Genetically Engineered Pigs to Study Cancer

Chair of Livestock Biotechnology, School of Life Sciences, Technische Universität München, 85354 Freising, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(2), 488; https://doi.org/10.3390/ijms21020488
Submission received: 18 December 2019
/
Revised: 8 January 2020
/
Accepted: 9 January 2020
/
Published: 13 January 2020
(This article belongs to the Special Issue Genetically Engineered Mice to Study Cancer)
Abstract
:Recent decades have seen groundbreaking advances in cancer research. Genetically engineered animal models, mainly in mice, have contributed to a better understanding of the underlying mechanisms involved in cancer. However, mice are not ideal for translating basic research into studies closer to the clinic. There is a need for complementary information provided by non-rodent species. Pigs are well suited for translational biomedical research as they share many similarities with humans such as body and organ size, aspects of anatomy, physiology and pathophysiology and can provide valuable means of developing and testing novel diagnostic and therapeutic procedures. Porcine oncology is a new field, but it is clear that replication of key oncogenic mutation in pigs can usefully mimic several human cancers. This review briefly outlines the technology used to generate genetically modified pigs, provides an overview of existing cancer models, their applications and how the field may develop in the near future.

1. Introduction
Human lifespan is continually increasing, as are expectations of health and well-being [1]. There is accordingly greater concern about age-related diseases such as cancer, cardiovascular diseases and diabetes [2]. The overall incidence of cancer is increasing [3] and certain cancers, e.g., pancreatic, urgently require improved diagnosis and treatment. However, the number of approvals for new cancer drugs is lower than for other diseases [4].
Animals have long been studied to gain insight into human diseases and remain an essential part of cancer research. Valuable data can be derived from non-mammalian species such as zebrafish, Danio rerio. While principally a model of development, zebrafish have been used to model cancers e.g., liver and pancreatic cancer [5,6,7]. Zebrafish have the interesting advantage that embryos and larvae are naturally transparent and transparent adults can be generated [8,9]. This facilitates the study and tracking of tumor angiogenesis [10], metastasis [11,12], and the evaluation of anti-angiogenic agents [13,14] in vivo. However, zebrafish are very different to humans in size, lifespan and especially environmental factors.
Mice are by far the most frequently used laboratory mammal, mainly because of the ease with which they can be housed, bred, and genetically modified [15]. They have provided a wealth of knowledge regarding the molecular and genetic bases of many human cancers and facilitated many proof-of-principle studies. Their short gestation time and relatively inexpensive upkeep are clear advantages over larger animal species, but their usefulness for preclinical research also has limitations.
They differ considerably from humans in size, lifespan and aspects of organ anatomy, e.g., pancreas and spleen [16,17] and in the ease with which murine cells undergo oncogenic transformation compared with human cells [18,19].
Furthermore, replication of human oncogenic mutations in mice can fail to recapitulate the human pathology [20], and murine tumors differ in important clinical features such as drug response, possibly due to differences in immunology and drug metabolism [21,22].
The murine innate and adaptive immune systems differ significantly from humans, leading to different responses in inflammatory disease that can affect cancer development [23,24].
Consequently, many of the drugs evaluated in mice fail in clinical trials [25]. There is thus a need for non-rodent species to provide complementary data and improve the predictive value of preclinical studies.
2. Large Mammals as Biomedical Models
Larger animals, such as dogs, cats, non-human primates, and pigs each share some similarities with humans and have been considered as research species. Dogs and cats spontaneously develop tumors [26,27], and their veterinary treatment has provided valuable information for cancer research, but the use of companion animals in systematic experimental research is not well accepted by the general public. Similarly, strict regulatory requirements and ethical concerns restrict the use of non-human primates. However, pigs have been domesticated for centuries as a food source and their humane, ethical use as experimental animals under regulated conditions raises fewer concerns [28]. Pigs share many similarities with humans in body size, organ size and architecture, physiology and pathophysiology, and are thus a valuable species for biomedical research [29,30]. Pigs have long been used to study the effect of nutrition, to assess new surgery procedures or improve organ transplantation and for the development of imaging modalities where human scale equipment can be used. Moreover, their relatively long lifespan of 12–15 years [31] allows longitudinal studies to be carried out to assess or validate novel biomarkers, treatment or imaging options, follow disease progression and regression in a single animal [32]. For drug trials, pigs also show similar pharmacokinetic responses to humans [33,34]. Thus, pigs are recognized as a useful animal model for translational medicine (Figure 1).
The use of pigs in biomedicine does have disadvantages relative to smaller species, mainly because of the space required for housing and the time needed for breeding. Veterinary care and handling are also rather different to small laboratory mammals such as rats and mice, but pigs are a very common agricultural species so veterinary expertise is widely available and husbandry procedures well established. Perhaps the greatest difficulty has been in engineering precise genetic modifications into pigs but, as outlined in the next section, this is rapidly changing.
3. Generation of Genetically Modified Pigs
Genetic modification of large mammals began when Palmiter and Brinster produced transgenic rabbits, sheep and pigs by microinjection of DNA into the pronuclei of fertilized oocytes [35]. This procedure however resulted in a low proportion of transgenic offspring, usually 1–5% [36]. The high lipid content of porcine oocytes also makes it difficult to visualize the pronuclei. The generation of excess non-transgenic animals, often greater than 95%, was ethically and practically undesirable. Furthermore, DNA microinjection as originally conceived, enables only the addition of transgenes at random locations in the host genome. This prompted a search for more efficient and versatile methods.
The tremendous success of gene targeting in ES cells [37,38] revolutionized reverse genetics in mice and since led to a profusion of modified mouse lines. The potential usefulness of an equivalent method of transferring predetermined genetic modifications from cultured cells to lines of pigs has long been evident. However, despite numerous attempts over decades, the isolation and culture of definitive porcine ES cells, i.e., capable of populating the germline, had been unsuccessful [39,40]. This may now change, porcine expanded pluripotent stem cells (EPSCs) have recently been reported capable of forming all three germ layers in chimeric animals, but germ line contribution is still elusive [41].
The development of nuclear transfer from somatic cells grown in culture [42] provided the first alternative means of generating genetically modified livestock. Using in vitro transfected primary cells as nuclear donors increased the proportion of transgenic animals to virtually 100% [43] and, importantly, enabled gene targeting in mammals other than mouse [44]. While first established in sheep, nuclear transfer was soon extended to pigs for random transgenesis [45] and targeted gene knockout [46]. However, the procedure is technically challenging and very few lines of viable gene-targeted pigs were generated during the subsequent decade.
The use of tailor-made highly-specific endonucleases has now raised the efficiency of gene targeting. The well-known RNA-guided CRISPR-Cas9 endonuclease system is currently the method of choice, superseding earlier tools such as zinc-finger nucleases and TALENs [47,48]. Gene editing was quickly used in pigs to effect genetic alterations, including gene inactivation via small insertion/deletions (indels) as a consequence of non-homologous end-joining [49,50,51], or sequence replacement via homology-directed repair [52].
Inspired by work originated in mice [53] transgenic pigs have recently been generated that express a Cas9 transgene placed at the ROSA26 locus in a Cre-dependent manner [54], or ubiquitously throughout the body (own unpublished work). This enables gene editing of somatic cells in vivo by local delivery of single or multiple guide RNAs with or without Cre-recombinase. In vivo genome editing is still in its early stages, but offers a very powerful tool for controlled and efficient genome modification in defined organs and cell types at any age. For example in modelling cancer, tumor-supportive and -suppressive genes can be modified in successive rounds of gene editing to mimic the accumulation of mutations that accompany the progression of tumor entities [55]. Somatic modification also allows otherwise lethal or deleterious mutations to be studied in particular tissues or organs free of the complicating effects in the rest of the organism.
In practical terms, Cas9-expressing animals can reduce the time involved in generating and breeding new lines carrying mutations of interest, a significant advantage for larger species.
Site-specific recombination systems e.g., Cre/loxP or FLP/FRT have long been established in the mouse as a means of removing transcriptional stop cassettes to activate Cas9 or latent oncogenic alleles at chosen locations and even at a chosen time [56]. At the time of writing no pig line has yet been established that expresses Cre-recombinase in a tissue-specific manner.
This is clearly a deficiency, but efforts are underway to establish both Cre transgenic animals and also devise efficient means of delivering Cre, as DNA or protein, directly to the organ of choice.
Transgenic pigs that express a Cre-responsive dual fluorescent reporter provide an important tool to establish and assess the success of these methods [57]. Regarding the delivery of guide RNAs, numerous methods from viral vectors to nanoparticles are currently being developed in the mouse and will undoubtedly soon be transferred to the large animals.
These technical advances are central to the success of modelling human diseases in pigs. Some examples include skin wound healing [58,59]; modelling neurodegenerative diseases such as Alzheimer’s [60,61] and Huntington’s disease [62]; cardiovascular diseases [63,64], diabetes [65,66]; and monogenic diseases including Duchenne muscular dystrophy and cystic fibrosis [67,68]. These will increasingly inform how pigs can best contribute to preclinical studies, much as happened with mice over the past decades.
4. Porcine Cancer Models
The value of pig cancer models obviously depends on how faithfully they represent human disease. Porcine cancer biology is still a new field, but indications are that pigs can correctly mimic human cancers. Spontaneous cancers occur only rarely in wild type pigs and, as in humans, arise mostly with age [20]. Similar to humans, oncogenic transformation of porcine cells is a rare event that requires multiple genetic alterations [69]. A fundamental question has been whether replication of human oncogenic mutation(s) in a pig has an equivalent effect on cell transformation and tumorigenesis. So far, this does appear to be the case. Adam and colleagues introduced sets of overexpressed oncogenic transgenes into porcine primary fibroblast cells, which were tumorigenic when returned to the donor animals by autologous transplantation [70]. Our group has systematically investigated the stages of sarcomagenesis in vitro and found that porcine mesenchymal stem cells (MSCs) resemble human MSCs in that they require perturbation of p53, KRAS and MYC signaling pathways with spontaneous Rb pathway inactivation and telomerase-independent immortalization steps to convert to a fully transformed phenotype [71]. This contrasts with murine MSCs that can be transformed by loss of p53 function alone [72].
These findings suggest basic similarity between porcine and human oncogenesis, but in vitro culture, randomly integrated overexpressed transgenes and engraftment of transformed cells can all be criticized as artificial non-physiological methods. In our view, the generation of autochthonous tumor entities by replication of oncogenic lesions in endogenous porcine genes is the ‘gold standard’ and cancer models generated in this way are likely to be the most representative of human disease. In the examples given below both types of models will be presented and compared.
