Nuclear P38: Roles in Physiological and Pathological Processes and Regulation of Nuclear Translocation

Department of Biological Regulation, Weizmann Institute of Science, Rehovot 7610001, Israel
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(17), 6102; https://doi.org/10.3390/ijms21176102
Submission received: 27 July 2020
/
Revised: 20 August 2020
/
Accepted: 21 August 2020
/
Published: 24 August 2020
(This article belongs to the Special Issue P38 Signaling Pathway)
Abstract
:The p38 mitogen-activated protein kinase (p38MAPK, termed here p38) cascade is a central signaling pathway that transmits stress and other signals to various intracellular targets in the cytoplasm and nucleus. More than 150 substrates of p38α/β have been identified, and this number is likely to increase. The phosphorylation of these substrates initiates or regulates a large number of cellular processes including transcription, translation, RNA processing and cell cycle progression, as well as degradation and the nuclear translocation of various proteins. Being such a central signaling cascade, its dysregulation is associated with many pathologies, particularly inflammation and cancer. One of the hallmarks of p38α/β signaling is its stimulated nuclear translocation, which occurs shortly after extracellular stimulation. Although p38α/β do not contain nuclear localization or nuclear export signals, they rapidly and robustly translocate to the nucleus, and they are exported back to the cytoplasm within minutes to hours. Here, we describe the physiological and pathological roles of p38α/β phosphorylation, concentrating mainly on the ill-reviewed regulation of p38α/β substrate degradation and nuclear translocation. In addition, we provide information on the p38α/β ’s substrates, concentrating mainly on the nuclear targets and their role in p38α/β functions. Finally, we also provide information on the mechanisms of nuclear p38α/β translocation and its use as a therapeutic target for p38α/β-dependent diseases.
1. Introduction
The p38 mitogen-activated protein kinase (p38MAPK, termed here p38) is a signaling protein kinase that operates within a signaling cascade to transmit extracellular signals to their intracellular targets. The p38 cascade is one of four similar cascades that are all key communication lines between the plasma membranes and the nucleus, and thereby, it is involved in fundamental cellular processes, including stress response, proliferation, differentiation and others [1,2,3]. The four MAPK cascades are extracellular signal-regulated kinase (ERK) 1/2 [4], c-Jun N-terminal kinase (JNK [5]), p38 [6], and ERK5 [7]. The MAPK cascades transmit signals via a sequential activation of protein kinases, which are organized in 3–5 tiers (MAP4K, MAP3K, MAPKK, MAPK, and MAPK activated protein kinases (MAPKAPKs also termed MKs)). Each of these tiers includes more than one kinase (e.g., 4 isoforms at the p38 tier), and the components involved, while the number of tiers may vary between cell lines or under different conditions (see the scheme of the MAPK cascades in ref [8]). In this review, we focus on p38 [9,10], whose cascade is composed of many kinases at the MAP4K and MAP3K levels, MKK3/6, and perhaps MKK4 at the MAPKK tier, p38α−δ at the MAPK tier, and several MKs at the next tier (MNK1/2, MSK1/2, MK2/3, and MK5). Interestingly, unlike the other MAPKs, p38 can also be activated via MKK-independent pathways, either by ZAP/LCK-mediated Tyr phosphorylation [11] or by interaction with TAB1 [12]. The downregulation/inactivation of the p38 cascade is regulated by various phosphatases, among them are several dual specificity phosphatases termed MAPK phosphatases (MKPs) that operate directly on the MAPKs [13]. As in all MAPK cascades, p38 transmits signals initiated by various agents, including cytokines and environmental queues, but it is known to operate mainly as a mediator of stress responses. Thus, the kinase is a key regulator of metabolic, oxidative, and endoplasmic reticulum (ER) stresses, but it plays an important role in other physiological processes such as cell cycle, senescence, differentiation, and several aspects of immunological processes.
Being responsible for the various distinct and even opposing fundamental cellular processes, the p38 cascade needs to be tightly regulated. Indeed, several regulatory mechanisms that determine the specificity of the cascade have been identified, including the duration and strength of the signals [13,14], which are controlled mainly by dual specificity phosphatases [15,16], scaffold proteins [17], and dynamic subcellular localization of the cascade’s components [18]. Importantly, the central roles of the cascade suggest that its dysregulation may cause various diseases. Indeed, p38 was shown to participate in the induction of pathologies such as inflammation-related diseases [19], autoimmune diseases [20], some types of cancer [6], and other pathologies, as specified later in this review. Interestingly, unlike other MAPKs, p38 demonstrates distinct and even opposing effects in different cancers, as it was shown to serve either as a tumor suppressor [21] or tumor promoter [22]. It was also shown that in some cases, it can perform both activities in different stages of cancer development [23]. Although all p38 isoforms have been implicated in the processes listed above, they can be divided into two somewhat distinct subgroups: p38α and p38β (p38α/β) versus p38γ and p38δ. In this review, we focus on p38α/β, mainly discussing the physiological and pathological roles of these protein kinases, providing information on nuclear p38α/βs and their substrates as well as the importance of their phosphorylation specificity. We also describe the mechanisms involved in the nuclear translocation of p38α/β and compare it to other mechanisms of nuclear shuttling. The fact that p38α/β has so many nuclear targets indicates that the prevention of their nuclear translocation may affect their physiological and pathological functions. Indeed, we show that prevention of the nuclear translocation can be used as a tool to combat inflammation and cancer.
