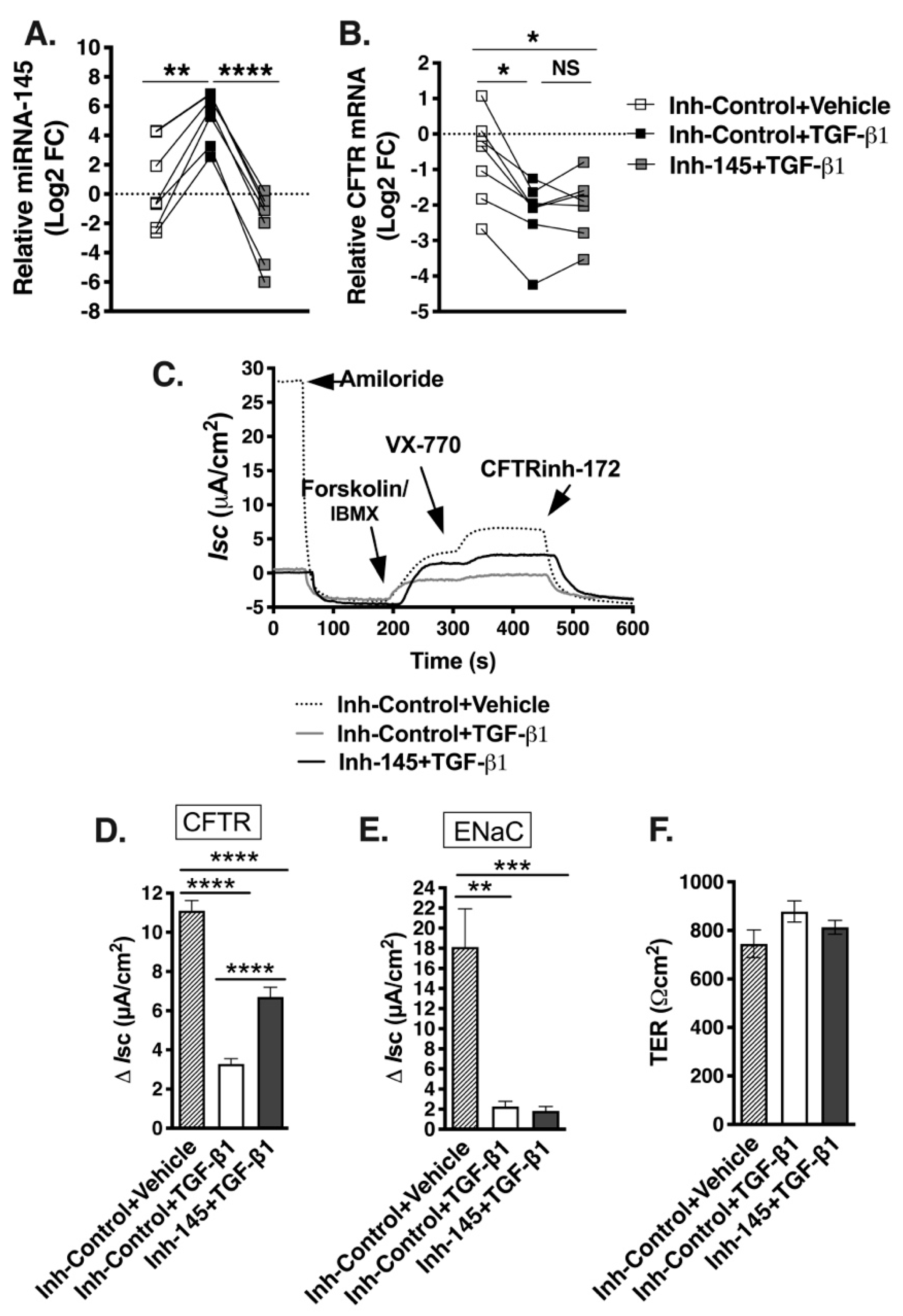
2.6. Anti-miR-145 Oligonucleotide Completely Blocks TGF-β1 Upregulation of miR-145 Expression but Only Partially Reverses TGF-β1 Inhibitory Effects on F508del-CFTR in Select HBE Cell Lines
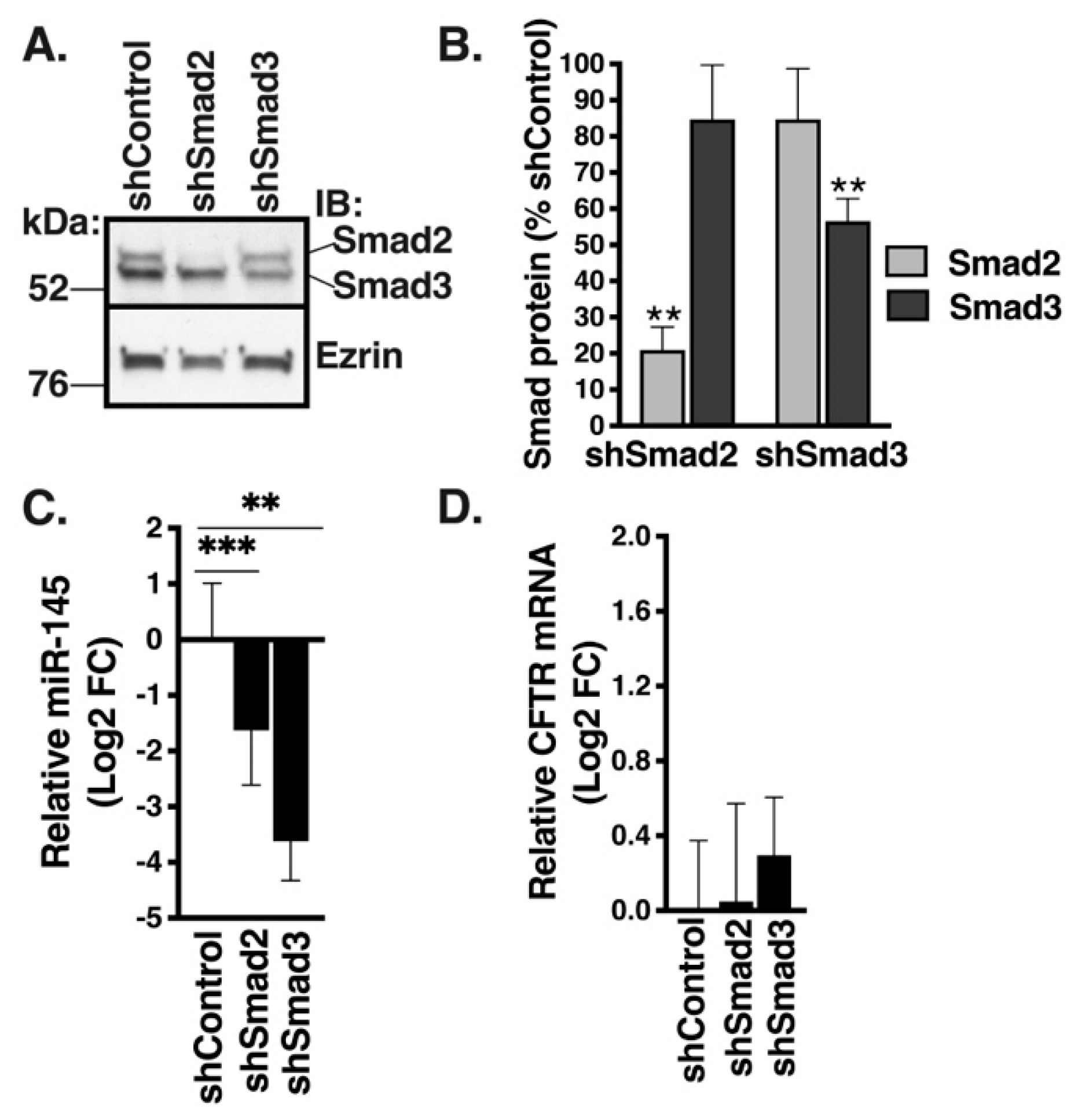
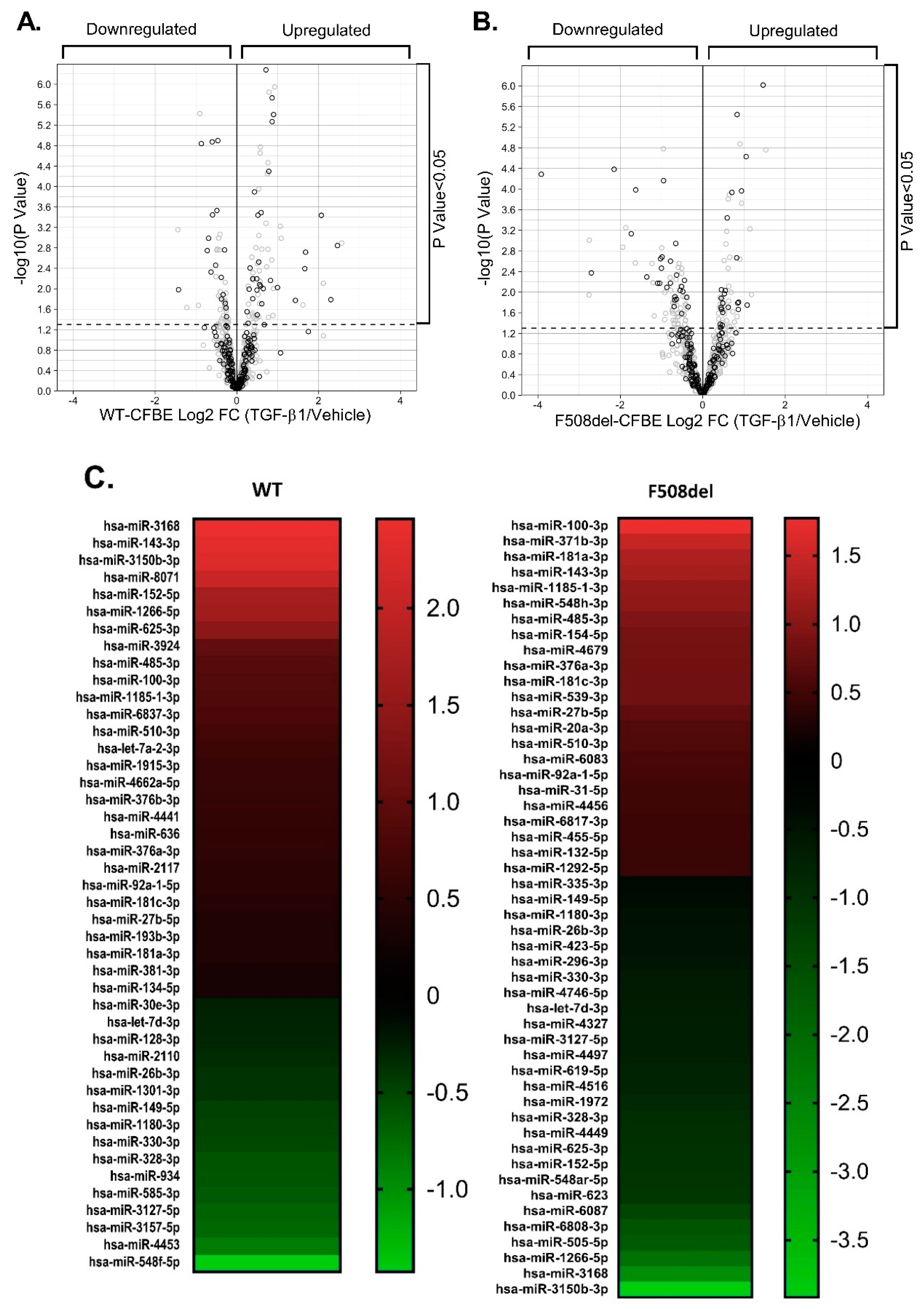
2.7. F508del-CFTR Modulates TGF-β1-Induced Changes in miRNA Landscape
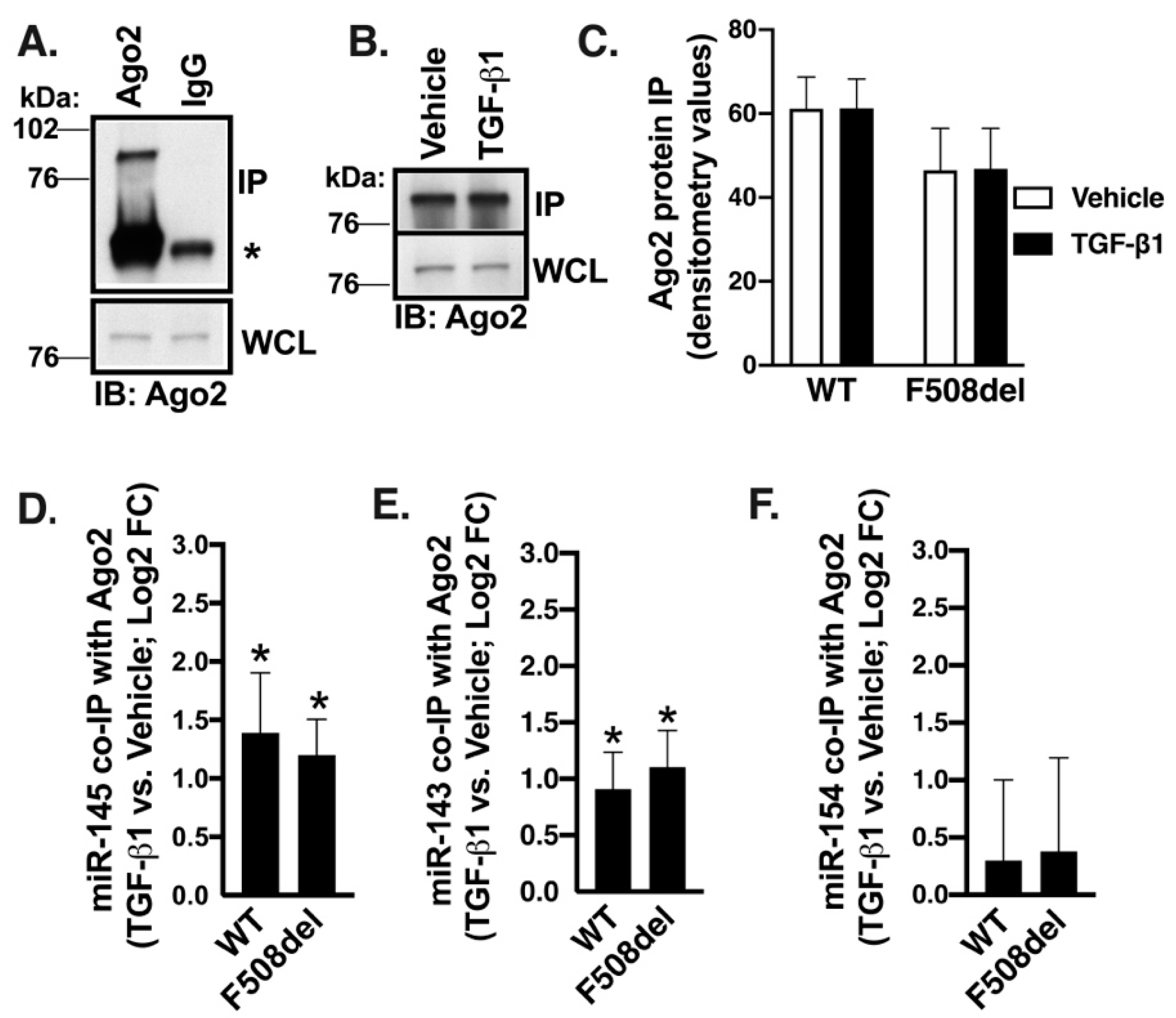
2.8. TGF-β1 Selectively Recruits Validated CFTR Inhibitors to RISC
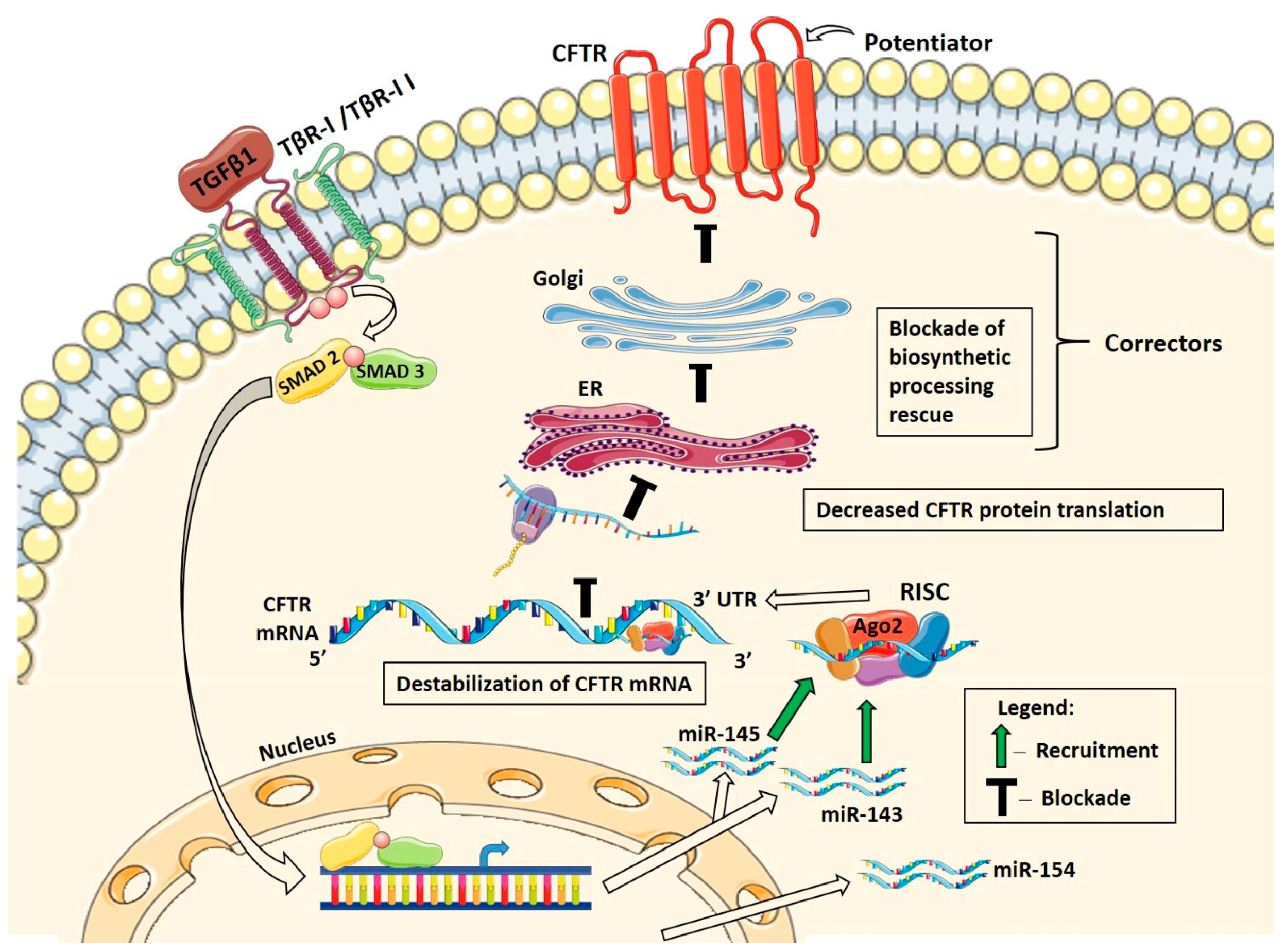
3. Discussion
4. Materials and Methods
4.1. Tissues, Cells and Cell Culture
4.2. Antibodies and Reagents
4.3. Western Blotting
4.4. Short Circuit Recordings
4.5. ASL Volume Measurements
4.6. CFTR Gene Promoter Reporter Assay
4.7. mRNA Degradation Measurements
4.8. In Situ Hybridization and lna™ Probes for miR-145
4.9. Immunohistochemistry
4.10. RNA Isolation
4.11. Real-Time Quantitative Reverse-Transcription PCR
4.12. miRNA qRT-PCR
4.13. miR-145 Inhibitor and Analog
4.14. RNA-Mediated Interference
4.15. Small RNA Library Preparation, Data Analysis, and miRNA Profiling
4.16. RISC IP Assay
4.17. Data and Code Availability
4.18. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ago2 | Argonaute 2 |
| AP | alkaline phosphatase |
| ASL | airway surface liquid |
| BSA | bovine serum albumin |
| cAMP | cyclic adenosine monophosphate |
| cDNA | complimentary DNA |
| CF | cystic fibrosis |
| CFFT | cystic fibrosis foundation therapeutics |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| CFTRinh-172 | thiazolidinone CFTR inhibitor 172 |
| COPD | chronic obstructive pulmonary disease |
| CRE | cAMP-responsive element |
| Ct | threshold cycle |
| DMSO | dimethyl sulfoxide |
| EMEM | Eagle’s minimal essential medium |
| ENaC | epithelial sodium channel |
| ER | endoplasmic reticulum |
| F508del | deletion of Phe508 |
| FEV1 | forced expiratory volume in one second |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GEO | gene expression omnibus |
| HBE | human bronchial epithelium |
| HEK | human embryonic kidney |
| IBMX | isobutylmethylxanthine |
| IHC | immunohistochemistry |
| IPF | idiopathic pulmonary fibrosis |
| IRB | institutional review board |
| Isc | short circuit current |
| ISH | in situ hybridization |
| LNA | locked nucleic acid |
| miR | microRNA |
| mRNA | messenger RNA |
| PAP | peroxidase, anti-peroxidase |
| RISC | RNA-induced silencing complex |
| rRNA | Ribosomal RNA |
| SNP | single nucleotide polymorphism |
| snRNA | small nuclear RNA |
| TGF-β1 | transforming growth factor β-1 |
| TER | trans-epithelial resistance |
| UMI | unique molecular identifiers |
| WT | wild type |
References
