Molecular Mechanisms of Premature Aging in Hemodialysis: The Complex Interplay between Innate and Adaptive Immune Dysfunction

 and
and 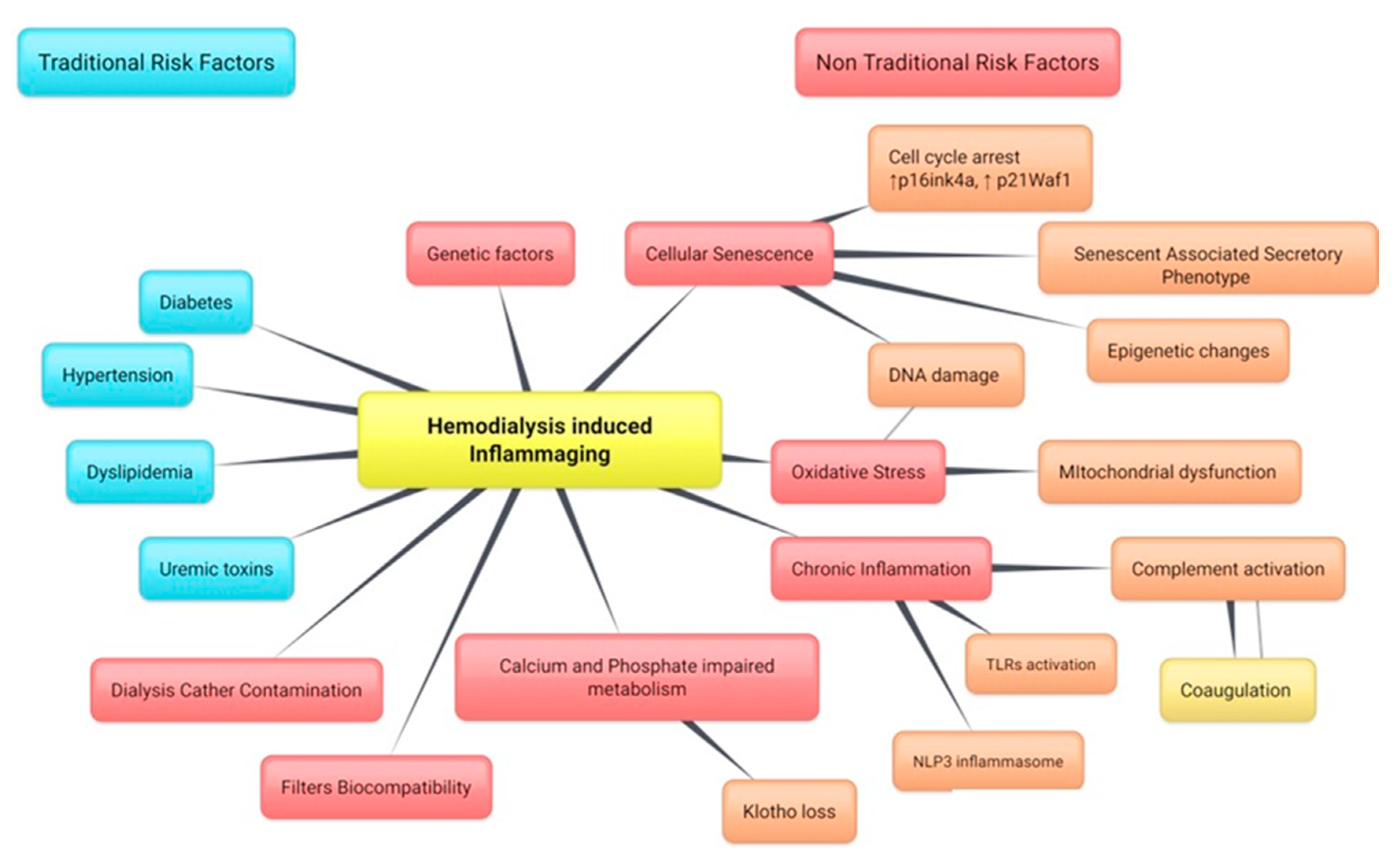{kind=link}
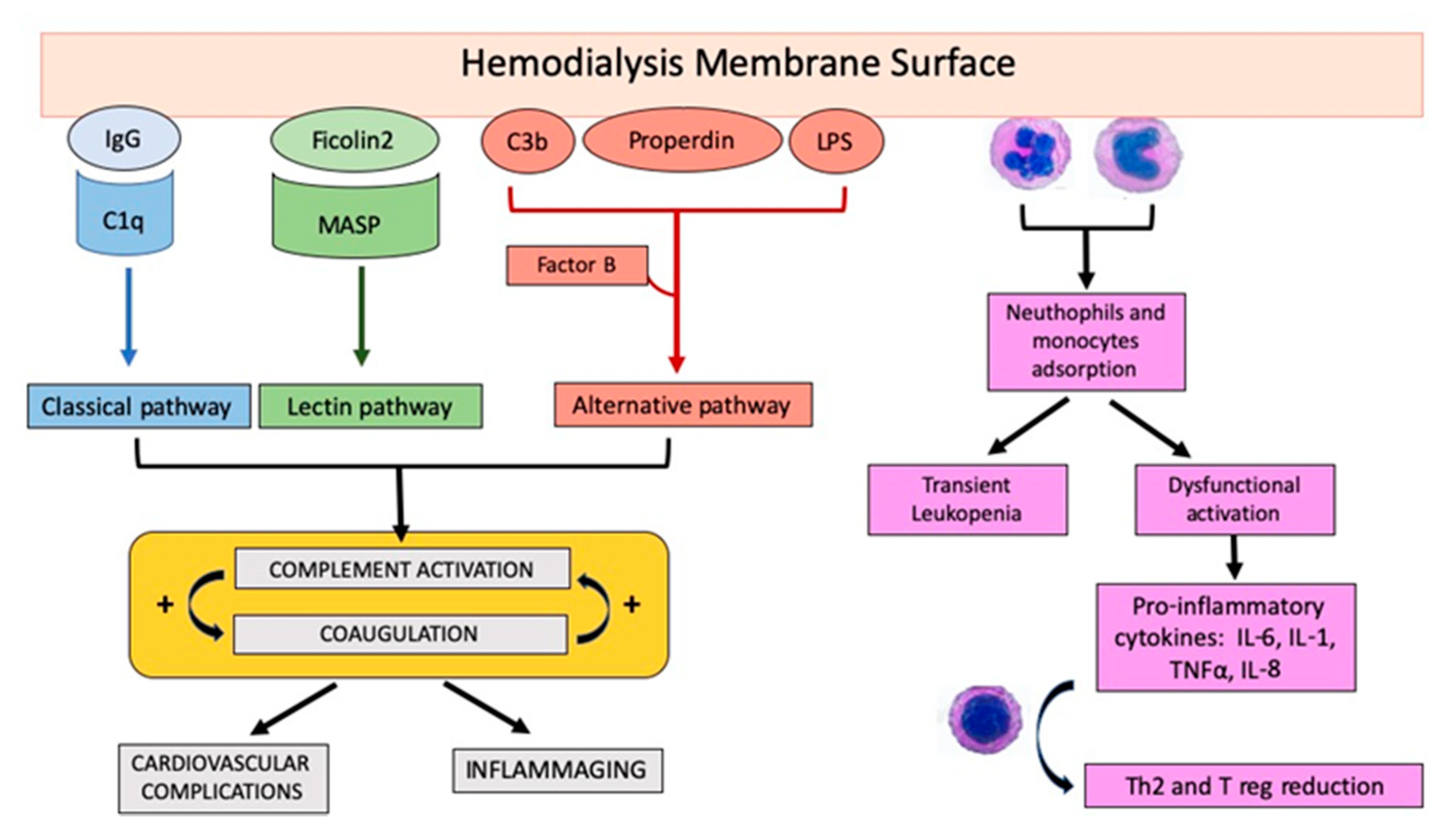{kind=link}
Abstract
:1. Introduction
2. The Link between Chronic Inflammation and Premature Renal Aging
3. Innate Immune Response during Hemodialysis
Cellular Compartments: Neutrophils and Monocytes
4. Humoral Compartments: PRR and Cytokines
5. The Role of Complement Activation during Hemodialysis
5.1. Alternative Pathway
5.2. Lectin Pathway
5.3. Classical Pathway
6. Short-Term Effect of Hemodialysis Treatment on Complement Activation
7. Alteration in Adaptive Immune Response during Hemodialysis
7.1. T Cells
7.2. B Cells
8. Future Directions
9. Modulation of Complement Activation during Dialysis
10. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| CKD | Chronic kidney disease |
| CRP | C-reactive Protein |
| CXCL1 | chemokine (C-X-C motif) ligand 1 |
| ESRD | end-stage renal disease |
| FGF23 | Fibroblast Growth Factor 23 |
| IL6 | Interleukin 6 |
| LPS | Lipopolysaccharide |
| MASP | Mannose-binding lectin (MBL)-associated serine protease |
| MBL | Mannose Binding Lectin, |
| MCP1 | Monocyte chemoattractant Protein-1 |
| MMP | Metalloproteinases |
| PAI-1 | Plasminogen Activator Inhibitor |
References
- Betjes, M.G.H. Immune Cell Dysfunction and Inflammation in End-Stage Renal Disease. Nat. Rev. Nephrol. 2013, 9, 255–652. [Google Scholar] [CrossRef]
- Recio-Mayoral, A.; Banerjee, D.; Streather, C.; CarlosKaski, J. Endothelial dysfunction, inflammation and atherosclerosis in chronic kidney disease—A cross-sectional study of predialysis, dialysis and kidney-transplantation patients. Atherosclerosis 2011, 216, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.; Vajdic, C.M.; van Leeuwen, M.; Amin, J.; Webster, A.C.; Chapman, J.R.; McDonald, S.P.; Grulich, A.E.; McCredie, M.R.E. The pattern of excess cancer in dialysis and transplantation. Nephrol. Dial. Transplant. 2009, 24, 3225–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallone, G.; Infante, B.; Grandaliano, G. Management and prevention of post-transplant malignancies in kidney transplant recipients. Clin. Kidney J. 2015, 8, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, B.; Wulf, S.; Bokbov, B.; Perico, N.; Cortinovis, M.; Courville de Vaccaro, K.; Flaxman, A.; Peterson, H.; Delossantos, A.; Haring, D.; et al. Maintenance Dialysis throughout the World in Years 1990 and 2010. J. Am. Soc. Nephrol. 2015, 26, 2621–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Renal Data System. USRDS 2015 Annual Data Report: Volume 2—Endstage Renal Disease [ESRD] in the United States; USRDS: Bethesda, MD, USA, 2016; Chapter 1. [Google Scholar]
- Lassalle, M.; Monnet, E.; Ayav, C. Annual Report Digest of the Renal Epidemiology Information Network (REIN) registry. Transpl. Int. 2019, 32, 892–902. [Google Scholar] [CrossRef]
- Gilg, J.; Caskey, F.; Fogarty, D. UK Renal Registry 18th Annual Report: Chapter 1 UK Renal Replacement Therapy Incidence in 2014: National and Centre-specific Analyses. Nephron 2016, 132, 9–40. [Google Scholar] [CrossRef]
- Kurella, M.; Covinsky, K.; Collins, J.; Chertow, G.M. Octogenarians and nonagenarians starting dialysis in the United States. Ann. Intern. Med. 2007, 146, 177–183. [Google Scholar] [CrossRef]
- Dai, L.; Golembiewska, E.; Lindholm, B.; Stenvinkel, P. End-Stage Renal Disease, Inflammation and Cardiovascular Outcomes. Contrib. Nephrol. 2017, 191, 32–43. [Google Scholar]
- Kooman, J.P.; Broers, N.J.H.; Usvyat, L.; Thijssen, S.; van der Sande, F.M.; Cornelis, T.; Levin, N.W.; Leunissen, K.M.L.; Kotanko, P. Out of control: Accelerated aging in uremia. Nephrol. Dial. Transplant. 2013, 28, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Temmar, M.; Lemke, H.D.; Tribouilloy, C.; Choukroun, G.; Vanholder, R.; Massy, Z.A. European Uremic Toxin Work Group [EUTox] Plasma interleukin-6 is independently with mortality in both hemodialysis and predialysis patient with cronic kidney disease. Kidney Int. 2010, 77, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Guo, D.; Perianayagam, M.C.; Tighiouart, H.; Jaber, B.L.; Pereira, B.J.G.; Balakrishnan, V.S. Plasma interleukin-6 predicts cardiovascular mortality in hemodialysis patients. Am. J. Kidney Dis. 2005, 45, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstädt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; Di Marco, G.S.; et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016, 90, 985–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, D.; Jameson, J. Kidney transplantation and the ageing immune system. Nat. Rev. Nephrol. 2012, 8, 700–708. [Google Scholar] [CrossRef]
- Margolick, J.B.; Ferrucci, L. Accelerating aging research: How can we measure the rate of biologic aging? Exp. Gerontol. 2015, 64, 78–80. [Google Scholar] [CrossRef] [Green Version]
- Kooman, J.P.; Dekker, M.J.; Usvyat, L.A.; Kotanko, P.; van der Sande, F.M.; Schalkwijk, C.G.; Shiels, P.G.; Stenvinkel, P. Inflammation and premature aging in advanced chronic kidney disease. Am. J. Ren. Physiol. 2017, 313, F938–F950. [Google Scholar] [CrossRef] [Green Version]