Table 1 provides an overview of genetically modified pig models for human cancers.
4.1. Porcine Models for Breast Cancer
Breast cancer is a common form of cancer and the leading cause of cancer-related death among women worldwide [85]. Despite tremendous advances in the past, incidence rates have been steadily increasing in the last decade [86]. Approximately 5–7% of all cases are diagnosed in women younger than 40 years old [87,88], whose disease progression is often more aggressive than in older women [89]. Breast cancer in young people is more often associated with germline mutations in the BRCA1/2 genes which constitute an increased familial risk for breast and ovarian cancer [90,91]. Indeed, the median ages at diagnosis for carriers of BRCA1 or BRCA2 mutations is 40 and 43 years [92]. BRCA1 and 2 are tumor suppressors that play an essential role in homologous repair of DNA breaks [93]. Thus, mutations in BRCA1/2 lead to genomic instability and predisposition to cancer [94].
Breast cancer was the subject of the first attempt to model a human cancer in genetically modified pigs. Nagashima and colleagues reported pigs carrying a v-Ha-ras oncogene with expression directed to mammary epithelium by a murine mammary tumor virus promoter, but observed no phenotype [73]. Transgene expression was detected in tissues such as lung and spleen, but was absent in mammary gland. This might have been due to a position effect of the randomly placed transgene cassette and/or silencing of the viral promoter by methylation. A later attempt aimed to inactivate the endogenous porcine BRCA1 locus, but heterozygous BRCA1 knockout piglets were inviable, whether due to the mutation or defects from nuclear transfer is not known [74]. Inactivation of BRCA1 in a porcine mammary cell line does result in a transformed phenotype resembling human breast cancer, suggesting that the pig is a suitable species to model breast cancer [95], but a representative pig model has yet to be produced.
Breast cancer remains such an important and common disease that further efforts are undoubtedly required. Indeed, the mammary gland is relatively accessible in living animals and would be a good candidate for in vivo gene editing using Cas9-expressing pigs, as described above. This would enable local inactivation or modification of key initiating genes such as BRCA1 and 2, and genes involved in disease progression e.g., TP53, and PIK3CA [96] without affecting overall animal development or viability.
4.2. Porcine Models for Colorectal Cancer
Colorectal cancer (CRC) is the third most common human cancer worldwide, and was the second leading cause of cancer-related deaths in 2018 [85]. While CRC incidence has decreased in patients older than 50 years, mostly due to routine screening, there has been an alarming increase in people under 50, and by 2030, colorectal cancer is expected to increase by more than 90 percent in people aged 20–34 years [97,98,99,100].
Colorectal cancer arises from the epithelial lining of the colon and rectum, with functional disruption of the tumor suppressor adenomatous polyposis coli (APC) the main event that initiates formation of adenomatous polyps [101,102]. As part of the β-catenin destruction complex, APC acts as a negative regulator of the Wnt pathway [103]. Loss or dysfunction leads to aberrant Wnt signaling resulting in increased proliferation and tumor formation [104]. Progression to cancer involves additional mutations and genomic instability [105].
In sporadic CRC, somatic APC mutations mainly occur in the mutation cluster region between codons 1281 and 1556 [106,107]. Germline mutations are found throughout the 5′ part of the gene, with two common hot spots at codons 1061 and 1309 [108], and are responsible for familial adenomatous polyposis (FAP), a hereditary predisposition for CRC that leads to the formation of large numbers of adenomatous polyps in the colon and rectum and adenomas at a young age [109]. If not removed in time, these premalignant lesions can turn into invasive adenocarcinoma.
Many mouse models have been generated to replicate human FAP. However, these have revealed that mutation of Apc alone is not sufficient to mimic the human phenotype in mice. The most commonly used ApcMin/+ mouse develops polyps predominantly in the small intestine rather than in the colon [110]. Successful FAP modelling in mice requires more complex modifications with tissue-specific Apc deletion and additional mutations [111,112,113].
Pigs have been generated that carry a translational stop codon at position 1311 in the endogenous porcine APC gene (APC1311), orthologous to a human APC1309 mutation responsible for a severe form of FAP [75]. Within their first year APC1311/+ pigs develop polyps in the colon and rectum (Figure 2) that show features typical of the human adenoma-carcinoma sequence, such as aberrant crypt foci and adenomas with low- and high-grade neoplasia and carcinoma in situ [75]. Porcine adenomas exhibit genetic and biochemical hallmarks of human FAP and sporadic CRC, such as loss of the wild type APC allele, β-catenin accumulation, high expression of its target gene c-MYC and mitogen-activated protein kinase (MAPK) pathway activation [75,114]. These results resemble the findings in human patients, where overexpression of c-MYC ensures tumor growth via metabolic reprogramming and survival of colon cancer stem cells [115,116].
An important advantage of initiating precancerous tumors from a single mutation such as APC1311 is that subsequent spontaneous events leading to cancer can be followed in detail, which is not possible in mice where additional engineered mutations are necessary to ‘force’ disease progression. For example, porcine polyps have been found to show microRNA dysregulation between low- and high-grade dysplasia [117], a natural feature of human cancer progression [118]. APC1311/+ pigs are also being used for preclinical studies, for example to evaluate the use of biodegradable fluorescent nanoparticles to visualize very early adenomas [119].
Others have also attempted to replicate colon cancer in pigs. Pigs with APC truncated by a premature stop codon at position 902 have been generated using TALENs, but no phenotype or polyposis has been reported [76].
Pigs with tissue-specific and 4-hydroxytamoxifen (4-OHT)-inducible expression of the oncogenic transgenes KRASG12D, cMYC, and SV40LT have been reported [77]. However, these pigs developed duodenal carcinoma rather than colon cancer, likely because the oncogenes were overexpressed from a random transgene and activation was regulated by the epithelial villin promoter which would be expected to drive expression throughout the whole intestine.
To date the APC1311/+ pigs are the only model carrying an endogenous mutation that leads to formation of polyposis in the colon and rectum. Due to their size and long lifespan, disease progression and mutation accumulation can be monitored by screening via colonoscopy. Ongoing studies are investigating the effects of diet and involvement of the microbiome in disease progression. One drawback of the porcine model is the slow progression to invasive cancer, as in humans. Artificial acceleration to late stage disease can however be engineered by introducing additional oncogenic mutations by breeding or in vivo genome editing of individual polyps.
4.3. Porcine Models for Pancreatic Cancer
Pancreatic cancer is the 11th most common cancer worldwide [85], but is a leading cause of cancer-related deaths due to the overwhelmingly poor prognosis [120]. Alarmingly, incidence is increasing and pancreatic cancer is expected to surpass colorectal cancer and breast cancer to become the second leading cause of cancer-related deaths in Germany and the United States by 2030 [121,122].
Pancreatic cancer mainly arises from the exocrine component, with less than 5% of all tumors developing from endocrine cells [123]. Pancreatic ductal adenocarcinoma (PDAC) accounts for more than 90% of exocrine malignancies. Several precursor lesions for PDAC have been described with proliferating epithelial lesions, pancreatic intraepithelial neoplasia (PanIN), the most prominent [124]. The main driver of PanIN formation is activation of the proto-oncogene KRAS, which is mutated in more than 90% of all PDAC cases, mostly a G to D amino acid substitution at codon 12 [125]. Progression of PanINs to PDAC is associated with accumulation of mutations in the tumor suppressor genes CDKN2A (p16), TP53, SMAD4 and BRCA1/2 [126].
PDAC was previously thought to derive solely from the epithelial lining of the pancreatic duct, but evidence now suggests acinar cells that undergo transdifferentiation to a ductal-like phenotype, acinar-to-ductal metaplasia (ADM). ADM is a reprogramming phenomenon that can be a consequence of stress and inflammation such as pancreatitis [127,128]. Indeed, PDAC has been shown to arise from both acinar and ductal cell types, but it is believed that acinar-derived PDAC develops via PanINs, while ductal-derived PDAC develops in a PanIN-independent manner [129,130].
The high morbidity of PDAC can be ascribed to several characteristics of the disease. Perhaps the most important is that it is aggressive and metastatic even at early stages, but is usually asymptomatic. Diagnoses tend to be made only when the tumor is advanced and unresectable due to complex vascular invasion and metastasis is in progress. Newly diagnosed patients thus have a five-year survival rate of only 9% [131]. While there are some reports of successful treatment of locally advanced PDAC by surgery and chemotherapy [132], such cases are rare. PDAC is also characterized by high intra-tumoral heterogeneity and plasticity that foster the emergence of drug-resistant populations that render most conventional therapies ineffective [133,134,135].
The search for better early diagnosis and effective treatments has motivated the generation of several mouse models to mimic human PDAC. The genetic requirements for PDAC development were defined in a series of key studies by Tuveson and colleagues. They reported that expression of KrasG12D directed by either the Ptf1a or Pdx1 promoters resulted in PanIN lesions, but these rarely developed to invasive carcinoma [136]. In contrast, combination of KrasG12D with deletion of Cdkn2a led to aggressive tumors that invaded other organs. This however resulted in death at 11 weeks, preventing the timely formation of distant metastases [137].
Most usefully it was shown that Pdx1-promoter-directed expression of Trp53R172H in combination with KrasG12D, orthologues of the most common mutations present in human PDAC, initiated the development of a widely metastatic PDAC in mice that recapitulated the main characteristics of the human disease [138].