- Tarran, R.; Button, B.; Boucher, R.C. Regulation of normal and cystic fibrosis airway surface liquid volume by phasic shear stress. Annu. Rev. Physiol. 2006, 68, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.B. Cystic fibrosis since 1938. Am. J. Resp. Crit. Care 2006, 173, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Lazrak, A.; Fu, L.; Bali, V.; Bartoszewski, R.; Rab, A.; Havasi, V.; Keiles, S.; Kappes, J.; Kumar, R.; Lefkowitz, E.; et al. Silent codon change I507-ATC->ATT contributes to the severity of the DeltaF508 CFTR channel dysfunction. FASEB J. 2013, 27, 4630–4645. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Denning, G.M.; Ostedgaard, L.S.; Cheng, S.H.; Smith, A.E.; Welsh, M.J. Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. J. Clin. Investig. 1992, 89, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Swiatecka-Urban, A.; Brown, A.; Moreau-Marquis, S.; Renuka, J.; Coutermarsh, B.; Barnaby, R.; Karlson, K.H.; Flotte, T.R.; Fukuda, M.; Langford, G.M.; et al. The short apical membrane half-life of rescued uF508-CFTR results from accelerated endocytosis uF508-CFTR in polarized human airway epithelial cells. J. Biol. Chem. 2005, 280, 36762–36772. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2012, 363, 1991–2003. [Google Scholar] [CrossRef]
- Polenakovik, H.M.; Sanville, B. The use of ivacaftor in an adult with severe lung disease due to cystic fibrosis (DeltaF508/G551D). J. Cyst. Fibros. 2013, 12, 530–531. [Google Scholar] [CrossRef]
- Accurso, F.J.; Van Goor, F.; Zha, J.; Stone, A.J.; Dong, Q.; Ordonez, C.L.; Rowe, S.M.; Clancy, J.P.; Konstan, M.W.; Hoch, H.E.; et al. Sweat chloride as a biomarker of CFTR activity: Proof of concept and ivacaftor clinical trial data. J. Cyst. Fibros. 2014, 13, 139–147. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Pilewski, J.; Griese, M.; Dong, Q.; Lee, P.-S. VX-661, an investigational CFTR corrector, in combination with ivacaftor, a CFTR potentiator, in patients with CF and homozygous for the F508Del-CFTR mutation: Interim analysis. J. Cyst. Fibros. 2013, 12, S14. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Hug, C.; Marigowda, G.; Tian, S.; Huang, X.; Stanojevic, S.; Milla, C.E.; Robinson, P.D.; Waltz, D.; Davies, J.C.; et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: A randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017, 5, 557–567. [Google Scholar] [CrossRef]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Edmondson, C.; Davies, J.C. Predicting the Future of Cystic Fibrosis Lung Disease: Gene Expression Holds Some of the Answers. Ann. Am. Thorac. Soc. 2018, 15, 556–557. [Google Scholar] [CrossRef]
- Drumm, M.L. Modifier genes and variation in cystic fibrosis. Respir Res. 2001, 2, 125–128. [Google Scholar] [CrossRef]
- Drumm, M.L.; Konstan, M.W.; Schluchter, M.D.; Handler, A.; Pace, R.; Zou, F.; Zariwala, M.; Fargo, D.; Xu, A.; Dunn, J.M.; et al. Genetic modifiers of lung disease in cystic fibrosis. N. Engl. J. Med. 2005, 353, 1443–1453. [Google Scholar] [CrossRef]
- Bremer, L.A.; Blackman, S.M.; Vanscoy, L.L.; McDougal, K.E.; Bowers, A.; Naughton, K.M.; Cutler, D.J.; Cutting, G.R. Interaction between a novel TGFB1 haplotype and CFTR genotype is associated with improved lung function in cystic fibrosis. Hum. Mol. Genet. 2008, 17, 2228–2237. [Google Scholar] [CrossRef] [Green Version]