2. Physiological Roles of Nuclear p38α/β
The p38α/β are best known for their involvement in stress signaling, and indeed, these kinases as well as JNKs were initially termed stress-activated protein kinases (SAPKs [24]). However, it was established that their activity is not confined to stress responses, and under some conditions, the p38α/β may participate in the regulation of other processes, such as proliferation, differentiation, immune response, migration, and apoptosis. In many cases, p38α/β mediate their effects by activating and regulating transcription factors. One interesting example is the modulation of endoplasmic reticulum (ER) stress in breast cancer cells, which is mediated by the p38α/β-dependent activation of the transcription factor XBP-1 that decreases the expression of the ER protein ERp29 [25]. Another example is the transcriptional inhibition of autophagy genes downstream of p38α/β in response to oxidative stress in HeLa cells [26]. Other stresses such as UV radiation translational inhibition and others were shown to operate by p38α/β−activated transcription factors, such as ATF, MEF2, Elk1, and p53 [3]. However, p38α/β can also affect other regulators (e.g., MKs, proteasome, EGFR [27]) to coordinate their signaling. Notable transcription factor-independent targets that exert p38α/β functions are cell cycle regulators that modulate (usually inhibiting) cell cycle progression. Thus, various stresses induce the downregulation of cyclinD, thereby arresting cells at G1 [28]. In addition, p38α/β cascades were shown to induce the expression of CDK inhibitors, activate p53, or inhibit the transcription factor E2F and the G2/M regulator Cdc25B phosphatase, all leading to the inhibition of cell cycle progression [6,29]. By contrast, in some systems, p38α/β seem to enhance proliferation. For example, such effects were detected in hematopoietic cells and in some cancer cell lines [30]. These differential effects may be mediated by changes in the duration of p38α/β signals, where transient signals lead to fibroblasts’ proliferation, while sustained signals induce cell cycle arrest [31]. However, the molecular mechanisms by which p38α/β are involved in proliferation have not been fully deciphered yet. Thus, the effects of p38α/β on the regulation of stress response or cell cycle progression are well-reviewed (e.g., [6,27,32,33]), and we will not elaborate on these effects. However, not less important are the roles of p38α/β in regulating protein degradation and the translocation of proteins upon stimulation. The molecular mechanisms involved are described in detail next.
2.1. 38α/β Regulation of Protein Degradation
The role of p38α/β in the regulation of protein degradation is widespread, mainly upon stress signals, and may involve several distinct mechanisms in both the cytoplasm and the nucleus. One such mechanism that mostly occurs in the nucleus involves phosphorylation of ubiquitin E3 ligases, such as Siah2, which is known to regulate PHD3 that further controls the stability of the transcription factor HIF1 α. p38α/β phosphorylate Siah2 on Ser24 and Thr29, thereby facilitating its activity towards degradation of PHD, and in turn destabilization of HIF1 α [34,35]. Similarly, p38α/β phosphorylate the E3 ligase Skp2 at Ser64, leading to enhanced degradation of the transcription factor Nkx3-1 and thereby blocking its effects on estrogen receptor-mediated gene expression [36]. Another mechanism involves the phosphorylation of the ubiquitination target which can either facilitate or inhibit the ubiquitination process. Examples for enhanced degradation are the phosphorylation of RBP-Jk at Thr339 which subsequently induces its degradation [37], or phosphorylation of p300 at Ser 1834 (together with AKT) that induces its degradation to allow DNA repair [38]. On the other hand, phosphorylation of the inflammation regulator TRIM9s at Ser76/80 stabilizes it, thereby causing a positive feedback loop for the degradation of the upstream MKK6 [39]. Interestingly, p38α/β may also regulate proteasomal activity and localization to govern protein stability in general. It was shown that osmotic stress inhibits proteasome by p38α/β-dependent phosphorylation of the proteasome subunit Rpn2 at Thr273, which is important for peptide degrading activity [40]. This inhibitory effect was supported by the finding that p38 inhibitors elevate proteasome activity under varying conditions [41]. In addition, p38α/β may regulate the subcellular localization of the proteasome by phosphorylating the proteasome-binding protein PI31. Consequently, this phosphorylation facilitates the association of the proteasome with the motor dynein complex, and regulates its transport on axons [42]. Other proteins whose stability is regulated by direct p38 phosphorylation are Cdt1, HBP1, p18Hamlet, Rb1, SRC3, CDC25A/B, CyclineD1/3, TACE, p53, Snail, Twist, Nav1.6, PGC1 α, HuR and Drosha [27,43]. Thus, p38α/β use various molecular mechanisms to regulate stimulation-dependent proteins stability.
2.2. p38α/β Regulation of Stimulated Nuclear Translocation
Another important process that is regulated by p38α/β is the dynamic change of protein localization upon stimulation. As described above regarding the regulation of protein degradation, the effect of p38α/β on nuclear translocation can be either global or specific to certain phosphorylated proteins. The global effects may be derived by p38α/β phosphorylation of either nuclear pore proteins or of karyopherins (importins/exportins). Indeed, it was shown that the nuclear pore proteins Nup62, Nup153, and Nup214 are phosphorylated by p38 (or ERK), and this phosphorylation inhibits the global nuclear protein shuttling initiated by viruses that affect the heart such as the encephalomyocarditis virus [44]. A similar effect was detected in cardiomyocytes of failing hearts in rats and humans, where p38α/β phosphorylation mediates the rearrangement of nuclear pores, leading to a decreased uptake of nuclear localization signal (NLS)-containing proteins [45]. As for karyopherins, it was shown that p38α/β regulate the expression of the beta-like importins (Imp) Imp7 and Imp8 [46], which are important for the nuclear translocation of various signaling proteins. Aside from the global changes, p38α/β is known to phosphorylate the translocating proteins themselves to mediate either nuclear accumulation or nuclear export. For example, the active SMAD3 phosphorylation by p38α/β upon TGF β stimulation reduces the rate of its nuclear translocation [47]. A similar effect was detected for FOXO3 α, which is phosphorylated by p38α/β at Ser7 to promote its nuclear localization [48]. The nuclear translocation of RhoA due to p38 phosphorylation upon LPS treatment and of actin upon TPA stimulation [49] was reported as well [50]. On the other hand, p38α/β phosphorylation may be responsible for the nuclear export of proteins; the most famous among them are its downstream MKs. It was shown that MK2/3 contain an NLS, which directs them to the nucleus of resting cells. Following phosphorylation by p38α/β, MK2/3 are exported to the cytoplasm, due to unmasking of the C-terminal NES of the MK2/3 (reviewed in [51]). Moreover, MK5 contains an NLS as well, and can be found in the nucleus under certain conditions. However, its export after p38α/β phosphorylation seems to be mediated not only by exposure of NES but also by anchoring to ERK3/4 [52]. Other proteins whose localization is directly regulated by p38α/β phosphorylation are retinoic acid receptor-γ, [53], androgen receptor [54], estrogen receptor-α [55], 5-lipoxygenase [56], the Hippo pathway transcription factor TEAD4 [57], as well as other proteins (NFATc4, Xbp1s, Drosha, CRTC2, HuR, Rabenosyn5, Lamin-B, FGFR1, PIP4K2B, EZH2, and Tripeptidyl-Peptidase II) as specified in previous reviews [27,43]. Thus, p38α/β use several distinct mechanisms for the regulation of nuclear translocation of proteins upon various stimulations.