The success of the mouse work has prompted efforts to generate similar models in pigs. Schook et al. have generated pigs with random transgenes containing Cre-inducible KRASG12D and TP53R167H mutations (orthologous to human TP53R175H) driven by the CAG-promoter [80]. Explanted cells from these animals transduced with adenovirus encoding Cre (AdCre) in vitro became transformed, while subcutaneous and intramuscular injection of AdCre led to tumor formation in vivo [80]. Interestingly, these pigs revealed marked intra-tumoral T-cell infiltration and an anti-tumor immune response regardless of the site of tumor formation [139]. These findings suggest that pig tumors are subject to strong immune surveillance, making them suitable to test possible immunotherapies. With regard to pancreatic cancer, AdCre administered to pancreatic duct cells in vitro render them immortal and capable of forming tumors when injected in immune-deficient mice [81]. Delivery of AdCre into the main porcine pancreatic duct in vivo gave rise to tumors that displayed features of human PDAC, e.g., a dense tumor stroma and E-cadherin expression. However, the pancreatic tumors also contained areas with neuroendocrine rather than PDAC phenotype [81], suggesting activation of the mutant transgenes in a variety of cell types. This is perhaps a consequence of non-specific viral transduction and the constitutively active CAG-promoter. Furthermore, these pigs showed no clinical symptoms, and tumors were not detectable by computer tomography one year after injection, but were found in the pancreatic duct upon resection [81]. The relevance of this model for human PDAC is thus not clear and perhaps illustrates the drawbacks of adding transgenes rather than modifying endogenous genes. Adding a transgene is necessarily artificial and can be non-physiological in some important respects. Normal gene dosage is disturbed because two endogenous non-mutant alleles are still in place. Transgenes placed at random locations are not subject to the normal regulatory influences and, depending on the constructs, can be expressed at artificially high levels.
Another group has reported a porcine model for pancreatic cancer based on overexpression of a multi-oncogene cassette consisting of KRASG12D, cMYC and SV40LT [82]. In contrast to the models of Schook and colleagues, oncogene expression was induced during embryogenesis using the murine Pdx1 promoter. None of the transgenic piglets survived long, most likely due to the cloning procedure. Pancreatic acinar cells of one piglet showed hyperplastic foci with colocalized oncogene expression and increased proliferation at day 45 after birth [82]. As Pdx1 is expressed throughout the whole pancreas during embryogenesis [140], and later becomes restricted to β-cells [141], oncogene activation would not be limited to acinar cells. The presence of hyperplastic foci in the acinar cell compartment could indicate PDAC development via ADM, but would require further investigation.
Another strategy used to generate a porcine PDAC model is orthotopic xenotransplantation of transformed cells. Explanted pancreatic ductal epithelial cells transformed by overexpression of KRASG12D and TP53R167H, and knock down of p16 and SMAD4 have been transplanted into the pancreas of immune-deficient mice. This resulted in the formation of metastatic tumors [142]. Implantation of ex vivo transformed cells into the pancreas of pigs has not yet been performed.
As this model uses transformed pancreatic ductal cells, the resulting tumors are likely to originate from this cell type [143], removing the uncertainty inherent in the transgenic models above. However, allogeneic implantation into pigs can lead to immune rejection and such tumors obviously originate in a quite different manner to spontaneous pancreatic cancers. Indeed, tumors from engrafted cells often derive from one or a few dominant cell clones and are thus less likely to recapitulate important features such as tumor heterogeneity.
Work towards another model has been based directly on Tuveson’s PDAC mice [138]. Schnieke and colleagues reported the first generation of pigs with latent KRASG12D and TP53R167H mutations engineered into the endogenous genes [78,79]. As in the equivalent murine alleles [136], expression is blocked by a floxed transcriptional stop cassette and activated by Cre recombination. Pigs carrying the latent KRASG12D and TP53R167H alleles in heterozygous form are viable and can be bred normally although, as described later, uninduced TP53R167H animals develop osteosarcoma in later life. This is a multi-component system that requires some means of specifically expressing Cre-recombinase in pancreas to activate the latent KRASG12D and TP53R167H alleles. This is not yet in place, but methods are being employed to deliver Cre locally into the porcine pancreas, and to use pancreatic promoters including PDX1 and PTF1A to direct expression. This work is aided by the use of a dual-fluorescent reporter pig that enables cells that have undergone Cre-recombination to be visualized by a switch in fluorescence [57]. While still incomplete this model has the advantage over the transgenic models that it should more closely mimic the events that cause spontaneous human PDAC.
4.4. Porcine Models for Osteosarcoma
Osteosarcoma (OS) is the major form of primary bone cancer, and is commonly located in the metaphyseal growth plates of the long bones of the extremities [144]. It predominantly affects young people and is highly malignant, requiring aggressive surgical resection and cytotoxic chemotherapy [145]. The 5-year survival rate has remained unchanged for decades, at ~60% for patients with primary OS and ~20% for patients with metastatic disease [146].
Most cases of human OS are sporadic, with identified risk factors that include rapid bone growth, exposure to radiation, and genetic diseases [144]. Increased incidence of OS is associated with Li-Fraumeni syndrome caused by germ line mutation of TP53 [147] and hereditary retinoblastoma caused by germ line mutation of RB1 [148,149].
Human OS displays high rates of chromosomal alterations and structural changes [150,151], and an overwhelming prevalence of mutations affecting p53 function [152,153]. While alterations have also been found in other genes including RB1, ATRX, and DLG2 [153], CDKN2A/B [154], PTEN [155], IGF1R [156], and several genes in the PI3K/mTOR pathway [155], defects affecting p53 predominate.
Three models of OS have been developed. Genetically engineered animal (mouse and pig) models, patient-derived primary tumor cells, and large breed dogs with spontaneous disease [28,157]. The patient-derived human cells and dog OS models are described in a recent review [157]. Of the genetically engineered models, OS development in mice was achieved by inactivating Trp53 and Rb1 [158]. Some Trp53 knockout mice developed OS, but the majority (75%) of mice homozygous for this mutation developed lymphomas [159,160]. Improved mouse OS models have been developed based on conditional Trp53 inactivation [161], and conditional activation of Trp53 hot spot mutations in the osteogenic lineage [162]. These show highly penetrant OS formation, mainly in the axial skeleton, a location rarely observed in human OS [163].
Two pig lines that develop OS have been described. Sieren et al. generated Yucatan minipigs that carry a R167H mutation in the endogenous TP53 gene that is ubiquitously expressed from the major P1 promoter [83]. They reported that heterozygous TP53R167H mutant pigs showed no tumor development even at 30 months of age, while those homozygotes that reached sexual maturity developed a variety of neoplastic lesions, including osteogenic tumors, lymphomas and renal tumors, broadly recapitulating the tumor spectrum observed in mice with the orthologous mutation.
As described in the section on pancreatic cancer, Schnieke and colleagues have generated pigs that carry a latent TP53R167H mutation in exon 5 that can be activated by Cre-recombination [78]. Observation of pigs over several years has revealed that pigs heterozygous and homozygous for the uninduced allele all develop OS [71]. Compared to the CRC model, the bone tumors develop rapidly in the homozygous animals, as early as six months old. Wilm’s tumors and lymphomas also occur occasionally. Porcine OS displays several similarities with the human disease. As in humans, it primarily affects the long bones, tumor cells show a highly abnormal karyotype and nuclei with atypical mitotic figures, and increased resistance to radiation [71]. The origin of human osteosarcoma is not well understood and these pigs provide a valuable resource to study the underlying mechanisms, an important step towards identifying possible drug targets. They also offer a resource to surgeons and clinicians seeking to improve surgical treatment and maintenance of this devasting condition.
5. Porcine Tumor Xenograft Models
Modelling human cancers by xenotransplantation of human cancer cells is well established in mice. Patient-derived xenograft (PDX) mouse models generated by subcutaneous or orthotopic grafting of human tumor samples into severe combined immunodeficient (SCID) mice [164,165] are currently preferred over xenografting of cell lines because those may have lost the original tumor heterogeneity during long periods in culture [166]. The PDX approach is thus a better predictor of human tumor behavior, and murine PDX models have been generated for colon, pancreatic and breast cancers [167,168,169].
Work is proceeding towards producing immunodeficient pigs that would enable a similar xenograft system. Nakayama and colleagues have generated immunodeficient pigs by removing the thymus and spleen in combination with drug immunosuppression [170]. Although effective, this approach is highly invasive and suitable for producing only small numbers of immunodeficient pigs. A better alternative is germline modification of genes required for B- and T-cell development, such as the X-linked interleukin-2 receptor gamma chain gene (IL2RG) or V(D)J recombination-activating genes (RAGs). Some groups have reported SCID pigs by disruption of IL2RG [171,172,173]. These animals lacked a thymus and showed loss or reduction of T- and NK-cells, but survival was poor, mostly due to infections such as pneumonia. Nevertheless, the pigs generated by Onishi and colleagues were subjected to allogeneic bone marrow transplantation and three survived for longer than 300 days [171]. Pigs with disruption of RAG1 and/or RAG2 also showed a SCID phenotype lacking T- and B-cells [174,175,176,177]. Kim and colleagues showed that injecting human iPS cells resulted in teratoma formation in RAG2-deficient pigs, demonstrating their value [175]. IL2RG and RAG2 double knockout pigs have also recently been reported [177].
The availability of SCID pigs will not only enable human cancer xenograft experiments, but will also allow transplantation of human immune cells to produce “humanized” pigs that can be used for drug and therapy testing [20]. However, immunodeficient pigs are necessarily more susceptible to infection and require pathogen-free housing.
Another means of avoiding immune rejection of tumor cells is based on xenografting into recipient animals in utero. Human cells have been injected into porcine fetuses before CD3+ lymphocytes populate the thymus [178,179]. Human hepatic cells injected into porcine fetal liver have been shown to engraft successfully [178], a process that can be used to tolerize a pig in preparation for further human cell transplantation after birth [20].
6. Future Perspectives and Challenges
Pigs are a relatively new species in which to study cancer. Their value for biomedical research will benefit from continuing increases in physiological, biochemical, immunological and genetic information, and the generation of new models is set to be simplified by improved techniques to modify the germline and somatic cells (Figure 3).
Future research will clearly involve the production of new pig lines, but there is also much to be gained by integrating the study of existing models with advanced culture systems. Tissues can be explanted and cultured as organoids, or grown in air–liquid interface culture systems. Culture systems that mimic the natural three-dimensional tissue organization and location in vivo will enable a wide range of manipulations and investigations to be carried out over relatively short timescales. These could include engineering panels of genetic modifications, coculture with microbiota, and exposure to drugs. Then, because pigs are long lived, explanted cells can be returned to the donor animal as autologous grafts and the effects studied in whole animals (Figure 3). For example, biopsy samples of adenomatous polyps from an APC1311 pig can be manipulated and characterized in vitro and then reimplanted to study cancer progression. Alternatively, in vivo genome editing of individual polyps can be performed by introducing different mutations into different polyps. Thus, both methods allow multiple experiments in a single animal to address complex questions. In addition, the Cas9-expressing pigs can be cross bred with APC, KRAS or TP53 mutant pigs to introduce additional mutations in later life.