- Cutting, G.R. Modifier genes in Mendelian disorders: The example of cystic fibrosis. Ann. N Y Acad. Sci. 2010, 1214, 57–69. [Google Scholar] [CrossRef]
- Roux, J.; Carles, M.; Koh, H.; Goolaerts, A.; Ganter, M.T.; Chesebro, B.B.; Howard, M.; Houseman, B.T.; Finkbeiner, W.; Shokat, K.M.; et al. Transforming growth factor β1 inhibits cystic fibrosis transmembrane conductance regulator-dependent cAMP-stimulated alveolar epithelial fluid transport via a phosphatidylinositol 3-kinase-dependent mechanism. J. Biol. Chem. 2010, 285, 4278–4290. [Google Scholar] [CrossRef] [PubMed]
- Snodgrass, S.M.; Cihil, K.M.; Cornuet, P.K.; Myerburg, M.M.; Swiatecka-Urban, A. Tgf-β1 Inhibits Cftr Biogenesis and Prevents Functional Rescue of DeltaF508-Cftr in Primary Differentiated Human Bronchial Epithelial Cells. PLoS ONE 2013, 8, e63167. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Harris, W.T.; Kortyka, S.; Kotha, K.; Ostmann, A.J.; Rezayat, A.; Sridharan, A.; Sanders, Y.; Naren, A.P.; Clancy, J.P. Tgf-β downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS ONE 2014, 9, e106842. [Google Scholar] [CrossRef] [PubMed]
- Lutful Kabir, F.; Ambalavanan, N.; Liu, G.; Li, P.; Solomon, G.M.; Lal, C.V.; Mazur, M.; Halloran, B.; Szul, T.; Gerthoffer, W.T.; et al. MicroRNA-145 Antagonism Reverses TGF-β Inhibition of F508del CFTR Correction in Airway Epithelia. Am. J. Respir. Crit. Care Med. 2018, 197, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Wilczynska, A.; Bushell, M. The complexity of miRNA-mediated repression. Cell Death Differ. 2015, 22, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Flores, O.; Kennedy, E.M.; Skalsky, R.L.; Cullen, B.R. Differential RISC association of endogenous human microRNAs predicts their inhibitory potential. Nucleic Acids Res. 2014, 42, 4629–4639. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Chotirmall, S.H.; McElvaney, N.G.; Greene, C.M. Regulation of cystic fibrosis transmembrane conductance regulator by microRNA-145, -223, and -494 is altered in DeltaF508 cystic fibrosis airway epithelium. J. Immunol. 2013, 190, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; Cialfi, S.; Cimino, G.; De Biase, R.V.; Dominici, C.; Quattrucci, S.; Pizzuti, A. Elevated levels of miR-145 correlate with SMAD3 down-regulation in cystic fibrosis patients. J. Cyst. Fibros. 2013, 12, 797–802. [Google Scholar] [CrossRef]
- Butz, H.; Racz, K.; Hunyady, L.; Patocs, A. Crosstalk between TGF-β signaling and the microRNA machinery. Trends Pharmacol. Sci. 2012, 33, 382–393. [Google Scholar] [CrossRef]
- Santos, J.I.; Teixeira, A.L.; Dias, F.; Gomes, M.; Nogueira, A.; Assis, J.; Medeiros, R. Restoring TGFβ1 pathway-related microRNAs: Possible impact in metastatic prostate cancer development. Tumour Biol. 2014, 35, 6245–6253. [Google Scholar] [CrossRef]
- Suzuki, H.I. MicroRNA Control of TGF-β Signaling. Int. J. Mol. Sci. 2018, 19, 1901. [Google Scholar] [CrossRef] [PubMed]