3. Role of Nuclear p38α/β in Pathologies
Abnormal activity and dysregulation of the p38α/β cascade are associated with a variety of diseases. Indeed, p38α/β were implicated in the induction and maintenance of several pathologies such as inflammation [19], cancer [6], and autoimmune diseases [20] mentioned above, but also Friedreich’s ataxia [58], Parkinson’s disease [59], Alzheimer’s disease [60], cardiac hypertrophy [61], hypoxic nephropathy [62], and diabetes [63]. In many cases, the role of p38α/β is not direct, but it is mediated by p38α/β-regulated inflammation, which in turn contributes to the development of the diseases. For example, Parkinson’s disease is induced in part by neuroinflammation associated with glial cells [61], and colorectal cancer often develops due to initial inflammatory disease of the colon [64]. Moreover, p38α was first identified due to its involvement in the production of pro-inflammatory cytokines upon endotoxin treatment, mainly via nuclear processes [65]. It was later found that p38α/β are involved in the production of pro-inflammatory cytokines, such as TNF-α, IL-1 β, IL-2 IL-6, IL-7, and IL-8, and also in the regulation of other inflammation mediators such as Cox2 [66,67]. Although many of these effects involve nuclear processes in some instances, it may be regulated also by translation, due to AU-rich elements (ARE) in the 3′ untranslated region of their mRNA. The presence of these elements is known to shorten the half-life of mRNA containing them or block their translation, mainly due to the phosphorylation of ARE binding proteins such as HuR [68] by MK2 downstream of p38α/β) [69]. The pro-inflammatory cytokines are known players in many inflammation-related diseases such as inflammatory bowel diseases (IBD), psoriasis asthma, rheumatoid arthritis, inflammation-induced cancer, and more [66]. However, the actual trigger for inflammation in some systems, either acute or chronic, is not known, but it still requires p38α/β for its mediation. The means by which p38α/β are involved in these processes and the upstream components involved need further investigation. Due to the involvement of p38α/β in several inflammatory diseases, it became clear that specific inhibitors of these kinases should become a beneficial therapeutic approach. Indeed, in the past decade, more than 20 specific inhibitors of p38α/β were developed and proven very effective in pre-clinical investigations, demonstrating good tolerability and efficacy in several mouse models [70,71]. However, when subjected to clinical trials, the effects were much less favorable. Apparently, except for one inhibitor, pirfenidone, which by itself demonstrated a weak and unselective effect, no durable therapeutic effects have been detected for any of the others tested. The reasons for these failures among the different drugs are numerous, and while in some cases they were toxic, the more common problem was that after an initial good response, there was a rebound effect that increased inflammation within weeks. The reason for this rebound is still not clear and is currently under investigation.
In the past decade, substantial research has been devoted to studying the role of p38α/β in cancer, confirming that these kinases can act either as tumor promotors, or more frequently, as tumor suppressors [6,30]. The tumor suppressor effects were corroborated by immortalized MKK3/6 knockout fibroblasts that were shown to have a higher tendency to develop xenografts in nude mice [72]. Moreover, it was shown that the expression of MKK3/6, or other components of the cascade, is reduced in many cancers [73]. The function of p38α/β as tumor suppressors usually affect early stages of tumor initiation, and it generally involves either an inhibition of cell cycle progression, enhanced apoptosis, senescence, or differentiation. Similar to the effects in non-transformed cells, the inhibition of cell cycle by p38α/β in tumors can be mediated by the direct or indirect phosphorylation of several nuclear substrates. Among them are CyclinD, whose inhibition may cause apoptosis in colorectal cancer cells [28], p53, that leads to the upregulation of p21Cip1/WAF1, GADD45, and 14-3-3 proteins to cause cell cycle arrest [74], RB1, which prevents the metastasis of prostate cancer [75], and others [6]. Additionally, it was shown that p38α/β facilitate the production of apoptotic cytokines such as TNF-α [76]. The terminal differentiation of cancer was detected in rhabdomyosarcoma cells overexpressing MKK3 or MKK6 [77], and premature senescence was related to p38α/β-induced phosphorylation of the transcription factor HBP1 [78]. Some other effects on tumor promotion, although less frequent, may be mediated via inflammation in relevant cancers. Other mechanisms that may initiate cancer by p38α/β are elevated migration/invasion, increased angiogenesis [6], or a direct effect on proliferation [79,80]. As mentioned above, p38α/β are central regulators of inflammation, which in many cases is involved in cancer initiation and progression [81]. Indeed, some of the specific p38α/β inhibitors that have been developed over the years, although having failed in inflammation-related clinical trials, were proven useful in treating cancers [82]. Interestingly, in some cancer cells, p38α/β may facilitate migration by several mechanisms, including an enhanced production of chemoattractants [83] or reduced expression of fibulin 3, which is a cell migration blocker [84]. Finally, the involvement of p38α/β in angiogenesis, which supplies blood vessels to the tumors and enhances their growth, was shown to occur in head and neck cancer [85]. Some reports have shown that the molecular mechanisms involved include expression of vascular endothelial growth factor (VEGFA) and hypoxia-inducible factor 1 α (HIF1 α [30]. Interestingly, in models of colorectal cancer, p38α/β may act as either tumor suppressors or promoters in different stages of cancer development [23].
4. p38α/β’s Substrates and their Phosphorylation Specificity
More than 150 direct substrates of p38α/β have been identified so far [27,43,86,87,88,89]. However, since their minimal consensus phosphorylation sequence on their substrates (Ser/Thr-Pro) is so limited, and estimated to be present on the surface of approximately 50% of the cellular proteins, the actual number is likely to increase. Moreover, the effects of p38α/β are propagated by their MKs, and therefore, the number of phosphorylated proteins downstream of p38α/β may reach several thousands. Direct substrates of p38α/β were categorized into several subgroups in a previous review [43], including DNA binding proteins, RNA binding proteins, Ser/Thr kinases, regulatory proteins, as well as membranal, endosomal, and structural proteins. Additional information has been accumulated over the years on the role of p38α/β in activating their downstream protein kinases (MKs), transcription factors, and other regulatory elements [6,9]. The phosphorylation of these substrates is important for orchestrating the various cellular processes that may be, under certain circumstances, opposing signals. For example, the stress-related transcription factors ATF2, MEF2s, and CHOP have long been known to transmit p38α/β stress signals, while Elk1 and cFos may transmit its downstream mitogenic signals [3]. These distinct effects raise the question as to how the specificity of p38 signals is regulated.