- Castellano, G.; Di Vittorio, A.; Dalfino, G.; Loverre, A.; Marrone, D.; Simone, S.; Schena, F.P.; Pertosa, G.; Grandaliano, G. Pentraxin 3 and complement cascade activation in the failure of arteriovenous fistula. Atherosclerosis 2010, 209, 241–247. [Google Scholar] [CrossRef]
- Simone, S.; Loverre, A.; Cariello, M.; Divella, C.; Castellano, G.; Gesualdo, L.; Pertosa, G.; Grandaliano, G. Arteriovenous fistula stenosis in hemodialysis patients is characterized by an increased adventitial fibrosis. J. Nephrol. 2014, 27, 555–562. [Google Scholar] [CrossRef]
- Crepin, T.; Legendre, M.; Courivaud, C.; Vauchy, C.; Laheurte, C.; Rebibou, J.M.; Saas, P.; Ducloux, D.; Bamoulid, J. Premature Immune Senescence and Chronic Kidney Disease: Update and Perspectives. Nephrol. Ther. 2019. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Ketteler, M.; Johnson, J.J.; Lindholm, B.; Pecoits-Filho, R.; Riella, M.; Heimbürger, O.; Cederholm, T.; Girndt, M. IL-10, IL-6, and TNF-central factors in the altered cytokine network of uremia—The good, the bad, and the ugly. Kidney Int. 2005, 67, 1216–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooman, J.P.; Kotanko, P.; Schols, A.M.W.J.; Shiels, P.; Stenvinkel, P. Chronic kidney disease and premature ageing. Nat. Rev. Nephrol. 2014, 10, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Stallone, G.; Infante, B.; Prisciandaro, C.; Grandaliano, G. mTOR and aging: An old fashioned dress. Int. J. Mol. Sci. 2019, 20, 2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steyers, C.M., III; Miller, F., Jr. Endothelial dysfunction in chronic inflammatory diseases. Int. J. Mol. Sci. 2014, 15, 11324–11349. [Google Scholar] [CrossRef] [Green Version]
- Sanchis, P.; Ho, C.Y.; Liu, Y.; Beltran, L.E.; Ahmad, S.; Jacob, A.P.; Furmanik, M.; Laycock, J.; Long, D.A.; Shroff, R.; et al. Arterial “Inflammaging” Drives Vascular Calcification in Children on Dialysis. Kidney Int. 2019, 95, 958–972. [Google Scholar] [CrossRef] [Green Version]
- Sepe, V.; Gregorini, M.; Rampino, T.; Esposito, P.; Coppo, R.; Galli, F.; Libetta, C. Vitamin E-Loaded Membrane Dialyzers Reduce Hemodialysis Inflammaging. BMC Nephrol. 2019, 20, 412. [Google Scholar] [CrossRef]
- Vanholder, R.; Ringoir, S.; Dhondt, A.; Hakim, R. Phagocytosis in uremic and hemodialysis patients: A prospective and cross-sectional study. Kidney Int. 1991, 39, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Colì, L.; Tumietto, F.; De Pascalis, A.; La Manna, G.; Zanchelli, F.; Isola, E.; Perna, C.; Raimondi, C.; De Sanctis, L.B.; Marseglia, C.D.; et al. Effect of dialysis membrane nature on intradialytic phagocytizing activity. Int. J. Artif. Organs 1999, 22, 74–80. [Google Scholar] [CrossRef]
- Moore, M.A.; Kaplan, D.S.; Picciolo, G.L.; Wallis, R.R.; Kowolik, M.J. Effect of cellulose acetate materials on the oxidative burst of human neutrophils. J. Biomed. Mater. Res. 2001, 55, 257–265. [Google Scholar] [CrossRef]
- Toren, M.; Goffinet, J.A.; Kaplow, L.S. Pulmonary Bed Sequestration of Neutrophils during Hemodialysis. Blood 1970, 36, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Hoenich, N.A.; Levett, D.; Fawcett, S.; Woffindin, C.; Kerr, D.N. Biocompatibility of hemodialysis membranes. J. Biomed. Eng. 1986, 8, 3–8. [Google Scholar] [CrossRef]
- Dell’Oglio, M.P.; Simone, S.; Ciccone, M.; Corciulo, R.; Gesualdo, M.; Zito, A.; Cortese, F.; Castellano, G.; Gigante, M.; Gesualdo, L.; et al. Neutrophil-dependent pentraxin-3 and reactive oxygen species production modulate endothelial dysfunction in haemodialysis patients. Nephrol. Dial. Transplant. 2017, 32, 1540–1549. [Google Scholar]
- Efthymios, P.M.; Spiridoula, M.; Periklis, K.; Andreas, C.; Sotiris, T.; Theodoros, E.; Evangelos, P.; Konstantinos, K.P.; Dimitrios, G.S. The Effect of Dialysis Modality and Membrane Performance on Native Immunity in Dialysis Patients. Prilozi 2019, 40, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Chonchol, M. Neutrophil dysfunction and infection risk in end-stage renal disease. Semin. Dial. 2006, 19, 291–296. [Google Scholar] [CrossRef]
- Craddock, P.R.; Fehr, J.; Dalmasso, A.P.; Brighan, K.H.; Jacob, H.S. Hemodialysis leukopenia. Pulmonary vascular leukostasis resulting from complement activation by dialyzer cellophane membrane. J. Clin. Investig. 1977, 59, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Martin-Rodriguez, S.; Caballo, C.; Gutierrez, G.; Vera, M.; Cruzado, J.M.; Cases, A.; Escolar, G.; Diaz-Ricart, M. TLR4 and NALP3 inflammasome in the development of endothelial dysfunction in uraemia. Eur. J. Clin. Investig. 2015, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wilson, C.; Passos, J.P.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef]