- Mayorga, M.E.; Penn, M.S. miR-145 is differentially regulated by TGF-β1 and ischaemia and targets Disabled-2 expression and wnt/β-catenin activity. J. Cell Mol. Med. 2012, 16, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Davis-Dusenbery, B.N.; Chan, M.C.; Reno, K.E.; Weisman, A.S.; Layne, M.D.; Lagna, G.; Hata, A. down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-β and bone morphogenetic protein 4. J. Biol. Chem. 2011, 286, 28097–28110. [Google Scholar] [CrossRef] [PubMed]
- Viart, V.; Bergougnoux, A.; Bonini, J.; Varilh, J.; Chiron, R.; Tabary, O.; Molinari, N.; Claustres, M.; Taulan-Cadars, M. Transcription factors and miRNAs that regulate fetal to adult CFTR expression change are new targets for cystic fibrosis. Eur. Respir. J. 2015, 45, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, X.; Wu, Y.; Fang, P.; Shi, H.; Xu, J.; Li, M. Effects of miRNA-145 on airway smooth muscle cells function. Mol. Cell. Biochem. 2015, 409, 135–143. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, L.; Sevinc, K.; Papazoglou, I.M.; Tildy, B.; Detillieux, K.; Halayko, A.J.; Chung, K.F.; Perry, M.M. Airway smooth muscle inflammation is regulated by microRNA-145 in COPD. FEBS Lett. 2016, 590, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-β on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Kang, H. Role of MicroRNAs in TGF-β Signaling Pathway-Mediated Pulmonary Fibrosis. Int. J. Mol. Sci. 2017, 18, 2527. [Google Scholar] [CrossRef]
- Gillen, A.E.; Gosalia, N.; Leir, S.H.; Harris, A. MicroRNA regulation of expression of the cystic fibrosis transmembrane conductance regulator gene. Biochem. J. 2011, 438, 25–32. [Google Scholar] [CrossRef]
- Ramachandran, S.; Karp, P.H.; Osterhaus, S.R.; Jiang, P.; Wohlford-Lenane, C.; Lennox, K.A.; Jacobi, A.M.; Praekh, K.; Rose, S.D.; Behlke, M.A.; et al. Post-transcriptional regulation of cystic fibrosis transmembrane conductance regulator expression and function by microRNAs. Am. J. Respir Cell Mol. Biol. 2013, 49, 544–551. [Google Scholar] [CrossRef]
- De Santi, C.G.S.; Swiatecka-Urban, A.; Greene, C.M. Identification of a novel functional miR-143-5p recognition element in the Cystic Fibrosis Transmembrane Conductance Regulator 3′UTR. AIMS Genet. 2018, 5, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.K.; Chinnapaiyan, S.; Rasmussen, L.; Raju, S.V.; Unwalla, H.J. A Neutralizing Aptamer to TGFBR2 and miR-145 Antagonism Rescue Cigarette Smoke- and TGF-β-Mediated CFTR Expression. Mol. Ther. 2019, 27, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Holleran, J.P.; Glover, M.L.; Peters, K.W.; Bertrand, C.A.; Watkins, S.C.; Jarvik, J.W.; Frizzell, R.A. Pharmacological rescue of the mutant cystic fibrosis transmembrane conductance regulator (CFTR) detected by use of a novel fluorescence platform. Mol. Med. 2012, 18, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Luz, S.; Cihil, K.M.; Brautigan, D.L.; Amaral, M.D.; Farinha, C.M.; Swiatecka-Urban, A. LMTK2 Mediated Phosphorylation Regulates CFTR Endocytosis in Human Airway Epithelial Cells. J. Biol. Chem. 2014, 289, 15080–15093. [Google Scholar] [CrossRef] [PubMed]