The use of animals in biomedical research can be controversial, but there are currently no other means of studying disease in the context of the whole organism, such as interactions with the immune system. Researchers have a duty to ensure that animals are used as efficiently as possible. This means minimizing the numbers necessary to generate genetic modifications, and ensuring that animal models produce high quality predictive information relevant to human patients. Recent years have seen significant progress and we are confident that this will continue.
Author Contributions
Conceptualization, D.K.; writing—original draft preparation, D.K.; writing—review and editing, D.K., A.K., A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cosco, T.D.; Howse, K.; Brayne, C. Healthy ageing, resilience and wellbeing. Epidemiol. Psychiatr. Sci. 2017, 26, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Jaul, E.; Barron, J. Age-Related Diseases and Clinical and Public Health Implications for the 85 Years Old and Over Population. Front. Public Health 2017, 5, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; Anderson, B.O.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Ignatius, M.S.; Hayes, M.N.; Moore, F.E.; Tang, Q.; Garcia, S.P.; Blackburn, P.R.; Baxi, K.; Wang, L.; Jin, A.; Ramakrishnan, A.; et al. tp53 deficiency causes a wide tumor spectrum and increases embryonal rhabdomyosarcoma metastasis in zebrafish. eLife 2018, 7. [Google Scholar] [CrossRef]
- Park, J.T.; Leach, S.D. Zebrafish model of KRAS-initiated pancreatic cancer. Anim. Cells Syst. 2018, 22, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Leng, X.; Wang, G.; Wan, X.; Cao, H. The construction of intrahepatic cholangiocarcinoma model in zebrafish. Sci. Rep. 2017, 7, 13419. [Google Scholar] [CrossRef] [Green Version]
- White, R.M.; Sessa, A.; Burke, C.; Bowman, T.; LeBlanc, J.; Ceol, C.; Bourque, C.; Dovey, M.; Goessling, W.; Burns, C.E.; et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008, 2, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Antinucci, P.; Hindges, R. A crystal-clear zebrafish for in vivo imaging. Sci. Rep. 2016, 6, 29490. [Google Scholar] [CrossRef] [Green Version]
- Nicoli, S.; Ribatti, D.; Cotelli, F.; Presta, M. Mammalian tumor xenografts induce neovascularization in zebrafish embryos. Cancer Res. 2007, 67, 2927–2931. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cao, Z.; Zhang, X.M.; Nakamura, M.; Sun, M.; Hartman, J.; Harris, R.A.; Sun, Y.; Cao, Y. Novel mechanism of macrophage-mediated metastasis revealed in a zebrafish model of tumor development. Cancer Res. 2015, 75, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Zhang, Y.; Lim, S.; Hosaka, K.; Yang, Y.; Pavlova, T.; Alkasalias, T.; Hartman, J.; Jensen, L.; Xing, X.; et al. A Zebrafish Model Discovers a Novel Mechanism of Stromal Fibroblast-Mediated Cancer Metastasis. Clin. Cancer Res. 2017, 23, 4769–4779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Gao, B.; Zhang, W.; Qian, Z.; Xiang, Y. Monitoring antiangiogenesis of bevacizumab in zebrafish. Drug Des. Dev. Ther. 2018, 12, 2423–2430. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Wang, G.; Xiao, Q.; Zhou, Y.; Wei, Y.; Gong, Z. Antiangiogenic effects of AA-PMe on HUVECs in vitro and zebrafish in vivo. Oncotargets Ther. 2018, 11, 1871–1884. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. 2017, 12, 187–215. [Google Scholar] [CrossRef] [Green Version]
- Dolensek, J.; Rupnik, M.S.; Stozer, A. Structural similarities and differences between the human and the mouse pancreas. Islets 2015, 7, e1024405. [Google Scholar] [CrossRef] [Green Version]
- Steiniger, B.S. Human spleen microanatomy: Why mice do not suffice. Immunology 2015, 145, 334–346. [Google Scholar] [CrossRef]
- Holliday, R. Neoplastic transformation: The contrasting stability of human and mouse cells. Cancer Surv. 1996, 28, 103–115. [Google Scholar]
- Rangarajan, A.; Weinberg, R.A. Opinion: Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef]
- Watson, A.L.; Carlson, D.F.; Largaespada, D.A.; Hackett, P.B.; Fahrenkrug, S.C. Engineered Swine Models of Cancer. Front. Genet. 2016, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Hagai, T.; Chen, X.; Miragaia, R.J.; Rostom, R.; Gomes, T.; Kunowska, N.; Henriksson, J.; Park, J.E.; Proserpio, V.; Donati, G.; et al. Gene expression variability across cells and species shapes innate immunity. Nature 2018, 563, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martignoni, M.; Groothuis, G.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Alvarez, C.E. Naturally occurring cancers in dogs: Insights for translational genetics and medicine. ILAR J. 2014, 55, 16–45. [Google Scholar] [CrossRef] [Green Version]
- MacEwen, E.G. Spontaneous tumors in dogs and cats: Models for the study of cancer biology and treatment. Cancer Metastasis Rev. 1990, 9, 125–136. [Google Scholar] [CrossRef]
- Perleberg, C.; Kind, A.; Schnieke, A. Genetically engineered pigs as models for human disease. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Flisikowska, T.; Kind, A.; Schnieke, A. Genetically modified pigs to model human diseases. J. Appl. Genet. 2014, 55, 53–64. [Google Scholar] [CrossRef]
- Lunney, J.K. Advances in swine biomedical model genomics. Int. J. Biol. Sci. 2007, 3, 179–184. [Google Scholar] [CrossRef]
- Hoffe, B.; Holahan, M.R. The Use of Pigs as a Translational Model for Studying Neurodegenerative Diseases. Front. Physiol. 2019, 10, 838. [Google Scholar] [CrossRef] [PubMed]
- Flisikowska, T.; Kind, A.; Schnieke, A. Pigs as models of human cancers. Theriogenology 2016, 86, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.J.; Kissinger, C.B.; McCain, R.R.; Cooper, B.R.; Marchant-Forde, J.N.; Vreeman, R.C.; Hannou, S.; Knipp, G.T. Assessment of juvenile pigs to serve as human pediatric surrogates for preclinical formulation pharmacokinetic testing. AAPS J. 2013, 15, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, M.J.; Farrell, D.E.; Howard, K.D.; Kawalek, J.C. Identification of multiple constitutive and inducible hepatic cytochrome P450 enzymes in market weight swine. Drug Metab. Dispos. Biol. Fate Chem. 2001, 29, 908–915. [Google Scholar]
- Hammer, R.E.; Pursel, V.G.; Rexroad, C.E., Jr.; Wall, R.J.; Bolt, D.J.; Ebert, K.M.; Palmiter, R.D.; Brinster, R.L. Production of transgenic rabbits, sheep and pigs by microinjection. Nature 1985, 315, 680–683. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Lv, D.; Ma, T.; Deng, S.; Yang, M.; Song, Y.; Zhang, X.; Zhang, J.; Fu, J.; Lian, Z.; et al. AANAT transgenic sheep generated via OPS vitrified-microinjected pronuclear embryos and reproduction efficiency of the transgenic offspring. PeerJ 2018, 6, e5420. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Thomas, K.R.; Capecchi, M.R. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 1987, 51, 503–512. [Google Scholar] [CrossRef]
- Nowak-Imialek, M.; Niemann, H. Pluripotent cells in farm animals: State of the art and future perspectives. Reprod. Fertil. Dev. 2012, 25, 103–128. [Google Scholar] [CrossRef]
- Blomberg, L.A.; Telugu, B.P. Twenty years of embryonic stem cell research in farm animals. Reprod. Domest. Anim. 2012, 47, 80–85. [Google Scholar] [CrossRef]
- Gao, X.; Nowak-Imialek, M.; Chen, X.; Chen, D.; Herrmann, D.; Ruan, D.; Chen, A.C.H.; Eckersley-Maslin, M.A.; Ahmad, S.; Lee, Y.L.; et al. Establishment of porcine and human expanded potential stem cells. Nat. Cell Biol. 2019, 21, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, K.H.; McWhir, J.; Ritchie, W.A.; Wilmut, I. Sheep cloned by nuclear transfer from a cultured cell line. Nature 1996, 380, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Schnieke, A.E.; Kind, A.J.; Ritchie, W.A.; Mycock, K.; Scott, A.R.; Ritchie, M.; Wilmut, I.; Colman, A.; Campbell, K.H. Human factor IX transgenic sheep produced by transfer of nuclei from transfected fetal fibroblasts. Science 1997, 278, 2130–2133. [Google Scholar] [CrossRef] [PubMed]
- McCreath, K.J.; Howcroft, J.; Campbell, K.H.; Colman, A.; Schnieke, A.E.; Kind, A.J. Production of gene-targeted sheep by nuclear transfer from cultured somatic cells. Nature 2000, 405, 1066–1069. [Google Scholar] [CrossRef]
- Park, K.W.; Cheong, H.T.; Lai, L.; Im, G.S.; Kuhholzer, B.; Bonk, A.; Samuel, M.; Rieke, A.; Day, B.N.; Murphy, C.N.; et al. Production of nuclear transfer-derived swine that express the enhanced green fluorescent protein. Anim. Biotechnol. 2001, 12, 173–181. [Google Scholar] [CrossRef]
- Dai, Y.; Vaught, T.D.; Boone, J.; Chen, S.H.; Phelps, C.J.; Ball, S.; Monahan, J.A.; Jobst, P.M.; McCreath, K.J.; Lamborn, A.E.; et al. Targeted disruption of the alpha1,3-galactosyltransferase gene in cloned pigs. Nat. Biotechnol. 2002, 20, 251–255. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Petersen, B. Basics of genome editing technology and its application in livestock species. Reprod. Domest. Anim. 2017, 52, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Hai, T.; Teng, F.; Guo, R.; Li, W.; Zhou, Q. One-step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell Res. 2014, 24, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, K.M.; Lee, K.; Benne, J.A.; Beaton, B.P.; Spate, L.D.; Murphy, S.L.; Samuel, M.S.; Mao, J.; O’Gorman, C.; Walters, E.M.; et al. Use of the CRISPR/Cas9 system to produce genetically engineered pigs from in vitro-derived oocytes and embryos. Biol. Reprod. 2014, 91, 78. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Rieblinger, B.; Hein, R.; Sfriso, R.; Zuber, J.; Fischer, A.; Klinger, B.; Liang, W.; Flisikowski, K.; Kurome, M.; et al. Viable pigs after simultaneous inactivation of porcine MHC class I and three xenoreactive antigen genes GGTA1, CMAH and B4GALNT2. Xenotransplantation 2019, e12560. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, L.; Du, Y.; Xie, F.; Li, L.; Liu, Y.; Liu, C.; Wang, S.; Zhang, S.; Huang, X.; et al. Efficient Generation of Gene-Modified Pigs Harboring Precise Orthologous Human Mutation via CRISPR/Cas9-Induced Homology-Directed Repair in Zygotes. Hum. Mutat. 2016, 37, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Jin, Q.; Ruan, D.; Yang, Y.; Liu, Q.; Wu, H.; Zhou, Z.; Ouyang, Z.; Liu, Z.; Zhao, Y.; et al. Cre-dependent Cas9-expressing pigs enable efficient in vivo genome editing. Genome Res. 2017, 27, 2061–2071. [Google Scholar] [CrossRef]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Schonhuber, N.; Seidler, B.; Schuck, K.; Veltkamp, C.; Schachtler, C.; Zukowska, M.; Eser, S.; Feyerabend, T.B.; Paul, M.C.; Eser, P.; et al. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat. Med. 2014, 20, 1340–1347. [Google Scholar] [CrossRef]