- Carrero, J.J.; Stenvinkel, P.; Fellstrom, B.; Qureshi, A.R.; Lamb, K.; Heimbürger, O.; Bárány, P.; Radhakrishnan, K.; Lindholm, B.; Soveri, I.; et al. Telomere attrition is associated with inflammation, low fetuin-A levels and high mortality in prevalent haemodialysis patients. J. Intern. Med. 2008, 263, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granata, S.; Masola, V.; Zoratti, E.; Scupoli, M.T.; Baruzzi, A.; Messa, M.; Sallustio, F.; Gesualdo, L.; Lupo, A.; Zaza, G. NLRP3 inflammasome activation in dialyzed chronic kidney disease patients. PLoS ONE 2015, 10, e0122272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watroba, M.; Szukiewicz, D. The role of sirtuins in aging and age-related diseases. Adv. Med. Sci. 2016, 61, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Sela, S.; Shurtz-Swirski, R.; Cohen-Mazor, M.; Mazor, R.; Chezar, J.; Shapiro, G.; Hassan, K.; Shkolnik, G.; Geron, R.; Kristal, B. Primed peripheral polymorphonuclear leukocyte: A culprit underlying chronic low-grade inflammation and systemic oxidative stress in chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Lin, K.Y.; Chen, Y.Q. Diverse functions of miR-125 family in different cell contexts. J. Hematol. Oncol. 2013, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Zawada, A.M.; Rogacev, K.; Muller, S.; Rotter, B.; Winter, P.; Fliser, D.; Heine, G.H. Massive analysis of cDNA Ends [MACE] and miRNA expression profiling identifies proatherogenic pathways in chronic kidney disease. Epigenetics 2014, 9, 161–172. [Google Scholar] [CrossRef]
- Nilsson, B.; Ekdahl, K.N.; Mollnes, T.E.; Lambris, J.D. The role of complement in biomaterial-induced inflammation. Mol. Immunol. 2007, 44, 82–94. [Google Scholar] [CrossRef]
- Craddock, P.R.; Fehr, J.; Brigham, K.L.; Kronenberg, R.S.; Jacob, H.S. Complement and Leukocyte-Mediated Pulmonary Dysfunction in Hemodialysis. N. Engl. J. Med. 1977, 296, 769–774. [Google Scholar] [CrossRef]
- Chenoweth, D.E.; Cheung, A.K.; Henderson, L.W. Anaphylatoxin formation during hemodialysis: Effects of different dialyzer membranes. Kidney Int. 1983, 24, 764–769. [Google Scholar] [CrossRef] [Green Version]
- Poothullil, J.; Shimizu, A.; Day, R.P.; Dolovich, J. Anaphylaxis from the product[s] of ethylene oxide gas. Ann. Intern. Med. 1975, 82, 58–60. [Google Scholar] [CrossRef]
- Villarroel, F.; Ciarkowski, A.A. A survey on hypersensitivity reactions in hemodialysis. Artif. Organs 1985, 9, 231–238. [Google Scholar] [CrossRef]
- Daugirdas, J.T.; Ing, T.S. First-use reactions during hemodialysis: A definition of subtypes. Kidney Int. 1988, 24, S37–S43. [Google Scholar]
- Nosé, Y. Hypersensitivity in hemodialysis: Only the tip of the iceberg. Artif. Organs 1984, 8, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Poppelaars, F.; da Costa, M.G.; Berger, S.P.; Assa, S.; Meter-Arkema, A.H.; Daha, M.R.; van Son, W.J.; Franssen, C.F.M.; Seelen, M.A.J. Strong predictive value of mannose-binding lectin levels for cardiovascular risk of hemodialysis patients. J. Transl. Med. 2016, 14, 236. [Google Scholar] [CrossRef] [Green Version]
- Lines, S.W.; Richardson, V.R.; Thomas, B.; Dunn, E.J.; Wright, M.J.; Carter, A.M. Complement and cardiovascular disease—The missing link in haemodialysis patients. Nephron 2016, 132, 5–14. [Google Scholar] [CrossRef]
- Poppelaars, F.; Faria, B.; da Costa, M.G.; Franssen, C.F.M.; van Son, W.J.; Berger, S.P.; Daha, M.R.; Seelen, M.A. The Complement System in Dialysis: A Forgotten Story? Front. Immunol. 2018, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mares, J.; Richtrova, P.; Hricinova, A.; Tuma, Z.; Moravec, J.; Lysak, D.; Matejovic, M. Proteomic profiling of blood-dialyzer interaction reveals involvement of lectin complement pathway in hemodialysis-induced inflammatory response. Proteom. Clin. Appl. 2010, 4, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Mares, J.; Thongboonkerd, V.; Tuma, Z.; Moravec, J.; Matejovic, M. Specific adsorption of some complement activation proteins to polysulfone dialysis membranes during hemodialysis. Kidney Int. 2009, 76, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Kourtzelis, I.; Markiewski, M.M.; Doumas, M.; Kourtzelis, I.; Markiewski, M.M.; Doumas, M.; Rafail, S.; Kambas, K.; Mitroulis, I.; Panagoutsos, S.; et al. Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood 2010, 116, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Buraczynska, M.; Ksiazek, P.; Zukowski, P.; Benedyk-Lorens, E.; Orlowska-Kowalik, G. Complement factor H gene polymorphism and risk of cardiovascular disease in end-stage renal disease patients. Clin. Immunol. 2009, 132, 285–290. [Google Scholar] [CrossRef]
- Girnt, M.; Heisel, O.; Kohler, H. Influence of Dialysis with Polyamide vs. Haemophan Haemodialysers on Monokines and Complement Activation During a 4-month Long-Term Study. Nephrol. Dial. Transplant. 1999, 14, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.K. Adsorption of Unactivated Complement Proteins by Hemodialysis Membranes. Am. J. Kidney Dis. 1989, 14, 472–477. [Google Scholar] [CrossRef]
- Hornum, M.; Bay, J.T.; Clausen, P.; Hornum, M.; Bay, J.T.; Clausen, P.; Hansen, J.M.; Mathiesen, E.R.; Feldt-Rasmussen, B.; Garred, P. High Levels of Mannose-Binding Lectin Are Associated with Lower Pulse Wave Velocity in Uraemic Patients. BMC Nephrol. 2014, 15, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoshita, H.; Ohsawa, I.; Onda, K.; Tamano, M.; Horikoshi, S.; Ohi, H.; Tomino, Y. An Analysis of Functional Activity via the Three Complement Pathways during Hemodialysis Sessions: A New Insight into the Association Between the Lectin Pathway and C5 Activation. Clin. Kidney J. 2012, 5, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mares, J.; Tuma, Z.; Moravec, J.; Pavlina, R.; Matejovic, M. Proteins Adsorbed to a Polysulfone Hemodialysis Membrane under Heparin and Citrate Anticoagulation Regimens. Artif. Organs 2019, 43, 1092–1103. [Google Scholar] [CrossRef]