- Astrand, A.B.; Hemmerling, M.; Root, J.; Wingren, C.; Pesic, J.; Johansson, E.; Garland, A.L.; Ghosh, A.; Tarran, R. Linking increased airway hydration, ciliary beating, and mucociliary clearance through ENaC inhibition. Am. J. Physiol Lung Cell Mol. Physiol. 2015, 308, L22–L32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstas, A.A.; Koch, J.P.; Korbmacher, C. cAMP-dependent activation of CFTR inhibits the epithelial sodium channel (ENaC) without affecting its surface expression. Pflugers Arch. 2003, 445, 513–521. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Poligone, E.G.; Stutts, M.J. CFTR regulation of ENaC. Methods Mol. Med. 2002, 70, 343–364. [Google Scholar] [PubMed]
- Frank, J.; Roux, J.; Kawakatsu, H.; Su, G.; Dagenais, A.; Berthiaume, Y.; Howard, M.; Canessa, C.M.; Fang, X.; Sheppard, D.; et al. Transforming growth factor-β1 decreases expression of the epithelial sodium channel alphaENaC and alveolar epithelial vectorial sodium and fluid transport via an ERK1/2-dependent mechanism. J. Biol. Chem. 2003, 278, 43939–43950. [Google Scholar] [CrossRef]
- Kramer, E.L.; Hardie, W.D.; Madala, S.K.; Davidson, C.; Clancy, J.P. Subacute TGFβ expression drives inflammation, goblet cell hyperplasia, and pulmonary function abnormalities in mice with effects dependent on CFTR function. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L456–L465. [Google Scholar] [CrossRef]
- Harvey, P.R.; Tarran, R.; Garoff, S.; Myerburg, M.M. Measurement of the airway surface liquid volume with simple light refraction microscopy. Am. J. Respir. Cell Mol. Biol. 2011, 45, 592–599. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Rab, A.; Twitty, G.; Stevenson, L.; Fortenberry, J.; Piotrowski, A.; Dumanski, J.P.; Bebok, Z. The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J. Biol. Chem. 2008, 283, 12154–12165. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Rab, A.; Fu, L.; Bartoszewska, S.; Collawn, J.; Bebok, Z. CFTR expression regulation by the unfolded protein response. Methods Enzymol. 2011, 491, 3–24. [Google Scholar] [PubMed]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, J.; Li, Y.; Li, X.; Yang, T.; Yang, Q.; Jiang, Y. Deregulation of serum microRNA expression is associated with cigarette smoking and lung cancer. Biomed. Res. Int. 2014, 2014, 364316. [Google Scholar] [CrossRef]
- Vejnar, C.E.; Zdobnov, E.M. MiRmap: Comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012, 40, 11673–11683. [Google Scholar] [CrossRef]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef]
- Harris, W.T.; Muhlebach, M.S.; Oster, R.A.; Knowles, M.R.; Noah, T.L. Transforming growth factor-β(1) in bronchoalveolar lavage fluid from children with cystic fibrosis. Pediatr. Pulmonol. 2009, 44, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.T.; Muhlebach, M.S.; Oster, R.A.; Knowles, M.R.; Clancy, J.P.; Noah, T.L. Plasma TGF-β in pediatric cystic fibrosis: Potential biomarker of lung disease and response to therapy. Pediatr. Pulmonol. 