- Li, S.; Flisikowska, T.; Kurome, M.; Zakhartchenko, V.; Kessler, B.; Saur, D.; Kind, A.; Wolf, E.; Flisikowski, K.; Schnieke, A. Dual fluorescent reporter pig for Cre recombination: Transgene placement at the ROSA26 locus. PLoS ONE 2014, 9, e102455. [Google Scholar] [CrossRef]
- Grossi, A.B.; Hyttel, P.; Jensen, H.E.; Leifsson, P.S. Porcine melanotic cutaneous lesions and lymph nodes: Immunohistochemical differentiation of melanocytes and melanophages. Vet. Pathol. 2015, 52, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Seaton, M.; Hocking, A.; Gibran, N.S. Porcine models of cutaneous wound healing. ILAR J. 2015, 56, 127–138. [Google Scholar] [CrossRef]
- Kragh, P.M.; Nielsen, A.L.; Li, J.; Du, Y.; Lin, L.; Schmidt, M.; Bogh, I.B.; Holm, I.E.; Jakobsen, J.E.; Johansen, M.G.; et al. Hemizygous minipigs produced by random gene insertion and handmade cloning express the Alzheimer’s disease-causing dominant mutation APPsw. Transgenic Res 2009, 18, 545–558. [Google Scholar] [CrossRef]
- Jakobsen, J.E.; Johansen, M.G.; Schmidt, M.; Liu, Y.; Li, R.; Callesen, H.; Melnikova, M.; Habekost, M.; Matrone, C.; Bouter, Y.; et al. Expression of the Alzheimer’s Disease Mutations AbetaPP695sw and PSEN1M146I in Double-Transgenic Gottingen Minipigs. J. Alzheimers Dis. 2016, 53, 1617–1630. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Tu, Z.; Liu, Z.; Fan, N.; Yang, H.; Yang, S.; Yang, W.; Zhao, Y.; Ouyang, Z.; Lai, C.; et al. A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell 2018, 173, 989–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, B.T.; Wang, X.J.; Rohret, J.A.; Struzynski, J.T.; Merricks, E.P.; Bellinger, D.A.; Rohret, F.A.; Nichols, T.C.; Rogers, C.S. Targeted disruption of LDLR causes hypercholesterolemia and atherosclerosis in Yucatan miniature pigs. PLoS ONE 2014, 9, e93457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Ouyang, H.; Wang, Y.; Pang, D.; Cong, N.X.; Wang, T.; Leng, B.; Li, D.; Li, X.; Wu, R.; et al. Characterization of a hypertriglyceridemic transgenic miniature pig model expressing human apolipoprotein CIII. FEBS J. 2012, 279, 91–99. [Google Scholar] [CrossRef]
- Renner, S.; Fehlings, C.; Herbach, N.; Hofmann, A.; von Waldthausen, D.C.; Kessler, B.; Ulrichs, K.; Chodnevskaja, I.; Moskalenko, V.; Amselgruber, W.; et al. Glucose intolerance and reduced proliferation of pancreatic beta-cells in transgenic pigs with impaired glucose-dependent insulinotropic polypeptide function. Diabetes 2010, 59, 1228–1238. [Google Scholar] [CrossRef] [Green Version]
- Renner, S.; Braun-Reichhart, C.; Blutke, A.; Herbach, N.; Emrich, D.; Streckel, E.; Wunsch, A.; Kessler, B.; Kurome, M.; Bahr, A.; et al. Permanent neonatal diabetes in INS(C94Y) transgenic pigs. Diabetes 2013, 62, 1505–1511. [Google Scholar] [CrossRef] [Green Version]
- Klymiuk, N.; Blutke, A.; Graf, A.; Krause, S.; Burkhardt, K.; Wuensch, A.; Krebs, S.; Kessler, B.; Zakhartchenko, V.; Kurome, M.; et al. Dystrophin-deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Hum. Mol. Genet. 2013, 22, 4368–4382. [Google Scholar] [CrossRef] [Green Version]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef] [Green Version]
- Schook, L.B.; Collares, T.V.; Darfour-Oduro, K.A.; De, A.K.; Rund, L.A.; Schachtschneider, K.M.; Seixas, F.K. Unraveling the swine genome: Implications for human health. Annu. Rev. Anim. Biosci. 2015, 3, 219–244. [Google Scholar] [CrossRef] [Green Version]
- Adam, S.J.; Rund, L.A.; Kuzmuk, K.N.; Zachary, J.F.; Schook, L.B.; Counter, C.M. Genetic induction of tumorigenesis in swine. Oncogene 2007, 26, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Saalfrank, A.; Janssen, K.P.; Ravon, M.; Flisikowski, K.; Eser, S.; Steiger, K.; Flisikowska, T.; Muller-Fliedner, P.; Schulze, E.; Bronner, C.; et al. A porcine model of osteosarcoma. Oncogenesis 2016, 5, e210. [Google Scholar] [CrossRef] [PubMed]
- Rubio, R.; García-Castro, J.; Gutiérrez-Aranda, I.; Paramio, J.; Santos, M.; Catalina, P.; Leone, P.E.; Menendez, P.; Rodríguez, R. Deficiency in p53 but not Retinoblastoma Induces the Transformation of Mesenchymal Stem Cells In vitro and Initiates Leiomyosarcoma In vivo. Cancer Res. 2010, 70, 4185–4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakawa, H.; Nagai, T.; Harasawa, R.; Yamagami, T.; Takahashi, J.; Ishikawa, K.-I.; Nomura, N.; Nagashima, H. Production of Transgenic Pig Carrying MMTV/v-Ha-ras. J. Reprod. Dev. 1999, 45, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Li, J.; Liu, Y.; Lin, L.; Du, Y.; Li, S.; Yang, H.; Vajta, G.; Callesen, H.; Bolund, L.; et al. High efficiency of BRCA1 knockout using rAAV-mediated gene targeting: Developing a pig model for breast cancer. Transgenic Res. 2011, 20, 975–988. [Google Scholar] [CrossRef]
- Flisikowska, T.; Merkl, C.; Landmann, M.; Eser, S.; Rezaei, N.; Cui, X.; Kurome, M.; Zakhartchenko, V.; Kessler, B.; Wieland, H.; et al. A porcine model of familial adenomatous polyposis. Gastroenterology 2012, 143, 1173–1175. [Google Scholar] [CrossRef]
- Tan, W.; Carlson, D.F.; Lancto, C.A.; Garbe, J.R.; Webster, D.A.; Hackett, P.B.; Fahrenkrug, S.C. Efficient nonmeiotic allele introgression in livestock using custom endonucleases. Proc. Natl. Acad. Sci. USA 2013, 110, 16526–16531. [Google Scholar] [CrossRef] [Green Version]
- Callesen, M.M.; Árnadóttir, S.S.; Lyskjaer, I.; Ørntoft, M.W.; Høyer, S.; Dagnaes-Hansen, F.; Liu, Y.; Li, R.; Callesen, H.; Rasmussen, M.H.; et al. A genetically inducible porcine model of intestinal cancer. Mol. Oncol. 2017, 11, 1616–1629. [Google Scholar] [CrossRef] [Green Version]
- Leuchs, S.; Saalfrank, A.; Merkl, C.; Flisikowska, T.; Edlinger, M.; Durkovic, M.; Rezaei, N.; Kurome, M.; Zakhartchenko, V.; Kessler, B.; et al. Inactivation and inducible oncogenic mutation of p53 in gene targeted pigs. PLoS ONE 2012, 7, e43323. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Edlinger, M.; Saalfrank, A.; Flisikowski, K.; Tschukes, A.; Kurome, M.; Zakhartchenko, V.; Kessler, B.; Saur, D.; Kind, A.; et al. Viable pigs with a conditionally-activated oncogenic KRAS mutation. Transgenic Res. 2015, 24, 509–517. [Google Scholar] [CrossRef]
- Schook, L.B.; Collares, T.V.; Hu, W.; Liang, Y.; Rodrigues, F.M.; Rund, L.A.; Schachtschneider, K.M.; Seixas, F.K.; Singh, K.; Wells, K.D.; et al. A Genetic Porcine Model of Cancer. PLoS ONE 2015, 10, e0128864. [Google Scholar] [CrossRef]
- Principe, D.R.; Overgaard, N.H.; Park, A.J.; Diaz, A.M.; Torres, C.; McKinney, R.; Dorman, M.J.; Castellanos, K.; Schwind, R.; Dawson, D.W.; et al. KRAS(G12D) and TP53(R167H) Cooperate to Induce Pancreatic Ductal Adenocarcinoma in Sus scrofa Pigs. Sci. Rep. 2018, 8, 12548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthelsen, M.F.; Callesen, M.M.; Ostergaard, T.S.; Liu, Y.; Li, R.; Callesen, H.; Dagnaes-Hansen, F.; Hamilton-Dutoit, S.; Jakobsen, J.E.; Thomsen, M.K. Pancreas specific expression of oncogenes in a porcine model. Transgenic Res 2017, 26, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Sieren, J.C.; Meyerholz, D.K.; Wang, X.-J.; Davis, B.T.; Newell, J.D., Jr.; Hammond, E.; Rohret, J.A.; Rohret, F.A.; Struzynski, J.T.; Goeken, J.A.; et al. Development and translational imaging of a TP53 porcine tumorigenesis model. J. Clin. Investig. 2014, 124, 4052–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCalla-Martin, A.C.; Chen, X.; Linder, K.E.; Estrada, J.L.; Piedrahita, J.A. Varying phenotypes in swine versus murine transgenic models constitutively expressing the same human Sonic hedgehog transcriptional activator, K5-HGLI2ΔN. Transgenic Res. 2010, 19, 869–887. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.H.; Anders, C.K.; Litton, J.K.; Ruddy, K.J.; Bleyer, A. Breast cancer in adolescents and young adults. Pediatric Blood Cancer 2018, 65, e27397. [Google Scholar] [CrossRef]
- Brenner, D.R.; Brockton, N.T.; Kotsopoulos, J.; Cotterchio, M.; Boucher, B.A.; Courneya, K.S.; Knight, J.A.; Olivotto, I.A.; Quan, M.L.; Friedenreich, C.M. Breast cancer survival among young women: A review of the role of modifiable lifestyle factors. Cancer Causes Control 2016, 27, 459–472. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadi, Z.; Lianos, G.D.; Ignatiadou, E.; Harissis, H.V.; Mitsis, M. Breast cancer in young women: An overview. Updates Surg. 2017, 69, 313–317. [Google Scholar] [CrossRef]
- De Silva, S.; Tennekoon, K.H.; Karunanayake, E.H. Overview of the genetic basis toward early detection of breast cancer. Breast Cancer 2019, 11, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Gewefel, H.; Salhia, B. Breast cancer in adolescent and young adult women. Clin. Breast Cancer 2014, 14, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of Type and Location of BRCA1 and BRCA2 Mutations with Risk of Breast and Ovarian Cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [Green Version]
- Donninger, H.; Hobbing, K.; Schmidt, M.L.; Walters, E.; Rund, L.; Schook, L.; Clark, G.J. A porcine model system of BRCA1 driven breast cancer. Front. Genet. 2015, 6, 269. [Google Scholar] [CrossRef] [Green Version]
- COSMIC Database. 2019. Available online: Cancer.sanger.ac.uk (accessed on 17 December 2019).