- Kishida, K.; Kishida, N.; Arima, M.; Nakatsuji, H.; Kobayashi, H.; Funahashi, T.; Shimomura, I. Serum C1q- binding adiponectin in maintenance hemodialysis patients. BMC Nephrol. 2013, 14, 50. [Google Scholar] [CrossRef] [Green Version]
- Zoccali, C.; Mallamaci, F.; Tripepi, G.; Benedetto, F.A.; Cutrupi, S.; Parlongo, S.; Malatino, L.S.; Bonanno, G.; Seminara, G.; Rapisarda, F.; et al. Adiponectin, metabolic risk factors, and cardiovascular events among patients with end-stage renal disease. J. Am. Soc. Nephrol. 2002, 13, 134–141. [Google Scholar]
- Rousseau, Y.; Carrero, M.P.; Poignet, J.L.; Kazatchkine, M.D.; Haeffner-Cavaillon, N. Dissociation between complement activation, integrin expression and neutropenia during hemodialysis. Biomaterials 1999, 20, 1959–1967. [Google Scholar] [CrossRef]
- Bergseth, G.; Lambris, J.D.; Mollnes, T.E.; Lappegard, K.T. Artificial surface-induced inflammation relies on complement factor 5: Proof from a deficient person. Ann. Thorac. Surg. 2011, 91, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Hempel, J.C.; Poppelaars, F.; da Costa, M.G.; Franssen, C.F.M.; de Vlaam, T.P.G.; Daha, M.R.; Berger, S.P.; Seelen, M.A.J.; Gaillard, C.A.J.M. Distinct in vitro complement activation by various intravenous iron preparations. Am. J. Nephrol. 2017, 45, 49–59. [Google Scholar] [CrossRef]
- Simone, S.; Rascio, F.; Castellano, G.; Divella, C.; Chieti, A.; Ditonno, P.; Battaglia, M.; Crovace, A.; Staffieri, F.; Oortwijn, B.; et al. Complement-dependent NADPH Oxidase Enzyme Activation in Renal Ischemia/Reperfusion Injury. Free Radic Biol. Med. 2014, 74, 263–273. [Google Scholar] [CrossRef]
- Lin, Y.P.; Yang, C.Y.; Liao, C.C.; Yu, W.C.; Chi, C.W.; Lin, C.H. Plasma protein characteristics of long-term hemodialysis survivors. PLoS ONE 2012, 7, e40232. [Google Scholar] [CrossRef] [Green Version]
- Hein, E.; Garred, P. The lectin pathway of complement and biocompatibility. Adv. Exp. Med. Biol. 2015, 865, 77–92. [Google Scholar]
- Fernandez-Fresnedo, G.; Ramos, M.A.; Gonzalez-Pardo, M.C.; de Francisco, A.L.; López-Hoyos, M.; Arias, M. B lymphopenia in uremia is related to an accelerated in vitro apoptosis and dysregulation of Bcl-2. Nephrol. Dial. Transplant. 2000, 15, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Nockher, W.A.; Scherberich, J.E. Expanded CD14+CD16+ monocyte subpopulation in patients with acute and chronic infections undergoing hemodialysis. Infect. Immun. 1998, 66, 2782–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, P.; Dayer, E.; Blanc, E.; Wauters, J.P. Early T cell activation correlates with expression of apoptosis markers in patients with end-stage renal disease. J. Am. Soc. Nephrol. 2002, 13, 204–212. [Google Scholar] [PubMed]
- Betjes, M.G.; Langerak, A.W.; van der Spek, A.; de Wit, E.A.; Litjens, N.H.R. Premature aging of circulating T cells in patients with end-stage renal disease. Kidney Int. 2011, 80, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, B.F.; Sempowski, G.D.; Wells, A.F.; Hale, L.P. The human thymus during aging. Immunol. Res. 2000, 22, 253–261. [Google Scholar] [CrossRef]
- Ferrando-Martınez, S.; Romero-Sanchez, M.C.; Solana, R.; Delgado, J.; de la Rosa, R.; Muñoz-Fernández, M.A.; Ruiz-Mateos, E.; Leal, M. Thymic function failure and C-reactive protein levels are independent predictors of all-cause mortality in healthy elderly humans. Age 2013, 35, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laan, M.; Haljasorg, U.; Kisand, K.; Salumets, A.; Peterson, P. Pregnancy-induced thymic involution is associated with suppression of chemokines essential for T-lymphoid progenitor homing. Eur. J. Immunol. 2016, 46, 2008–2017. [Google Scholar] [CrossRef] [Green Version]
- Hamazaki, Y.; Sekai, M.; Minato, N. Medullary thymic epithelial stem cells: Role in thymic epithelial cell maintenance and thymic involution. Immunol. Rev. 2016, 271, 38–55. [Google Scholar] [CrossRef]
- Mackall, C.L.; Fry, T.J.; Morgan, P.; Galbraith, A.; Gress, L.E. IL-7 increases both thymic-dependent and thymic-independent T-cell regeneration after bone marrow transplantation. Blood 2001, 97, 1491–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, M.L.; Bonan, N.B.; Dias, G.; Brehm, F.; Steiner, T.M.; Souza, W.M.; Stinghen, A.E.M.; Barreto, F.C.; Elifio-Esposito, S.; Pecoits-Filho, R.; et al. p-Cresyl sulfate affects the oxidative burst, phagocytosis process, and antigen presentation of monocyte-derived macrophages. Toxicol. Lett. 2016, 263, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Crepin, T.; Legendre, M.; Courivaid, C.; Vauchy, C.; Laheurte, C.; Rebibou, J.M.; Saas, P.; Ducloux, D.; Bamoulid, J. Uremia-induced immune senescence and clinical outcomes in chronic kidney disease patients. Nephrol. Dial. Transplant. 2018, 35, 1–9. [Google Scholar]