2012, 46, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Brazova, J.; Sismova, K.; Vavrova, V.; Bartosova, J.; Macek, M., Jr.; Lauschman, H.; Sediva, A. Polymorphisms of TGF-β1 in cystic fibrosis patients. Clin. Immunol. 2006, 121, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, R.; Sandford, A.; Taylor, C.; Huang, B.; Frangolias, D.; Wang, Y.; Sang, R.; Pereira, L.; Sun, L.; Berthiaume, Y.; et al. Complex two-gene modulation of lung disease severity in children with cystic fibrosis. J. Clin. Investig. 2008, 118, 1040–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dransfield, M.T.; Wilhelm, A.M.; Flanagan, B.; Courville, C.; Tidwell, S.L.; Raju, S.V.; Gaggar, A.; Steele, C.; Tang, L.P.; Liu, B.; et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 2013, 144, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Tanaka, M.; Takami, K.; Ohtoshi, T.; Ito, K.; Satoh, M.; Okada, Y.; Yamasawa, F.; Nakahara, K.; Umeda, A. Increased expression of transforming growth factor-β1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am. J. Respir. Crit. Care Med. 2001, 163, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Morty, R.E.; Konigshoff, M.; Eickelberg, O. Transforming growth factor-β signaling across ages: From distorted lung development to chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2009, 6, 607–613. [Google Scholar] [CrossRef]
- Mak, J.C.; Chan-Yeung, M.M.; Ho, S.P.; Chan, K.S.; Choo, K.; Yee, K.S.; Chau, C.H.; Cheung, A.H.; Ip, M.S.; Members of Hong Kong Thoracic Society COPD Study Group. Elevated plasma TGF-β1 levels in patients with chronic obstructive pulmonary disease. Respir. Med. 2009, 103, 1083–1089. [Google Scholar] [CrossRef]
- Sailland, J.; Grosche, A.; Baumlin, N.; Dennis, J.S.; Schmid, A.; Krick, S.; Salathe, M. Role of Smad3 and p38 Signalling in Cigarette Smoke-induced CFTR and BK dysfunction in Primary Human Bronchial Airway Epithelial Cells. Sci. Rep. 2017, 7, 10506. [Google Scholar] [CrossRef]
- Pruliere-Escabasse, V.; Fanen, P.; Dazy, A.C.; Lechapt-Zalcman, E.; Rideau, D.; Edelman, A.; Escudier, E.; Coste, A. TGF-β 1 downregulates CFTR expression and function in nasal polyps of non-CF patients. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L77–L83. [Google Scholar] [CrossRef]
- Unwalla, H.J.; Ivonnet, P.; Dennis, J.S.; Conner, G.E.; Salathe, M. Transforming growth factor-β1 and cigarette smoke inhibit the ability of β2-agonists to enhance epithelial permeability. Am. J. Respir Cell Mol. Biol. 2015, 52, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Janas, M.M.; Wang, B.; Harris, A.S.; Aguiar, M.; Shaffer, J.M.; Subrahmanyam, Y.V.; Behlke, M.A.; Wucherpfennig, K.W.; Gygi, S.P.; Gagnon, E.; et al. Alternative RISC assembly: Binding and repression of microRNA-mRNA duplexes by human Ago proteins. RNA 2012, 18, 2041–2055. [Google Scholar] [CrossRef] [PubMed]