As all other MAPKs, activated p38α/β execute their functions through the phosphorylation of downstream proteins. To the best of our knowledge, unlike ERK [90], no phosphorylation-independent effects of p38α/β have been identified. The full (Pro-Xaa-Ser/Thr-Pro) or minimal (Ser/Thr-Pro) consensus phosphorylation sites of all MAPKs are similar to each other [91]. Therefore, the interaction with the residues in the phosphorylation site is not sufficient to provide p38α/β specificity to its substrates. Rather, the specificity is mostly achieved by docking motifs that are localized on the substrates (D, DEF) that interact with specific docking sites on p38α/β (CD, Hydrophobic pocket). Thus, a docking motif that is found in many substrates of p38α/β is a D domain with consensus sequence Arg/Lys2-Xaa2-6-Φaa-Xaa-Φaa (where Φaa is a hydrophobic residue). The D-domains in the substrates bind to their counterpart docking site on p38α/β termed common docking motif (CD), which is composed of three negatively charged residues and at least two hydrophobic residues [92]. The CD domain and the catalytic site of p38α/β are located in distinct regions of the kinases, which allows the phosphorylation to occur at a fixed distance from the substrates D domain [93]. Binding of the D domain to the CD occurs in many cases when p38α/β are inactive, indicating that it may be responsible for a pre-activation association, which facilitates the rates of phosphorylation. Interestingly, similar to the consensus phosphorylation sites, also the D–CD domains interactions are nearly identical among all MAPKs, and therefore, p38α/β specificity requires extra determinants. Indeed, it was shown that in some cases, p38, ERK, and JNK may interact with distinct residues within the D-domain [94], and that two hydrophobic residues in the domain may determine specificity to some extent [95].
However, the differences between the hydrophobic residues cannot fully explain the specificity of p38α/β phosphorylation. Rather, this is likely explained by a second docking site on p38α/β substrates termed DEF (docking site for ERK, also known as FXF), which consists of two Phe residues separated by one residue and is often followed by a Pro residue [96]. The DEF domain binds the hydrophobic pocket in p38α/β, which is located at several residues C-terminal of the activatory Thr-Gly-Tyr phosphorylation site of the kinases. Structural studies show that the hydrophobic pocket is formed only upon phosphorylation, and therefore, unlike the interaction of the D-domain, only active p38α/β molecules bind to the DEF motif. Finally, it was also shown that p38, but not p38α, possesses intrinsic autophosphorylation activity, which may by itself elevate the basal activity of the kinase [97]. Thus, the combination of the three substrate interaction motifs together with intrinsic kinase activity contribute to the substrate specificity and affinity required for the proper p38α/β’s functions under various conditions. In addition, other regulators that affect signaling specificity may contribute to the kinase activity, including the level of substrates’ expression and their stability after stimulation as well as compartmentalization, scaffold proteins, or distinct phosphatases in a given cell. These regulating elements may also lead to sustained rather than transient activation of p38α/β, which is another specificity determinant. This is because the sustained phosphorylation results in the substrate’s phosphorylation at later stages after stimulation [98], as seen in cases of cell cycle facilitation versus senescence [31,99]. Taken together, these effects regulate the outcome of the p38α/β signal that are important both for the physiological and pathological fates of the cells.
5. Subcellular Localization of p38α/β and their Substrates
One of the mechanisms that determines the ability of MAPKs to phosphorylate distinct substrates upon varying conditions or cell lines is the cellular localization of the substrates’ phosphorylation [100]. Indeed, the large number of p38α/β’s substrates was shown to localize in various distinct compartments, including the nucleus, cytoplasm, cytoskeletal elements, and other sites [86]. In many cases, the distribution of the substrates is dynamic and changed upon stimulation, or in different cell lines. As mentioned above, some of these changes can be regulated by p38α/β, but others are regulated by other signaling proteins. The dynamic changes in localization raise the question of where the phosphorylation by p38α/β is actually taking place. Unexpectedly, the transcription factors that serve as substrates and function within the nucleus are almost always cytoplasmic in resting cells and translocate to the nucleus upon stimulation. As mentioned above, the translocations of some of these transcription factors are regulated by p38α/β. For example, the transcription factor ATF2 is phosphorylated by p38α/β to facilitate its dimerization with other AP-1 transcription factors, which enhances nuclear translocation [101]. Additionally, p38α/β phosphorylate the transcription factor Xbp1s at Thr48 and Ser61 enhancing migration to the nucleus, thus regulating glucose homeostasis in obesity [102]. The nuclear translocation of the transcription factor MEF2A is regulated by p38α/β as well [103], leading to the expression of neonatal myosin heavy chain in C2C12 myoblasts. Thus, counterintuitively, the phosphorylation of most, if not all, transcription factors by p38α/β may take place primarily in the cytoplasm, although some phosphorylations can also occur in the nucleus upon translocation. Similar effects were shown for other effectors of p38α/β, which translocate to the nucleus upon stimulation. Importantly, the localization of some substrates might be cell-type or condition-dependent [86], but others are less variable, and they are either constantly localized in the cytoplasm (e.g., keratin-8), in the nucleus (e.g., histones), or in the nucleus of resting cells followed by export upon stimulation (e.g., MKs). A list of various p38α/β substrates, which includes several transcription factors but mostly other proteins (Table 1), indicates that about 30% of the p38α/β substrates are confined to the nucleus under most/all conditions, and therefore, their phosphorylation upon stimulation should be nuclear. Almost all these proteins were shown to participate in the regulation of cancer and inflammation at least under some conditions. These effects are supported by our recent findings that the inhibition of nuclear p38α/β translocation prevents DSS-induced colitis and DSS/AOM-induced colon cancer [80], confirming the importance of nuclear p38α/β. However, it should be noted that the involvement of nuclear p38α/β in cancer and inflammation was not always confirmed, and the effects are sometimes cell line-dependent. Taken together, the information included in Table 1, as well as our recent study, clearly indicate that p38α/β are involved in either positive or negative regulations of cancer and inflammation.