- Van Riemsdijk, I.C.; Baan, C.C.; Loonen, E.H.; Knoop, C.J.; Betonico, G.N.; Niesters, N.H.; Zietse, R.; Weimar, W. T cells activate the tumor necrosis factor-α system during hemodialysis, resulting in tachyphylaxis. Kidney Int. 2001, 59, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litjens, N.H.; van Druningen, C.J.; Betjes, M.G.H. Progressive loss of renal function is associated with activation and depletion of naive T lymphocytes. Clin. Immunol. 2006, 118, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Gollapudi, S.; Pahl, M.V.; Vaziri, M.D. Naive and central memory Tcell lymphopenia in end-stage renal disease. Kidney Int. 2006, 70, 371–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrikx, T.K.; van Gurp, E.A.; Mol, W.M.; Schoordijk, W.; Sewgobind, V.D.; Ijzermans, J.N.; Weimar, W.; Baan, C.C. End-stage renal failure and regulatory activities of CD4+CD25bright+FoxP3+ Tcells. Nephrol. Dial. Transplant. 2009, 24, 1969–1978. [Google Scholar] [CrossRef] [Green Version]
- Kruger, S.; Muller-Steinhardt, M.; Kiechner, H.; Kreft, B. A 5 year follow-up on antibody response after diphtheria and tetanus vaccination in hemodialysis patients. Am. J. Kidney Dis. 2001, 38, 1264–1270. [Google Scholar] [CrossRef] [Green Version]
- Pahl, M.V.; Gollapoudi, S.; Sepassi, L.; Gollapudi, P.; Elahimehr, R.; Vaziri, N.D. Effect of end-stage renal disease on Blymphocyte subpopulations, IL7, BAFF and BAFF receptor expression. Nephrol. Dial. Transplant. 2010, 25, 205–212. [Google Scholar] [CrossRef]
- Beaman, M.; Michael, J.; MacLennan, I.C.; Adu, D. Tcellindependent and Tcelldependent antibody responses in patients with chronic renal failure. Nephrol. Dial. Transplant. 1989, 4, 216–221. [Google Scholar] [CrossRef]
- Litjens, N.H.R.; Huisman, M.; van der Dorpel, M.; Betjes, M.G. Impaired immune responses and antigen-specific memory CD4+ T cells in hemodialysis patients. J. Am. Soc. Nephrol. 2008, 19, 1483–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobler, C.C.; McDonald, S.P.; Marks, G.B. Risk of tuberculosis in dialysis patients: A nationwide cohort study. PLoS ONE 2011, 6, e29563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weltevrede, M.; Eilers, R.; de Melker, H.E.; van Baarle, D. Cytomegalovirus persistence and T-cell immunosenescence in people aged fifty and older: A systematic review. Exp. Gerontol. 2016, 77, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Shu, K.H.; Yang, F.J.; Chou, T.Y.; Chen, P.M.; Lay, F.Y.; Pan, S.Y.; Lin, C.J.; Litjens, N.H.R.; Betjes, M.G.H.; et al. A comprehensive characterization of aggravated aging-related changes in T lymphocytes and monocytes in end-stage renal disease: The iESRD study. Immun. Ageing 2018, 15, 27. [Google Scholar] [CrossRef]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients—A prospective cohort study. Nephrol. Dial. Transplant. 2012, 27, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Meijers, R.W.J.; Litjens, N.H.R.; de Wit, E.A.; Langerak, A.W.; van der Spek, A.; Baan, C.C.; Weimar, W.; Betjes, M.G.H. Cytomegalovirus contributes partly to uremia-associated premature immunological aging of T cell compartment. Clin. Exp. Immunol. 2013, 174, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Kuijpers, T.W.; Vossen, M.T.; Gent, M.R.; Davin, J.C.; Roos, M.T.; Wertheim-van Dillen, P.M.; Weel, J.F.; Baars, P.A.; van Lier, R.A. Frequencies of circulating cytolytic, CD45RA+C27-, CD8+ T lymphocytes depend on infection with CMV. J. Immunol. 2003, 170, 4342–4348. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Langerak, A.W.; Baan, C.C.; Litjens, N.H.R.; Betjes, M.G.H. Latency for Cytomegalovirus impacts T-cell ageing significantly in elderly end-stage renal disease patients. Clin. Exp. Immunol. 2016, 186, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Olson, N.C.; Doyle, M.F.; Jenny, N.S.; Huber, S.A.; Psaty, B.M.; Kronmal, R.A.; Tracy, R.P. Decreased naive and increased memory CD4[+] T cells are associated with subclinical atherosclerosis: The multi-ethnic study of atherosclerosis. PLoS ONE 2013, 8, e71498. [Google Scholar] [CrossRef]
- Van de Berg, P.J.; Yong, S.L.; Remmerswaal, E.B.M.; van Lier, R.A.W.; ten Berge, I.J.M. Cytomegalovirus-induced effector T cells cause endothelial cell damage. Clin. Vaccine Immunol. 2012, 19, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.J.; Shu, K.H.; Chen, H.Y.; Chen, I.Y.; Lay, F.Y.; Chuang, Y.F.; Wu, C.S.; Tsai, W.C.; Peng, Y.S.; Hsu, S.P.; et al. Anti-cytomegalovirus IgG antibody titer is positively associated with advanced T cell differentiation and coronary artery disease in end-stage renal disease. Immun. Ageing 2018, 15, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appay, V.; Sauce, D. Naive T cells: The crux of cellular immune aging? Exp. Gerontol. 2014, 54, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Montoya-Ortiz, G. Immunosenescence, aging, and systemic lupus erythematous. Autoimmune Dis. 2013, e267078. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraccioli, G.; Gremese, E. B cell activating factor [BAFF] and BAFF receptors: Fakes and facts. Clin. Exp. Immunol. 2017, 190, 291–292. [Google Scholar] [CrossRef] [Green Version]
- Esposito, P.; Rampino, T.; Gregorini, M.; Gabanti, E.; Bianzina, S.; Dal Canton, A. Mechanisms Underlying sCD40 Production in Hemodialysis Patients. Cell. Immunol. 2012, 278, 10–15. [Google Scholar] [CrossRef]
- Contin, C.; Pitard, V.; Delmas, Y.; Pelletier, N.; Defrance, T.; Moreau, J.F.; Merville, P.; Déchanet-Merville, J. Potential role of soluble CD40 in the humoral immune response impairment of uraemic patients. Immunology 2003, 110, 131–140. [Google Scholar] [CrossRef]