- Arvey, A.; Larsson, E.; Sander, C.; Leslie, C.S.; Marks, D.S. Target mRNA abundance dilutes microRNA and siRNA activity. Mol. Syst. Biol. 2010, 6, 363. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.N.; Habermacher, R.; Martine, U.; Closs, E.I.; Filipowicz, W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 2006, 125, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Karginov, F.V.; Hannon, G.J. Remodeling of Ago2-mRNA interactions upon cellular stress reflects miRNA complementarity and correlates with altered translation rates. Genes Dev. 2013, 27, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Mayya, V.K.; Duchaine, T.F. On the availability of microRNA-induced silencing complexes, saturation of microRNA-binding sites and stoichiometry. Nucleic Acids Res. 2015, 43, 7556–7565. [Google Scholar] [CrossRef] [Green Version]
- Saunders, M.A.; Liang, H.; Li, W.H. Human polymorphism at microRNAs and microRNA target sites. Proc. Natl. Acad. Sci. USA 2007, 104, 3300–3305. [Google Scholar] [CrossRef] [Green Version]
- Amato, F.; Seia, M.; Giordano, S.; Elce, A.; Zarrilli, F.; Castaldo, G.; Tomaiuolo, R. Gene mutation in microRNA target sites of CFTR gene: A novel pathogenetic mechanism in cystic fibrosis? PLoS ONE 2013, 8, e60448. [Google Scholar] [CrossRef]
- Ji, Z.; Lee, J.Y.; Pan, Z.; Jiang, B.; Tian, B. Progressive lengthening of 3’ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 7028–7033. [Google Scholar] [CrossRef]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3’-UTR landscape across seven tumour types. Nat. Commun. 2014, 5, 5274. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Pal, J.K. Role of 5’- and 3’-untranslated regions of mRNAs in human diseases. Biol. Cell 2009, 101, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Myerburg, M.M.; Harvey, P.R.; Heidrich, E.M.; Pilewski, J.M.; Butterworth, M.B. Acute regulation of the epithelial sodium channel in airway epithelia by proteases and trafficking. Am. J. Respir. Cell Mol. Biol. 2010, 43, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Eickmeier, O.; Boom, L.V.; Schreiner, F.; Lentze, M.J.; Ngampolo, D.; Schubert, R.; Zielen, S.; Schmitt-Grohe, S. Transforming Growth Factor β 1 Genotypes in Relation to TGF β 1, Interleukin-8, and Tumor Necrosis Factor Alpha in Induced Sputum and Blood in Cystic Fibrosis. Mediators Inflamm. 2013, 2013, 913135. [Google Scholar] [CrossRef] [PubMed]
- Cihil, K.M.; Ellinger, P.; Fellows, A.; Stolz, D.B.; Madden, D.R.; Swiatecka-Urban, A. Disabled-2 protein facilitates assembly polypeptide-2-independent recruitment of cystic fibrosis transmembrane conductance regulator to endocytic vesicles in polarized human airway epithelial cells. J. Biol. Chem. 2012, 287, 15087–15099. [Google Scholar] [CrossRef]
- Manzanares, D.; Krick, S.; Baumlin, N.; Dennis, J.S.; Tyrrell, J.; Tarran, R.; Salathe, M. Airway Surface Dehydration by Transforming Growth Factor β (TGF-β) in Cystic Fibrosis Is Due to Decreased Function of a Voltage-dependent Potassium Channel and Can Be Rescued by the Drug Pirfenidone. J. Biol. Chem. 2015, 290, 25710–25716. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, G.J. In situ detection of microRNAs in paraffin embedded, formalin fixed tissues and the co-localization of their putative targets. Methods 2010, 52, 307–315. [Google Scholar] [CrossRef]