- Campos, F.G. Colorectal cancer in young adults: A difficult challenge. World J. Gastroenterol. 2017, 23, 5041–5044. [Google Scholar] [CrossRef]
- Siegel, R.L.; Fedewa, S.A.; Anderson, W.F.; Miller, K.D.; Ma, J.; Rosenberg, P.S.; Jemal, A. Colorectal Cancer Incidence Patterns in the United States, 1974–2013. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Connell, L.C.; Mota, J.M.; Braghiroli, M.I.; Hoff, P.M. The Rising Incidence of Younger Patients with Colorectal Cancer: Questions about Screening, Biology, and Treatment. Curr. Treat. Options Oncol. 2017, 18, 23. [Google Scholar] [CrossRef]
- Bailey, C.E.; Hu, C.Y.; You, Y.N.; Bednarski, B.K.; Rodriguez-Bigas, M.A.; Skibber, J.M.; Cantor, S.B.; Chang, G.J. Increasing disparities in the age-related incidences of colon and rectal cancers in the United States, 1975–2010. JAMA Surg. 2015, 150, 17–22. [Google Scholar] [CrossRef]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Rowan, A.J.; Lamlum, H.; Ilyas, M.; Wheeler, J.; Straub, J.; Papadopoulou, A.; Bicknell, D.; Bodmer, W.F.; Tomlinson, I.P. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc. Natl. Acad. Sci. USA 2000, 97, 3352–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. Wnt Signaling and Colorectal Cancer. Curr. Colorectal Cancer Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, A.H.; Cheng, K.H.; Wong, A.S.; Ng, S.S.; Ma, B.B.; Chan, C.M.; Tsui, N.B.; Chan, L.W.; Yung, B.Y.; Wong, S.C. Current and future molecular diagnostics in colorectal cancer and colorectal adenoma. World J. Gastroenterol. 2014, 20, 3847–3857. [Google Scholar] [CrossRef]
- Mori, Y.; Nagse, H.; Ando, H.; Horii, A.; Ichii, S.; Nakatsuru, S.; Aoki, T.; Miki, Y.; Mori, T.; Nakamura, Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum. Mol. Genet. 1992, 1, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Cheadle, J.P.; Krawczak, M.; Thomas, M.W.; Hodges, A.K.; Al-Tassan, N.; Fleming, N.; Sampson, J.R. Different combinations of biallelic APC mutation confer different growth advantages in colorectal tumours. Cancer Res. 2002, 62, 363–366. [Google Scholar]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Half, E.; Bercovich, D.; Rozen, P. Familial adenomatous polyposis. Orphanet J. Rare Dis. 2009, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Zeineldin, M.; Neufeld, K.L. Understanding phenotypic variation in rodent models with germline Apc mutations. Cancer Res. 2013, 73, 2389–2399. [Google Scholar] [CrossRef] [Green Version]
- Jackstadt, R.; Sansom, O.J. Mouse models of intestinal cancer. J. Pathol. 2016, 238, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Tetteh, P.W.; Kretzschmar, K.; Begthel, H.; van den Born, M.; Korving, J.; Morsink, F.; Farin, H.; van Es, J.H.; Offerhaus, G.J.; Clevers, H. Generation of an inducible colon-specific Cre enzyme mouse line for colon cancer research. Proc. Natl. Acad. Sci. USA 2016, 113, 11859–11864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, K.; Tamai, Y.; Horiike, S.; Oshima, M.; Taketo, M.M. Colonic polyposis caused by mTOR-mediated chromosomal instability in Apc+/Δ716 Cdx2+/− compound mutant mice. Nat. Genet. 2003, 35, 323–330. [Google Scholar] [CrossRef]
- Flisikowska, T.; Stachowiak, M.; Xu, H.; Wagner, A.; Hernandez-Caceres, A.; Wurmser, C.; Perleberg, C.; Pausch, H.; Perkowska, A.; Fischer, K.; et al. Porcine familial adenomatous polyposis model enables systematic analysis of early events in adenoma progression. Sci. Rep. 2017, 7, 6613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, K.; Yachida, S.; Sugimoto, M.; Oshima, M.; Nakagawa, T.; Akamoto, S.; Tabata, S.; Saitoh, K.; Kato, K.; Sato, S.; et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. USA 2017, 114, E7697–E7706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.L.; Wang, P.; Lu, M.Z.; Zhang, S.D.; Zheng, L. c-Myc maintains the self-renewal and chemoresistance properties of colon cancer stem cells. Oncol. Lett. 2019, 17, 4487–4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachowiak, M.; Flisikowska, T.; Bauersachs, S.; Perleberg, C.; Pausch, H.; Switonski, M.; Kind, A.; Saur, D.; Schnieke, A.; Flisikowski, K. Altered microRNA profiles during early colon adenoma progression in a porcine model of familial adenomatous polyposis. Oncotarget 2017, 8, 96154–96160. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Rogalla, S.; Flisikowski, K.; Gorpas, D.; Mayer, A.T.; Flisikowska, T.; Mandella, M.J.; Ma, X.; Casey, K.M.; Felt, S.A.; Saur, D.; et al. Biodegradable Fluorescent Nanoparticles for Endoscopic Detection of Colorectal Carcinogenesis. Adv. Funct. Mater. 2019, 1904992. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Quante, A.S.; Ming, C.; Rottmann, M.; Engel, J.; Boeck, S.; Heinemann, V.; Westphalen, C.B.; Strauch, K. Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer Med. 2016, 5, 2649–2656. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, T.J.; Hua, K.; Singh, A. Molecular Pathogenesis of Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Adsay, N.V.; Albores–Saavedra, J.; Compton, C.; Garrett, E.S.; Goodman, S.N.; Kern, S.E.; Klimstra, D.S.; Klöppel, G.; Longnecker, D.S.; et al. Pancreatic Intraepithelial Neoplasia: A New Nomenclature and Classification System for Pancreatic Duct Lesions. Am. J. Surg. Pathol. 2001, 25, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtaugh, L.C.; Keefe, M.D. Regeneration and repair of the exocrine pancreas. Annu. Rev. Physiol. 2015, 77, 229–249. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.M.M.; Sancho, R.; Messal, H.A.; Nye, E.; Spencer-Dene, B.; Stone, R.K.; Stamp, G.; Rosewell, I.; Quaglia, A.; Behrens, A. Duct- and Acinar-Derived Pancreatic Ductal Adenocarcinomas Show Distinct Tumor Progression and Marker Expression. Cell Rep. 2017, 21, 966–978. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, J.; Yokoyama, Y.; Kokuryo, T.; Ebata, T.; Nagino, M. Cells of origin of pancreatic neoplasms. Surg. Today 2018, 48, 9–17. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Yokota, T.; Takano, S.; Yoshitomi, H.; Kagawa, S.; Furukawa, K.; Takayashiki, T.; Kuboki, S.; Suzuki, D.; Sakai, N.; Nojima, H.; et al. Successful treatment of a locally advanced unresectable pancreatic cancer patient with interstitial pneumonitis by conversion surgery following gemcitabine plus nab-paclitaxel chemotherapy: A case report. Mol. Clin. Oncol. 2019, 10, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef]
- Pedersen, K.; Bilal, F.; Bernadó Morales, C.; Salcedo, M.T.; Macarulla, T.; Massó-Vallés, D.; Mohan, V.; Vivancos, A.; Carreras, M.J.; Serres, X.; et al. Pancreatic cancer heterogeneity and response to Mek inhibition. Oncogene 2017, 36, 5639–5647. [Google Scholar] [CrossRef] [PubMed]
- Biancur, D.E.; Kimmelman, A.C. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F., III; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Overgaard, N.H.; Principe, D.R.; Schachtschneider, K.M.; Jakobsen, J.T.; Rund, L.A.; Grippo, P.J.; Schook, L.B.; Jungersen, G. Genetically Induced Tumors in the Oncopig Model Invoke an Antitumor Immune Response Dominated by Cytotoxic CD8β+ T Cells and Differentiated γδ T Cells Alongside a Regulatory Response Mediated by FOXP3+ T Cells and Immunoregulatory Molecules. Front. Immunol. 2018, 9, 1301. [Google Scholar] [CrossRef] [Green Version]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab 2014, 19, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Remmers, N.; Cox, J.L.; Grunkemeyer, J.A.; Aravind, S.; Arkfeld, C.K.; Hollingsworth, M.A.; Carlson, M.A. Generation of tumorigenic porcine pancreatic ductal epithelial cells: Toward a large animal model of pancreatic cancer. bioRxiv 2018, 267112. [Google Scholar] [CrossRef] [Green Version]
- Bailey, K.L.; Carlson, M.A. Porcine Models of Pancreatic Cancer. Front. Oncol. 2019, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Durfee, R.A.; Mohammed, M.; Luu, H.H. Review of Osteosarcoma and Current Management. Rheumatol. Ther. 2016, 3, 221–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, K.A.; Barkauskas, D.A.; Krailo, M.D.; Meyers, P.A.; Schwartz, C.L.; Ebb, D.H.; Seibel, N.L.; Grier, H.E.; Gorlick, R.; Marina, N. Outcome for adolescent and young adult patients with osteosarcoma: A report from the Children’s Oncology Group. Cancer 2012, 118, 4597–4605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ognjanovic, S.; Olivier, M.; Bergemann, T.L.; Hainaut, P. Sarcomas in TP53 germline mutation carriers: A review of the IARC TP53 database. Cancer 2012, 118, 1387–1396. [Google Scholar] [CrossRef]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef]