The ability of p38α/β to phosphorylate substrates in the cytoplasm as well as in the nucleus and the role of nuclear p38α/β in the regulation of cancer and inflammation diverted attention toward the subcellular localization of these kinases. Thus, it was initially shown that p38α/β may be localized in the cytoplasm of resting cells, and similarly to ERK [104], it may translocate to the nucleus upon stimulation [105,106]. In that case, the kinases remain in the nucleus for minutes to hours, after which they are exported back to the cytoplasm. Surprisingly, in other systems, p38α/β were detected in the nucleus of resting cells [107] and were exported out of the nucleus shortly upon stimulation [108]. The nuclear localization in resting cells likely occurs because of continued stress signals in these cells, or due to the expression of specific nuclear anchors that attract p38α/β to the nucleus. However, this localization seems to occur in only a limited number of systems, whereas, in most cases, p38α/β are localized in the cytoplasm of resting cells. As for the mechanisms that regulate the dynamic subcellular localization, similarly to ERKs [109], it was shown that the cytoplasmic localization of p38α/β is mediated by various cytoplasmic anchoring proteins such as PTP-SL [106], keratins [110], and others [111]. Upon stimulation, the p38α/β detach from their anchors by mechanisms that may [112] or may not [113] require the activating TGY phosphorylation. Then, p38α/β translocate to the nucleus via the nuclear pore and can stay there for minutes to hours, either in the nucleoplasm, bound to chromatin [114], or nuclear proteins [108]. Interestingly, the mechanism of export either in cells with constant nuclear p38α/β, or in later stages upon stimulation-induced translocation, involves binding to the nuclear export signal (NES)-containing p38α/β’s substrate MK2 but probably not to those containing MK3 and MK5 [108,112].
6. Mechanism of Nuclear p38α/β Translocation and the Effect of its Inhibition
Although it is clear that p38α/β shuttle to the nucleus, either upon or without stimulation, the mechanism that regulates this is not fully understood. Similarly to ERK and JNK, p38α/β do not contain the canonical nuclear localization signals (NLS). In addition, they do not seem to interact with the classical Impα/β [111], or use passive diffusion for their nuclear shuttling. On the other hand, our group has recently shown that the nuclear translocation is mediated by three β-like importins, Imp3, 7, and 9 [115]. Thus, we found that upon stimulation, p38α/β as well as JNK1/2 are released from their cytoplasmic anchoring proteins and interact with either Imp7 or Imp9, each one in a complex with Imp3 (see the schematic representation in Figure 1). Then, the trimers formed (Imp7/Imp3/kinase or Imp9/Imp3/kinase) are shuttled to the nuclear pore, where Imp3 remains, while Imp7 or Imp9 escort the shuttling p38α/β or JNK1/2 into the nucleus [111]. In the nucleus, the small GTPase Ran dissociates the importins from p38α/β, and the latter are freed to execute their functions. This mechanism has some similarity to the translocation of ERK, which is detached from its anchoring protein upon stimulation and interacts with Imp7 that escorts it to the nucleus [18,116,117]. The Imp7 binding site of p38α/β resides in the N terminus of the kinase, and it is composed of at least nine residues: PERYQNLSP. Indeed, deletion of this region or substitution of its residues to Ala residues prevents the interaction [80]. Interestingly, the nuclear translocation of p38α/β may require HSP70 interaction in the nucleus [118], rely on microtubules and dynein, which may indicate that the translocation is helped by a trafficking machinery [112] and might involve SUMOylation of the kinases [119]. However, the mechanisms by which these components participate in the translocation and how they are related to the importins involved are not clear.
In previous studies on the nuclear translocation of ERK, our group designed a myristoylated peptide (EPE peptide) based on the ERK interaction site with Imp7 [120,121]. When added to cells, the peptide completely abolished the interaction between ERK and Imp7, and it prevented both stimulated and non-stimulated translocation of the kinase, indicating that the great majority of the translocation is mediated by Imp7, but not by passive diffusion [122] under both conditions. The potency of the peptide varied between cell lines, completely abolishing the proliferation of melanoma and other ERK-addicted cancer cell lines, but not the growth of non-transformed cells [120,123]. The peptide also diminished the growth of melanoma xenografts, better than the Raf inhibitor vemurafenib. Based on the successful development of the EPE peptide, we developed a myristoylated peptide targeting the binding site of p38α/β to Imp7/9, which we termed the PERY peptide. As expected [80], the peptide completely abolished the interaction of p38α/β to both Imp7 and Imp9, preventing the nuclear translocation of the kinases and the phosphorylation of nuclear targets. Since p38α/β signaling is involved in the proliferation of only limited types of cancers [6], the PERY peptide inhibited the proliferation of some breast cancer and melanoma cells but not of other cancers. In most cases, the effects were similar to those of the commercial p38 activity inhibitors, indicating that the effect on inflammation and cancer is mediated mainly by the nuclear p38α/β. Importantly, the peptide inhibited DSS-induced colon inflammation (colitis-like) and impressively, it demonstrated a significant inhibitory effect on a model of colitis-associated colon cancer [80]. Its effect was stronger when added throughout the DSS treatment than when added during the last two cycles of the treatment, and it was much stronger when compared to the effect of the commercial p38 activity inhibitor. Moreover, we have shown that the effect is mediated by macrophages [80] and not via enterocytes or other colon cells. Hence, these results clearly indicate that the nuclear translocation plays a role in the induction of inflammation and not the proliferation of colon cells. Taken together, we demonstrated that the nuclear translocation of p38α/β (and JNK) is important for the induction of inflammation, which may further lead to the development of some cancers. Additionally, it can be involved in the induction of proliferation of other cancer types such as triple negative breast cancers. Moreover, the results demonstrate that inhibiting the nuclear translocation of p38α/β may serve as a therapeutic strategy to combat various cancers as well as inflammatory diseases (Table 1).