- Jorgensen, S.; Baker, A.; Moller, S.; Nielsen, B.S. Robust one-day in situ hybridization protocol for detection of microRNAs in paraffin samples using LNA probes. Methods 2010, 52, 375–381. [Google Scholar] [CrossRef]
- Elmen, J.; Lindow, M.; Silahtaroglu, A.; Bak, M.; Christensen, M.; Lind-Thomsen, A.; Hedtjarn, M.; Hansen, J.B.; Hansen, H.F.; Straarup, E.M.; et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008, 36, 1153–1162. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef]
- Barberan-Soler, S.; Vo, J.M.; Hogans, R.E.; Dallas, A.; Johnston, B.H.; Kazakov, S.A. Decreasing miRNA sequencing bias using a single adapter and circularization approach. Genome Biol. 2018, 19, 105. [Google Scholar] [CrossRef] [PubMed]
- Comincini, S.; Allavena, G.; Palumbo, S.; Morini, M.; Durando, F.; Angeletti, F.; Pirtoli, L.; Miracco, C. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol. Ther. 2013, 14, 574–586. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TISSUE | MIR-145 | TβR-I | TβR-II |
|---|---|---|---|
| CONTROL-1 * | 0 | +2 | +3 |
| CONTROL-2 | 0 | +2 | +3 |
| CONTROL-3 | 0 | +2 | +3 |
| F508del-1 | +3 | +2 | +2 |
| F508del-2 | +1 | +2 | +3 |
| F508del-3 * | +2 | +2 | +3 |
| COPD-1 * | +3 | +1 | +3 |
| COPD-2 | +3 | +2 | +3 |
| IPF-1 | +3 | +1 | +3 |
| MIRNA (N) | WT-CFBE CELLS | F508DEL-CFBE CELLS | ||
|---|---|---|---|---|
| UPREGULATED | 119 | Log2 FC Range 0.25 to 2.6 | 75 | Log2 FC Range 0.38 to 2.3 |
| p Value Range * to **** | p Value Range * to **** | |||
| DOWNREGULATED | 50 | Log2 FC Range −1.44 to −0.2 | 110 | Log2 FC Range −3.9 to −0.36 |
| p Value Range * to **** | p Value Range * to **** | |||
| TOTAL DYSREGULATED | 169 | / | 185 | p Value **** (Fisher’s exact test) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitash, N.; Mu, F.; Donovan, J.E.; Myerburg, M.M.; Ranganathan, S.; Greene, C.M.; Swiatecka-Urban, A. Transforming Growth Factor-β1 Selectively Recruits microRNAs to the RNA-Induced Silencing Complex and Degrades CFTR mRNA under Permissive Conditions in Human Bronchial Epithelial Cells. Int. J. Mol. Sci. 2019, 20, 4933. https://doi.org/10.3390/ijms20194933
Mitash N, Mu F, Donovan JE, Myerburg MM, Ranganathan S, Greene CM, Swiatecka-Urban A. Transforming Growth Factor-β1 Selectively Recruits microRNAs to the RNA-Induced Silencing Complex and Degrades CFTR mRNA under Permissive Conditions in Human Bronchial Epithelial Cells. International Journal of Molecular Sciences. 2019; 20(19):4933. https://doi.org/10.3390/ijms20194933
Chicago/Turabian StyleMitash, Nilay, Fangping Mu, Joshua E. Donovan, Michael M. Myerburg, Sarangarajan Ranganathan, Catherine M. Greene, and Agnieszka Swiatecka-Urban. 2019. "Transforming Growth Factor-β1 Selectively Recruits microRNAs to the RNA-Induced Silencing Complex and Degrades CFTR mRNA under Permissive Conditions in Human Bronchial Epithelial Cells" International Journal of Molecular Sciences 20, no. 19: 4933. https://doi.org/10.3390/ijms20194933