- Kokubo, K.; Kurihara, Y.; Kobayashi, K.; Tsukao, H.; Kobayashi, H. Evaluation of the biocompatibility of dialysis membranes. Blood Purif. 2015, 40, 293–297. [Google Scholar] [CrossRef]
- Ronco, C.; Brendolan, A.; Nalesso, F.; Zanella, M.; De Cal, M.; Corradi, V.; Virzì, G.M.; Ferrari, F.; Garzotto, F.; Lorenzin, A.; et al. Prospective, randomized, multicenter, controlled trial [TRIATHRON 1] on a new antithrombogenic hydrophilic dialysis membrane. Int. J. Artif. Organs 2017, 40, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Oshihara, W.; Ueno, Y.; Fujeda, H. A new Polysulphone membrane dialyzer, NV, with low fouling and antithrombotic properties. Contrib. Nephrol. 2017, 189, 222–229. [Google Scholar]
- Yamaka, T.; Ichikawa, K.; Saito, M. Biocompatibility of the new anticoagulant dialyzer Toraylight NV. Sci. Postprint 2014, 1, e00020. [Google Scholar] [CrossRef]
- Hidaka, S.; Kobayashi, H.; Maesato, K. Hydrophilic polymer-coated polysulfone membrane improves endothelial function of hemodialysis patients: A pilot study. J. Clin. Neph. Res. 2015, 2, 1020. [Google Scholar]
- Rodriguez-Ribera, L.; Corredor, Z.; Silva, I.; Díaz, J.M.; Ballarín, J.; Marcos, R.; Pastor, S.; Coll, E. Vitamin E Coated Dialysis Membrane reduce the levels of oxidative genetic damage in hemodialysis patients. Mutat. Res. 2017, 815, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, C.A.; Wolley, M. The rationale for expanded hemodialysis therapy [HDx]. Contrib. Nephrol. 2017, 191, 142–152. [Google Scholar]
- Aoike, I. Clinical significance of protein adsorbable membranes—Long-term clinical effects and analysis using a proteomic technique. Nephrol. Dial. Transplant. 2007, 22, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Yu, X. The evolving pattern of uremia: Unmet clinical needs in dialysis. Contrib. Nephrol. 2017, 191, 1–7. [Google Scholar]
- Boschetti de Fierro, A.; Beck, W.; Hildwein, H.; Krause, B.; Storr, M.; Zweigart, C. Membrane innovation in dialysis. Contrib. Nephrol. 2017, 191, 100–114. [Google Scholar]
- Freitas Lima, L.C.; Braga, V.A.; do Soccorro de Franca Silva, M.; Cruz, J.C.; Sousa Santos, S.H.; de Oliveira Monteiro, M.M.; Balarini, C.M. Adipokines, diabetes and atherosclerosis: An inflammatory association. Front. Physiol. 2015, 6, 304. [Google Scholar] [CrossRef]
- Kato, S.; Chmielewski, M.; Honda, H.; Pecoits-Filho, R.; Matsuo, S.; Yuzawa, Y.; Tranaus, A.; Stenvinkel, P.; Lindholm, B. Aspects of immune dysfunction in end-stage renal disease. Clin. J. Am. Soc. 2008, 3, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- MacDougall, I.C.; Cooper, A.C. Erythropoietin resistance: The role of inflammation and proinflammatory cytokines. Nephrol. Dial. Transplant. 2002, 17, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Zickler, D.; Schindler, R.; Willy, K.; Pawlak, M.; Storr, M.; Hulko, M.; Boehler, T.; Glomb, M.A.; Liehr, K.; Henning, C.; et al. Medium cut-off (MCO) membranes reduce inflammation in chronic dialysis patient: A randomized controlled clinical trial. PLoS ONE 2017, 12, e0169024. [Google Scholar] [CrossRef] [PubMed]
- Hakim, R.M.; Fearon, D.T.; Lazarus, J.M. Biocompatibility of dialysis membranes: Effects on chronic complement activation. Kidney Int. 1984, 26, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonomini, M.; Fiederling, B.; Bucciarelli, T.; Manfrini, V.; Di Ilio, C.; Albertazzi, A. A new polymethylmethacrylate membrane for hemodialysis. Int. J. Artif. Organs 1996, 19, 232–239. [Google Scholar] [CrossRef]
- Hata, H.; Nishi, K.; Oshihara, W.; Arai, J.; Shimizu, K.; Kawakita, T.; Nakamura, M.; Mitsuya, H. Adsorption of Bence-Jones protein to polymethylmethacrylate membrane in primary amyloidosis. Amyloid 2009, 16, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Oshihara, W.; Nagao, H.; Megano, H.; Arai, J.; Koide, M.; Takada, M. Trial use of a polymethylmethacrylate membrane for the removal of free immunoglobulin light chains in dialysis patients. Nephrol. Dial. Transplant. 2010, 3, i3–i7. [Google Scholar] [CrossRef] [Green Version]
- Sensi, M.; Pricci, F.; Andreani, D.; Di Mario, U. Advanced non-enzymatic glycation end-products [AGE]: Their relevance to aging and the pathogenesis of late diabetic complications. Diabetes Res. 1991, 16, 1–9. [Google Scholar] [PubMed]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef] [Green Version]
- Dozio, E.; Ambrogi, F.; de Cal, M.; Vianello, E.; Ronco, C.; Corsi Romanelli, M.M. Role of the soluble receptor for advanced glycation end products [sRAGE] as a prognostic factor for mortality in hemodialysis and peritoneal dialysis patients. Mediat. Inflamm. 2018, 15, e1347432. [Google Scholar] [CrossRef] [Green Version]
- Ritz, E.; Deppish, R.; Nawroth, P. Toxicity of uremia: Does it come of AGE? Nephrol. Dial. Transplant. 1994, 9, 1–2. [Google Scholar]
- Kimura, A.; Naka, T.; Kishimoto, T. IL-6-dependent and independent pathways in the development of interleukin 17-producing T helper cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12099–12104. [Google Scholar] [CrossRef] [Green Version]