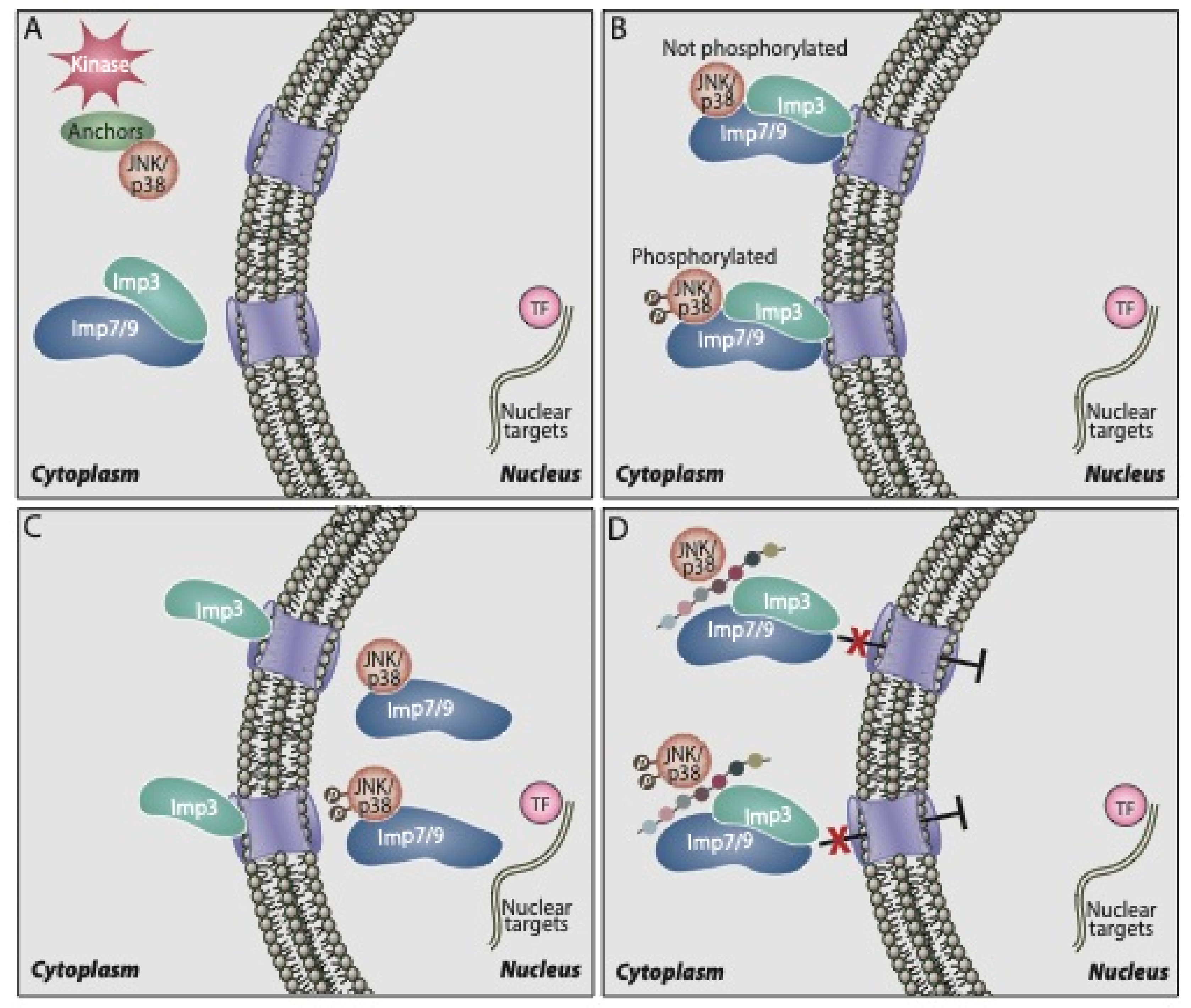{kind=link}
Table 1.
Nuclear substrates of p38α/β and their role in cancer and inflammation. More than 120 substrates of p38α/β were found in several reviews [27,43,86,87,88,89], and the translocation of each one of them was inspected in various databases. Substrates with constant (> 80%) nuclear localization in all cell lines described are shown under “Mostly Nuclear Proteins”, while proteins that are mostly nuclear in resting cells but are exported to the cytoplasm after stimulation are shown under “Nuclear Export”. The role of the p38α/β phosphorylation, as well as their general involvement in cancer or inflammation (independent of the phosphorylation in the nucleus) is described for each nuclear substrate. ND—not determined.
Table 1.
Nuclear substrates of p38α/β and their role in cancer and inflammation. More than 120 substrates of p38α/β were found in several reviews [27,43,86,87,88,89], and the translocation of each one of them was inspected in various databases. Substrates with constant (> 80%) nuclear localization in all cell lines described are shown under “Mostly Nuclear Proteins”, while proteins that are mostly nuclear in resting cells but are exported to the cytoplasm after stimulation are shown under “Nuclear Export”. The role of the p38α/β phosphorylation, as well as their general involvement in cancer or inflammation (independent of the phosphorylation in the nucleus) is described for each nuclear substrate. ND—not determined.