- Hansen, M.F.; Koufos, A.; Gallie, B.L.; Phillips, R.A.; Fodstad, O.; Brogger, A.; Gedde-Dahl, T.; Cavenee, W.K. Osteosarcoma and retinoblastoma: A shared chromosomal mechanism revealing recessive predisposition. Proc. Natl. Acad. Sci. USA 1985, 82, 6216–6220. [Google Scholar] [CrossRef] [Green Version]
- Bridge, J.A.; Nelson, M.; McComb, E.; McGuire, M.H.; Rosenthal, H.; Vergara, G.; Maale, G.E.; Spanier, S.; Neff, J.R. Cytogenetic findings in 73 osteosarcoma specimens and a review of the literature. Cancer Genet. Cytogenet. 1997, 95, 74–87. [Google Scholar] [CrossRef]
- Selvarajah, S.; Yoshimoto, M.; Ludkovski, O.; Park, P.C.; Bayani, J.; Thorner, P.; Maire, G.; Squire, J.A.; Zielenska, M. Genomic signatures of chromosomal instability and osteosarcoma progression detected by high resolution array CGH and interphase FISH. Cytogenet. Genome Res. 2008, 122, 5–15. [Google Scholar] [CrossRef]
- Plummer, S.J.; Santibanez-Koref, M.; Kurosaki, T.; Liao, S.; Noble, B.; Fain, P.R.; Anton-Culver, H.; Casey, G. A germline 2.35 kb deletion of p53 genomic DNA creating a specific loss of the oligomerization domain inherited in a Li-Fraumeni syndrome family. Oncogene 1994, 9, 3273–3280. [Google Scholar] [PubMed]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohseny, A.B.; Tieken, C.; van der Velden, P.A.; Szuhai, K.; de Andrea, C.; Hogendoorn, P.C.; Cleton-Jansen, A.M. Small deletions but not methylation underlie CDKN2A/p16 loss of expression in conventional osteosarcoma. Genes Chromosomes Cancer 2010, 49, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [Green Version]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef]
- Castillo-Tandazo, W.; Mutsaers, A.J.; Walkley, C.R. Osteosarcoma in the Post Genome Era: Preclinical Models and Approaches to Identify Tractable Therapeutic Targets. Curr. Osteoporos. Rep. 2019, 17, 343–352. [Google Scholar] [CrossRef]
- Walia, M.K.; Castillo-Tandazo, W.; Mutsaers, A.J.; Martin, T.J.; Walkley, C.R. Murine models of osteosarcoma: A piece of the translational puzzle. J. Cell Biochem. 2018, 119, 4241–4250. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef]
- Quist, T.; Jin, H.; Zhu, J.F.; Smith-Fry, K.; Capecchi, M.R.; Jones, K.B. The impact of osteoblastic differentiation on osteosarcomagenesis in the mouse. Oncogene 2015, 34, 4278–4284. [Google Scholar] [CrossRef] [Green Version]
- Mutsaers, A.J.; Ng, A.J.; Baker, E.K.; Russell, M.R.; Chalk, A.M.; Wall, M.; Liddicoat, B.J.; Ho, P.W.; Slavin, J.L.; Goradia, A.; et al. Modeling distinct osteosarcoma subtypes in vivo using Cre:lox and lineage-restricted transgenic shRNA. Bone 2013, 55, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, M.V.; Ghivizzani, S.C.; Gibbs, C.P. Animal models in osteosarcoma. Front. Oncol. 2014, 4, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Li, X.; Liu, P.; Li, M.; Luo, F. Patient-derived xenograft mouse models: A high fidelity tool for individualized medicine. Oncol. Lett. 2019, 17, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Yada, E.; Wada, S.; Yoshida, S.; Sasada, T. Use of patient-derived xenograft mouse models in cancer research and treatment. Future Sci. OA 2018, 4, Fso271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murayama, T.; Gotoh, N. Patient-Derived Xenograft Models of Breast Cancer and Their Application. Cells 2019, 8, 621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig, I.; Chicote, I.; Tenbaum, S.P.; Arques, O.; Herance, J.R.; Gispert, J.D.; Jimenez, J.; Landolfi, S.; Caci, K.; Allende, H.; et al. A personalized preclinical model to evaluate the metastatic potential of patient-derived colon cancer initiating cells. Clin. Cancer Res. 2013, 19, 6787–6801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, E.; Hong, S.M.; Yoo, H.J.; Kim, M.B.; Won, J.S.; An, S.; Shim, I.K.; Chang, S.; Hoffman, R.M.; Kim, S.C. Genetic and metabolic comparison of orthotopic and heterotopic patient-derived pancreatic-cancer xenografts to the original patient tumors. Oncotarget 2018, 9, 7867–7881. [Google Scholar] [CrossRef]
- Merino, D.; Weber, T.S.; Serrano, A.; Vaillant, F.; Liu, K.; Pal, B.; Di Stefano, L.; Schreuder, J.; Lin, D.; Chen, Y.; et al. Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat. Commun. 2019, 10, 766. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Mukae, Y.; Kitsuka, T.; Arai, K.; Nakamura, A.; Uchihashi, K.; Toda, S.; Matsubayashi, K.; Oyama, J.-I.; Node, K.; et al. Development of an immunodeficient pig model allowing long-term accommodation of artificial human vascular tubes. Nat. Commun. 2019, 10, 2244. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Iwamoto, M.; Saito, Y.; Fuchimoto, D.; Sembon, S.; Suzuki, M.; Mikawa, S.; Hashimoto, M.; Aoki, Y.; Najima, Y.; et al. Il2rg Gene-Targeted Severe Combined Immunodeficiency Pigs. Cell Stem Cell 2012, 10, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.T.; Cho, B.; Ryu, J.; Ray, C.; Lee, E.J.; Yun, Y.J.; Ahn, S.; Lee, J.; Ji, D.Y.; Jue, N.; et al. Biallelic modification of IL2RG leads to severe combined immunodeficiency in pigs. Reprod. Biol. Endocrinol. 2016, 14, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Nakano, K.; Matsunari, H.; Matsuda, T.; Maehara, M.; Kanai, T.; Kobayashi, M.; Matsumura, Y.; Sakai, R.; Kuramoto, M.; et al. Generation of interleukin-2 receptor gamma gene knockout pigs from somatic cells genetically modified by zinc finger nuclease-encoding mRNA. PLoS ONE 2013, 8, e76478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Guo, X.; Fan, N.; Song, J.; Zhao, B.; Ouyang, Z.; Liu, Z.; Zhao, Y.; Yan, Q.; Yi, X.; et al. RAG1/2 Knockout Pigs with Severe Combined Immunodeficiency. J. Immunol. 2014, 193, 1496–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Kwon, D.N.; Ezashi, T.; Choi, Y.J.; Park, C.; Ericsson, A.C.; Brown, A.N.; Samuel, M.S.; Park, K.W.; Walters, E.M.; et al. Engraftment of human iPS cells and allogeneic porcine cells into pigs with inactivated RAG2 and accompanying severe combined immunodeficiency. Proc. Natl. Acad. Sci. USA 2014, 111, 7260–7265. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Sendai, Y.; Yamazaki, S.; Seki-Soma, M.; Hirose, K.; Watanabe, M.; Fukawa, K.; Nakauchi, H. Generation of recombination activating gene-1-deficient neonatal piglets: A model of T and B cell deficient severe combined immune deficiency. PLoS ONE 2014, 9, e113833. [Google Scholar] [CrossRef]
- Suzuki, S.; Iwamoto, M.; Hashimoto, M.; Suzuki, M.; Nakai, M.; Fuchimoto, D.; Sembon, S.; Eguchi-Ogawa, T.; Uenishi, H.; Onishi, A. Generation and characterization of RAG2 knockout pigs as animal model for severe combined immunodeficiency. Vet. Immunol. Immunopathol. 2016, 178, 37–49. [Google Scholar] [CrossRef]
- Fisher, J.E.; Lillegard, J.B.; McKenzie, T.J.; Rodysill, B.R.; Wettstein, P.J.; Nyberg, S.L. In utero transplanted human hepatocytes allow postnatal engraftment of human hepatocytes in pigs. Liver Transplant. 2013, 19, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Sinkora, M.; Sinkora, J.; Rehakova, Z.; Butler, J.E. Early ontogeny of thymocytes in pigs: Sequential colonization of the thymus by T cell progenitors. J. Immunol. 2000, 165, 1832–1839. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The pig as a biomedical model. Pigs can help translate basic research findings into new medical drugs and procedures; ‘bridging the gap between bench and bedside’.
Figure 1.
The pig as a biomedical model. Pigs can help translate basic research findings into new medical drugs and procedures; ‘bridging the gap between bench and bedside’.

Figure 2.
Phenotype of adenomatous polyposis coli (APC)1311 pigs. Polyp progression during the first 24 months.
Figure 2.
Phenotype of adenomatous polyposis coli (APC)1311 pigs. Polyp progression during the first 24 months.

Figure 3.