- Zaza, G.; Granata, S.; Rascio, F.; Pontrelli, P.; Dell’Oglio, M.P.; Cox, S.N.; Pertosa, G.B.; Grandaliano, G.; Lupo, A. A specific immune transcriptomic profile discriminates chronic kidney disease patients in predialysis from hemodialyzed patients. BMC Med. Genom. 2013, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Hua, G.; Zhang, X.; Tong, R.; DU, X.; Li, Z. Regulatory T cells/T-helper cell 17 functional imbalance in uraemic patients on maintenance haemodialysis: A pivotal link between microinflammation and adverse cardiovascular events. Nephrology 2010, 15, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yuan, X.; Deng, L.; Xu, W.; Zheng, Y.; Yue, C.; Zhang, G.; Xie, F.; Yang, Y.H.; Gantier, M.P.; et al. Imbalanced frequencies of Th17 and Treg cells in acute coronary syndromes are mediated by IL-6-STAT3 signaling. PLoS ONE 2013, 8, e72804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, F.; Benedetti, S.; Floridi, A.; Canestrari, F.; Piroddi, M.; Buoncristiani, E.; Buoncristiani, U. Glycoxidation and inflammatory markers in patients on treatment with PMMA-based protein-leaking dialyzers. Kidney Int. 2005, 67, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Abe, M.; Hamano, T.; Wada, A.; Nakai, S.; Masakane, I. Renal Data Registry Committee; Japanese Society for Dialysis Therapy. Effect of dialyzer membrane materials on survival in chronic hemodialysis patients: Results from the annual survey of the Japanese Nationwide Dialysis Registry. PLoS ONE 2017, 12, e0184424. [Google Scholar] [CrossRef] [Green Version]
- Horl, W.H.; Schollmeyer, P.; Rautenberg, W.; Neumann, S. Different complement and granulocyte activation in patients dialyzed with PMMA dialyzers. Clin. Nephrol. 1986, 25, 304–307. [Google Scholar]
- Contin-Bordes, C.; Lacraz, A.; de Precigout, V. Potential role of the soluble form of CD40 in deficient immunologica function of dialysis patients: New findings of its amelioration using polymethylmethacrylate [PMMA] membranes. NDT Plus 2010, 3, i20–i27. [Google Scholar]
- Lin, H.H.; Liu, Y.L.; Liu, J.H.; Chou, C.Y.; Yang, Y.F.; Kuo, H.L.; Huang, C.C. Uremic pruritus, cytokines, and polymethylmethacrylate artificial kidney. Artif. Organs 2008, 32, 468–472. [Google Scholar] [CrossRef]
- Aucella, F.; Vigilante, M.; Gesuete, A. Review: The effect of polymethylmethacrylate dialysis membranes on uraemic pruritus. Nephrol. Dial. Transplant. 2010, 3, i8–i11. [Google Scholar] [CrossRef] [Green Version]
- Kreusser, W.; Reiermann, S.; Vogelbusch, G.; Bartual, J.; Schulze-Lohoff, E. Effect of different synthetic membranes on laboratory parameters and survival in chronic haemodialysis patients. Nephrol. Dial. Transplant. 2010, 3, i12–i19. [Google Scholar] [CrossRef] [Green Version]
- Nakai, S.; Iseki, K.; Itami, N.; Ogata, S.; Kazama, J.J.; Kimata, N.; Shigematsu, T.; Shinoda, T.; Shoji, T.; Suzuki, K.; et al. Overview of regular dialysis treatment in Japan. AS of December 31, 2009. Ther. Apher. Dial. 2012, 16, 11–53. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Humano, T.; Wada, A.; Nakai, S.; Masakane, I. High-performance membrane dialyzers and mortality in hemodialysis patients: A 2-year cohort study from the annual survey of the Japanese renal data registry. Am. J. Nephrol. 2017, 46, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Meijers, R.W.; Litjens, N.H.R.; de Wit, E.A.; Langerak, A.W.; van der Spek, A.; Baan, C.C.; Weimar, W.; Betjes, M.G. Uremia causes premature ageing of the T cell compartment in end-stage renal disease patients. Immun. Ageing 2012, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducloux, D.; Legendre, M.; Bamoulid, J.; Rebibou, J.M.; Saas, P.; Courivaud, C.; Crepin, T. ESRD-associated immune phenotype depends on dialysis modality and iron status: Clinical implications. Immun. Ageing 2018, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Rostoker, G. When should iron supplementation in dialysis patients be avoided, minimized or withdrawn? Semin. Dial. 2019, 32, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seelen, M.A.; Roos, A.; Wieslander, J.; Mollnes, T.E.; Sjöholm, A.G.; Wurzner, R.; Loos, M.; Tedesco, F.; Sim, R.B.; Garred, P.; et al. Functional analysis of the classical, alternative, and MBL pathways of the complement system: Standardization and validation of a simple ELISA. J. Immunol. Methods 2005, 296, 187–198. [Google Scholar] [CrossRef]
- Huang, S.; Sandholm, K.; Jonsson, N.; Nilsson, A.; Wieslander, A.; Grundström, G.; Hancock, V.; Ekdahl, K.N. Low concentrations of citrate reduce complement and granulocyte activation in vitro in human blood. Clin. Kidney J. 2015, 8, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Böhler, J.; Schollmeyer, P.; Bressel, B.; Dobos, G.; Hörl, W.H. Reduction of granulocyte activation during hemodialysis with regional citrate anticoagulation: Dissociation of complement activation and neutropenia from neutrophil degranulation. J. Am. Soc. Nephrol. 1996, 7, 234–241. [Google Scholar]
- Opatrný, K.; Richtrova, P.; Polanska, K.; Wirth, J.; Sefrna, F.; Brandl, M.; Falkenhagen, D. Citrate anticoagulation control by ionized calcium levels does not prevent hemostasis and complement activation during hemodialysis. Artif. Organs 2007, 31, 200–207. [Google Scholar] [CrossRef]
- Gabutti, L.; Ferrari, N.; Mombelli, G.; Keller, F.; Marone, C. The favorable effect of regional citrate anticoagulation on interleukin-1beta release is dissociated from both coagulation and complement activation. J. Nephrol. 2004, 17, 819–825. [Google Scholar]