| Localization | p38-phosphorylated Protein | Role of Phosphorylation | Involvement in Cancer | Involvement in Inflammation |
|---|---|---|---|---|
| Cyclin D3 | Targets cyclin D3 for proteasomal degradation [124]. | Together with CDK6 regulates cell metabolism to promote cancer [125]. | Together with CDK6 phosphorylates NFκB to induce inflammatory gene expression [126]. | |
| Mostly Nuclear Proteins | E47 | Promotes MyoD/E47 association and muscle-gene transcription [127]. | Induces EMT and therefore may facilitate tumor formation [128]. | Required for the efficient recruitment of GR (anti-inflammatory) to chromatin [129]. |
| FBP2 (KSRP) | Controls stability of myogenic transcripts [130]. | Regulates c-Fos RNA stability and therefore cancers [131]. | Induce pro-inflammatory genes upon resveratrol treatment [132]. | |
| FBP3 | Controls prothrombin expression [133]. | May regulate Myc expression [134]. | May be involved in thrombin-induced inflammation [133]. | |
| H2AX | Chromatin remodeling. Involved in G2 checkpoint that protects cells from DNA breaks [135]. | Phosphorylation of Ser139 by RSK (the same site phosphorylated by p38) inhibits cell transformation [136]. | Colonocytes from ulcerative colitis patients showed an increase in H2AX content. Not necessarily related to phosphorylation [137]. | |
| H3 | Related to chromatin remodeling and chromosome condensation [138]. | p38 phosphorylation of Ser10 causes aggressive gastric cancer [139]. | p38-dependent H3 phosphorylation may mark promoters for increased NFκB recruitment and inflammation [140]. | |
| HBP1 | Stabilizes the proteins that leads to cell cycle inhibition [141]. | Inhibits cell cycle and functions as a tumor suppressor [78]. | Promote vascular inflammation in atherogenesis [142]. | |
| Id2 | Regulates transcription, cell cycle, and differentiation [143]. | Participate in VHL inactivation in cancer [144]. | Maintains regulatory T cell to suppress inflammatory diseases [145]. | |
| IWS1 | Likely regulates RNA processing and export [89]. | Regulates trimethylation of Histone H3 that may lead to cancer [146]. | ND | |
| JDP2 | Phosphorylation at Thr148 likely leads to proteasomal degradation (as with JNK [147]). | Implicated in progression and suppression of different cancers [148]. | Involved in liver inflammation [149]. | |
| MEF2d | Regulates recruitment of proteins to specific genes [150]. | Enhances proliferation migration and invasion in pancreatic cancer [151]. | Regulates IL-10 production in microglia to protect neuronal cells from inflammation-induced death [152]. | |
| Mnk2b | Induces activation [153]. | Mnk2b is oncogenic, by enhancing eIF4E phosphorylation [154]. | MNK2 is involved in adipose tissue inflammation (possibly both isoforms) [155]. | |
| MSK1 | Induces activation [156]. | Induces the transcription of immediate-early oncogenes [32]. | Activation of the pro-inflammatory NF-κB signaling pathway through MSK1 in microglial cells [157]. | |
| MSK2 | Induces activation [158]. | Induces the transcription of immediate-early oncogenes [32]. | Plays a role in limiting Toll-like receptor-driven inflammation [159]. | |
| P18Hamlet (Znhit1) | Stimulates p53-dependent apoptosis [160]. | Regulates p53 and therefore cancer [160]. | May affect p53-dependent inflammation [160,161]. | |
| P53 | Regulates apoptosis [162]. | Tumor suppressor [161]. | Suppressor of inflammation and autoimmunity [161]. | |
| PGC-1α | Regulates cytokine-induced energy expenditure [163]. | PGC-1α expression is altered in tumors and metastasis in relation to modifications in cellular metabolism [164]. | Connects oxidative stress and mitochondrial metabolism with inflammatory response and metabolic syndrome [165]. | |
| PPARalpha | Plays a role in cardiac metabolic stress response [166]. | Modulates metabolic pathways and attenuates kidney tumor growth [167]. | Exerts a major anti-inflammatory action in human liver [168]. | |
| Ranbp2 | Probably regulates SUMOylation and myotube formation [89]. | Involved in inflammatory myofibroblastic tumor formation [169]. | Inflammatory myofibroblastic tumor with RANBP2 and ALK gene rearrangement [169]. | |
| Rb1 | Mediates Fas-effects on inactivation of Rb1, independent of CDKs [170]. | Functions as a tumor suppressor. Inactivation induces retinoblastoma and other cancers [171]. | RB inactivation enhances pro-inflammatory signaling that can lead to cancer [172]. | |
| RNF2 | Modulates the expression of transcription factors and histone 2B acetylation [173]. | Monoubiquitinates H2AK119 at the promoter of LTBP2, thus regulates TGFβ signaling to induce melanoma [174]. | Inhibit interferon-dependent responses that may include inflammation [175]. | |
| Rpn2 | Negatively regulates proteasome activity [40]. | Promotes metastasis of hepatocellular carcinoma [176]. | Downregulated the inflammatory-associated JAK1/STAT3 pathway [177]. | |
| RUNX2 | Increases transcriptional activity [178]. | Abnormally expressed in prostatecancerand associates with metastatic disease [179]. | May have a role in the inflammatory remodeling of the collagen matrix [180]. | |
| SPF45 | Regulates alternative splicing site utilization [181], which may lead to multidrug resistance phenotypes [182]. | The phosphorylation inhibits proliferation and therefore may block cancer [181]. | Highly expressed in lung’s inflammatory cells, which might be involved in their function [182]. | |
| SRC3 | Controls the dynamics of interactions with RARalpha to facilitate gene activation [183]. | Promotes breast and prostate cancer cell proliferation and survival [184]. | Regulates inflammation during wound healing [185]. | |
| AHNAK | Probably induces its differentiation-related activity [89]. | Promotes metastasis through TGF-β-mediated EMT [186]. | Silencing of AHNAK in dental pulp cells led to reduced inflammation-related proteins [187]. | |
| Nuclear Export | c/EBPalpha | Inhibits enhancer activity [188]. | Suppresses tumor metastasis and growth in gastric cancer [189]. | Interacts with NF-κB to regulate inflammation [190]. |
| c/EBPbeta | Activates enhancer activity [191]. | Regulates tumor progression [192]. | Induces inflammation and ER stress [193]. | |
| ERalpha | Induces activation and nuclear export [55]. | Functions as an oncogene in breast cancer [194]. | Abnormal ERalpha signaling leads to inflammation [195]. | |
| MK2 | Induces activation [196]. | Plays a role in the induction of lung cancer [197]. Activates cancer-related proteins (Cdc25B/C, Plk1, and TSC2) [198]. | Plays a role in inflammatory pulmonary diseases [197]. Regulates inflammatory cytokines, transcript stability, and critical cellular processes [69]. | |
| MK3 | Induces activation [199]. | Leads to pancreatic cancer growth [200]. | Induces TNF biosynthesis and inflammation [201]. | |
| MK5 | Induces activation [202]. | Induces breast cancer [203]. | Phosphorylates HSP27 to induce inflammation [204]. | |
| MRF4 | Reduces transcriptional activity [205]. | May regulate hairy cell leukemia (HCL) [206]. | ND | |
| NFATc4 | Activation and nuclear export [207]. | Correlates with decreased proliferation and poor prognosis of ovarian cancer [208]. | Involved in the secretion of inflammatory factors [209]. | |
| NR4A | Regulates dopamine synthesis genes [210]. | Has both tumor suppressor and oncogenic functions in different cells [211]. | May contribute to the cellular processes that control inflammation [212]. | |
| Pax6 | Elevates transcriptional activity [213]. | Induces cell proliferation in lung cancer [214]. | ND |
7. Concluding Remarks
The response of cells to stress and other extracellular stimuli leads to the activation of several signaling pathways, including primarily those of p38α/β and JNK. The p38α/β signaling cascade is well-known for its ability to transmit stress signals to various targets within the cells. Thus, it regulates the activity of many transcription factors involved in stress response, and it was shown to also regulate cell cycle, RNA processes, and cytoskeletal elements. Here, we discussed in detail its involvement in the degradation and nuclear translocation of many proteins, which is an effect that may be in both cases either global, by affecting executing enzymes, or individual by acting specifically on degrading/translocating molecules. Regarding protein degradation, the global effects may be mediated by regulating proteasome or ubiquitin ligases, while the phosphorylation of individual substrates may either stabilize them or enhance their degradation. As for nuclear translocation, the global effect is possibly mediated by regulating importins or nuclear pore proteins (NUPs), while the individual effects can be mediated by either reduced or enhanced binding to the translocation machinery or to anchoring proteins. Being such central signaling pathways, the dysregulation of the p38α/β cascade results in pathologies, and indeed, almost all constantly nuclear targets were shown to play a role in the regulation of cancer and inflammation.