Porcine cancer models. These can be established by different methods: e.g., introduction of oncogenic germline mutations or via genome editing in organs of a Cas9 pig. Biopsies taken early in life can be oncogenically transformed and reimplanted into the same pig. If isolated from a pig with latent oncogenic mutations, these can be activated in vitro prior to implantation. Human tumor samples can also be implanted in immune deficient animals.
Figure 3.
Porcine cancer models. These can be established by different methods: e.g., introduction of oncogenic germline mutations or via genome editing in organs of a Cas9 pig. Biopsies taken early in life can be oncogenically transformed and reimplanted into the same pig. If isolated from a pig with latent oncogenic mutations, these can be activated in vitro prior to implantation. Human tumor samples can also be implanted in immune deficient animals.


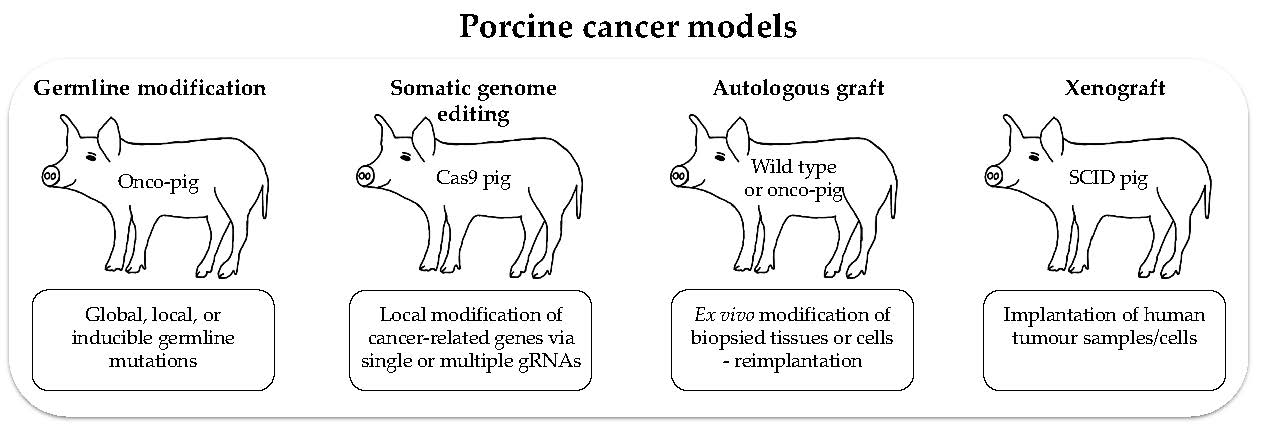{kind=link}
{kind=link}
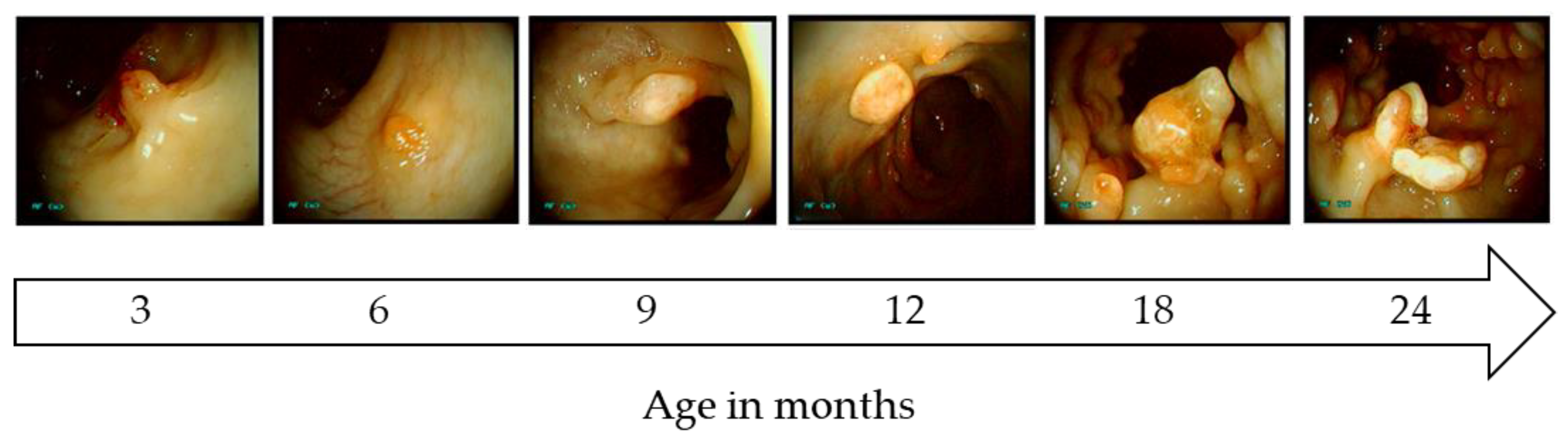{kind=link}
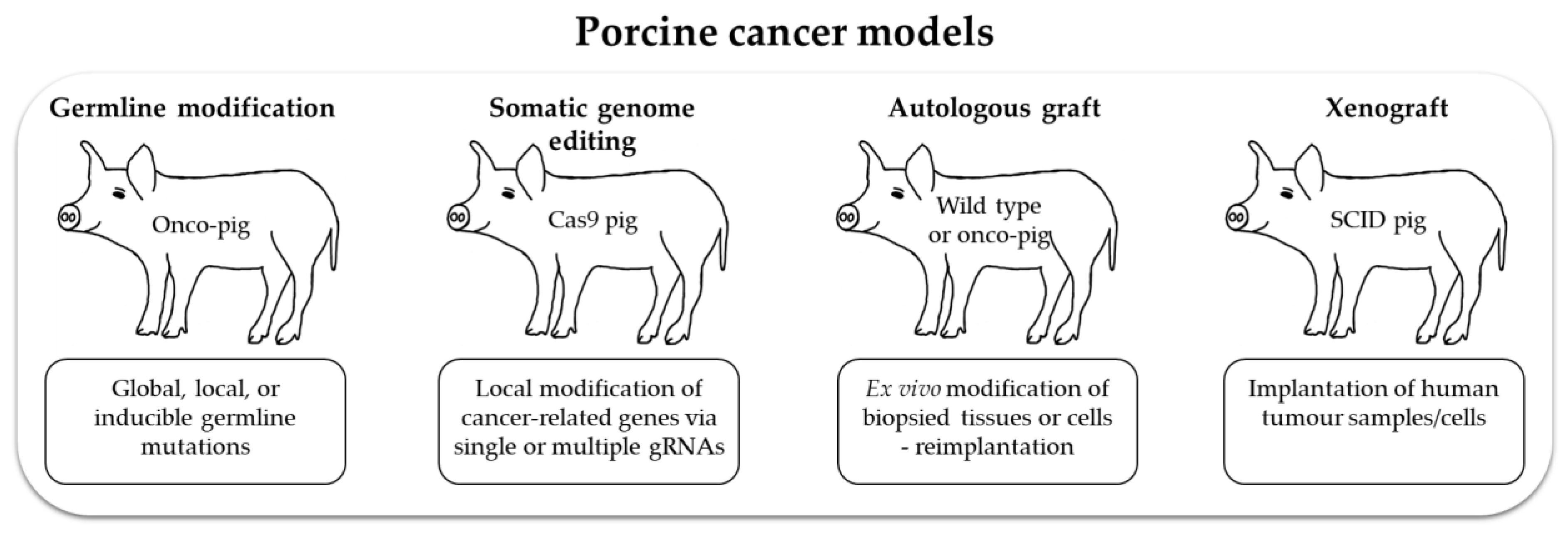{kind=link}
Table 1.
Providing an overview of genetically modified pig models for human cancers.
| Human Cancer | Genetic Modification | Generated By | Comments | Reference |
|---|---|---|---|---|
| Breast Cancer | MMTV/v-Ha-ras (transgene) | Microinjection | No phenotype | [73] |
| Heterozygous BRCA1 knockout (endogenous gene) | Gene targeting via AAV + SCNT | No survival of born piglets | [74] | |
| Colorectal Cancer | Heterozygous APC1311 truncation mutation (endogenous gene) | Gene targeting + SCNT | Colonic polyposis | [75] |
| Heterozygous APC902 truncation mutation (endogenous gene) | TALENs + Chromatin transfer | No phenotype | [76] | |
| Flp-inducible KRASG12D + cMYC+SV40LT (transgenes) | Random integration + SCNT | Villin-driven; Duodenal carcinoma | [77] | |
| Pancreatic Cancer | Cre-inducible TP53R167H + KRASG12D mutation (endogenous genes) | Gene targeting + SCNT | Pancreas-specific activation intended | [78,79] |
| Cre-inducible TP53R167H + KRASG12D mutation (transgenes) | Random integration + SCNT | AdCre delivery into duct led to tumor formation | [80,81] | |
| Flp-inducible KRASG12D + cMYC+SV40LT (transgenes) | Random integration + SCNT | Pdx1-driven; Hyperplastic foci of acinar cells | [82] | |
| Osteosarcoma | Hetero- and homozygous knockout of TP53 major transcript (endogenous gene) | Gene targeting + SCNT | OS primarily affecting long bones | [71] |
| Homozygous TP53R167H mutation (endogenous gene) | Gene targeting via AAV + SCNT | Various lesions, e.g., osteogenic tumors, lymphomas and renal tumors | [83] | |
| Other Cancers | Human Gli2 transcriptional activator K5-hGli2ΔN (transgene) | Random integration + SCNT | Basal cell carcinoma-like lesions; infection | [84] |
| Cre-inducible TP53R167H + KRASG12D mutation (endogenous genes) | Gene targeting + SCNT | Suitable for diverse cancers, e.g., lung cancer | [78,79] |
AAV: adeno-associated virus; SCNT: somatic cell nuclear transfer; AdCre: adenovirus encoding Cre.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kalla, D.; Kind, A.; Schnieke, A. Genetically Engineered Pigs to Study Cancer. Int. J. Mol. Sci. 2020, 21, 488. https://doi.org/10.3390/ijms21020488
AMA Style
Kalla D, Kind A, Schnieke A. Genetically Engineered Pigs to Study Cancer. International Journal of Molecular Sciences. 2020; 21(2):488. https://doi.org/10.3390/ijms21020488
Chicago/Turabian StyleKalla, Daniela, Alexander Kind, and Angelika Schnieke. 2020. "Genetically Engineered Pigs to Study Cancer" International Journal of Molecular Sciences 21, no. 2: 488. https://doi.org/10.3390/ijms21020488
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.