- Poppelaars, F.; Damman, J.; de Vrij, E.L.; Burgerhof, J.G.; Saye, J.; Daha, M.R.; Leuvenink, H.G.; Uknis, M.E.; Seelen, M.A. New insight into the effects of heparinoids on complement inhibition by C1-inhibitor. Clin. Exp. Immunol. 2016, 184, 378–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtzelis, I.; Rafail, S.; DeAngelis, R.A.; Foukas, P.G.; Ricklin, D.; Lambris, J.D. Inhibition of biomaterial-induced complement activation attenuates the inflammatory host response to implantation. FASEB J. 2013, 27, 2768–2776. [Google Scholar] [CrossRef] [Green Version]
- Lazar, H.L.; Bokesch, P.M.; van Lenta, F.; Fitzgerald, C.; Emmett, C.; Marsh, H.C., Jr.; Ryan, U. OBE and the TP10 Cardiac Surgery Study Group. Soluble human complement receptor 1 limits ischemic damage in cardiac surgery patients at high risk requiring cardiopulmonary bypass. Circulation 2004, 110, 274–279. [Google Scholar] [CrossRef] [Green Version]
- Reis, E.S.; DeAngelis, R.A.; Chen, H.; Resuello, R.R.; Ricklin, D.; Lambris, J.D. Therapeutic C3 inhibitor Cp40 abrogates complement activation induced by modern hemodialysis filters. Immunobiology 2015, 220, 476–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, H.; Ricklin, D.; Lambris, J.D. Recent developments in low molecular weight complement inhibitors. Mol. Immunol. 2009, 47, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busse, P.J.; Christiansen, S.C. Hereditary Angioedema. N. Engl. J. Med. 2020, 19, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Butani, L.; Calogiuri, G. Hypersensitivity reactions in patients receiving hemodialysis. Ann. Allergy Asthma Immunol. 2017, 118, 680–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, M.; Cohn, D.M. The Role of Complement in Hereditary Angioedema. Transfus. Med. Rev. 2019, 33, 243–247. [Google Scholar] [CrossRef]
- Lepelley, M.; Bernardeau, C.; Defendi, F.; Crochet, J.; Mallaret, M.; Bouillet, L. Update on bradykinin-mediated angioedema in 2020. Therapie 2020, 75, 195–205. [Google Scholar] [CrossRef]
- Cicardi, M.; Banerji, A.; Bracho, F.; Malbrán, A.; Rosenkranz, B.; Riedl, M.; Bork, K.; Lumry, W.; Aberer, W.; Bier, H.; et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N. Engl. J. Med. 2010, 363, 532–541. [Google Scholar] [CrossRef] [Green Version]
- Krieter, D.H.; Fink, E.; Bönner, G.; You, H.M.; Eisenhauer, T. Anaphylactoid reactions during haemodialysis in sheep are associated with bradykinin release. Nephrol. Dial. Transplant. 1995, 10, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Maheut, H.; Lacour, F. Using AN69 ST membrane: A dialysis centre experience. Nephrol. Dial. Transplant. 2001, 16, 1519–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuraw, B.; Yasothan, U.; Kirkpatrick, P. Ecallantide. Nat. Rev. Drug Discov. 2010, 9, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Lumry, W.; Vegh, A.; Williams, A.H.; Schmalbach, T. Critical role of kallikrein in hereditary angioedema pathogenesis: A clinical trial of ecallantide, a novel kallikrein inhibitor. J. Allergy Clin. Immunol. 2007, 120, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Kanchongkittiphon, W.; Kabil, N.; Bacharier, L.B.; Kitcharoensakkul, M. Ecallantide: An alternative treatment of refractory angioedema in adolescents with systemic lupus erythematosus. J. Allergy Clin. Immunol. Pract. 2020, 8, 1115–1116. [Google Scholar] [CrossRef]
- Banerji, A.; Riedl, M.A.; Bernstein, J.A.; Cicardi, M.; Longhurst, H.J.; Zuraw, B.L.; Busse, P.J.; Anderson, J.; Magerl, M.; Martinez-Saguer, I.; et al. HELP Investigators. Effect of Lanadelumab Compared With Placebo on Prevention of Hereditary Angioedema Attacks: A Randomized Clinical Trial. JAMA 2018, 320, 2108–2121. [Google Scholar] [CrossRef] [Green Version]
- Wedi, B. Lanadelumab to treat hereditary angioedema. Drugs Today 2019, 55, 439–448. [Google Scholar] [CrossRef]
- Kohl, J. Drug evaluation: The C5a receptor antagonist PMX-53. Curr. Opin. Mol. Ther. 2006, 8, 529–538. [Google Scholar]
- Wu, Y.Q.; Qu, H.; Sfyroera, G.; Tzekou, A.; Kay, B.K.; Nilsson, B.; Nilsson Ekdahl, K.; Ricklin, D.; Lambris, J.D. Protection of non-self surfaces from complement attack by factor H-binding peptides: Implications for therapeutic medicine. J. Immunol. 2001, 186, 4269–4277. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losappio, V.; Franzin, R.; Infante, B.; Godeas, G.; Gesualdo, L.; Fersini, A.; Castellano, G.; Stallone, G. Molecular Mechanisms of Premature Aging in Hemodialysis: The Complex Interplay between Innate and Adaptive Immune Dysfunction. Int. J. Mol. Sci. 2020, 21, 3422. https://doi.org/10.3390/ijms21103422
Losappio V, Franzin R, Infante B, Godeas G, Gesualdo L, Fersini A, Castellano G, Stallone G. Molecular Mechanisms of Premature Aging in Hemodialysis: The Complex Interplay between Innate and Adaptive Immune Dysfunction. International Journal of Molecular Sciences. 2020; 21(10):3422. https://doi.org/10.3390/ijms21103422
Chicago/Turabian StyleLosappio, Vincenzo, Rossana Franzin, Barbara Infante, Giulia Godeas, Loreto Gesualdo, Alberto Fersini, Giuseppe Castellano, and Giovanni Stallone. 2020. "Molecular Mechanisms of Premature Aging in Hemodialysis: The Complex Interplay between Innate and Adaptive Immune Dysfunction" International Journal of Molecular Sciences 21, no. 10: 3422. https://doi.org/10.3390/ijms21103422