One of the hallmarks of many stimulations is the rapid and robust nuclear translocation of p38α/β. This translocation is essential for the regulation of targets that are only localized in the nucleus, although it can enhance the phosphorylation of proteins that are phosphorylated in the cytoplasm and translocate to the nucleus upon stimulation (e.g., transcription factors). We found that the translocation is mediated by the binding of p38α/β with either Imp7 or Imp9, which further bind individually to Imp3. Then, the complex moves to the NUPs, where Imp3 stays, while Imp7 or Imp9 shuttles the p38α/β into the nucleus. In the nucleus, p38 is freed from the importins by Ran and then, it is able to execute its nuclear functions. The duration of p38α/β residence in the nucleus may vary between cells and conditions, after which the kinases are exported out of the nucleus by their NES-containing substrate, MK2. A few years ago, our group developed the PERY peptide that completely prevents the nuclear translocation of p38α/β and thereby prevents the growth of some cancer cells—particularly DSS-induced colon inflammation and inflammation-induced cancer. Thus, the nuclear translocation of p38α/β can serve as a good target for inflammation and cancer, and inhibitors of this kinase translocation should be further developed for clinical use.
Author Contributions
All three authors contributed to the preparation of this review. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. Rony Seger is an incumbent of the Yale S. Lewine and Ella Miller Lewine professorial chair for cancer research.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| DEF | docking site for ERK |
| ERK | extracellular signal-regulated kinase |
| Imp | importin |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| MAPKAPK | MAPK-activated protein kinase (also known as MK) |
| MKK | MAPK kinase |
| NUP | nuclear pore protein |
References
- Peti, W.; Page, R. Molecular basis of MAP kinase regulation. Protein Sci. 2013, 22, 1698–1710. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eblen, S.T. Extracellular-Regulated Kinases: Signaling From Ras to ERK Substrates to Control Biological Outcomes. Adv. Cancer Res. 2018, 138, 99–142. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Kang, E.; Seo, J.; Cho, S. Phosphorylation Dynamics of JNK Signaling: Effects of Dual-Specificity Phosphatases (DUSPs) on the JNK Pathway. Int. J. Mol. Sci. 2019, 20, 6157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Limon, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef] [Green Version]
- Tubita, A.; Lombardi, Z.; Tusa, I.; Dello Sbarba, P.; Rovida, E. Beyond Kinase Activity: ERK5 Nucleo-Cytoplasmic Shuttling as a Novel Target for Anticancer Therapy. Int. J. Mol. Sci. 2020, 21, 938. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Shiryaev, A.; Moens, U. Mitogen-activated protein kinase p38 and MK2, MK3 and MK5: Menage a trois or menage a quatre? Cell. Signal. 2010, 22, 1185–1192. [Google Scholar] [CrossRef]
- Salvador, J.M.; Mittelstadt, P.R.; Guszczynski, T.; Copeland, T.D.; Yamaguchi, H.; Appella, E.; Fornace, A.J., Jr.; Ashwell, J.D. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol. 2005, 6, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Ge, B.; Gram, H.; Di Padova, F.; Huang, B.; New, L.; Ulevitch, R.J.; Luo, Y.; Han, J. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science 2002, 295, 1291–1294. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.S.; Nam, H.; Morrison, R.F. Dual-specificity phosphatases regulate mitogen-activated protein kinase signaling in adipocytes in response to inflammatory stress. Cell. Signal. 2019, 53, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Patterson, K.I.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Seger, R. The molecular Mechanism of MAPK/ERK inactivation. Curr. Genom. 2004, 5, 385–393. [Google Scholar] [CrossRef]
- Wang, X.D.; Zhao, C.S.; Wang, Q.L.; Zeng, Q.; Feng, X.Z.; Li, L.; Chen, Z.L.; Gong, Y.; Han, J.; Li, Y. The p38-interacting protein p38IP suppresses TCR and LPS signaling by targeting TAK1. EMBO Rep. 2020. [Google Scholar] [CrossRef]
- Flores, K.; Yadav, S.S.; Katz, A.A.; Seger, R. The Nuclear Translocation of Mitogen-Activated Protein Kinases: Molecular Mechanisms and Use as Novel Therapeutic Target. Neuroendocrinology 2019, 108, 121–131. [Google Scholar] [CrossRef]
- Kumar, S.; Boehm, J.; Lee, J.C. p38 MAP kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003, 2, 717–726. [Google Scholar] [CrossRef]
- Mavropoulos, A.; Orfanidou, T.; Liaskos, C.; Smyk, D.S.; Billinis, C.; Blank, M.; Rigopoulou, E.I.; Bogdanos, D.P. p38 mitogen-activated protein kinase (p38 MAPK)-mediated autoimmunity: Lessons to learn from ANCA vasculitis and pemphigus vulgaris. Autoimmun. Rev. 2013, 12, 580–590. [Google Scholar] [CrossRef]
- Dolado, I.; Swat, A.; Ajenjo, N.; De Vita, G.; Cuadrado, A.; Nebreda, A.R. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007, 11, 191–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomas-Loba, A.; Manieri, E.; Gonzalez-Teran, B.; Mora, A.; Leiva-Vega, L.; Santamans, A.M.; Romero-Becerra, R.; Rodriguez, E.; Pintor-Chocano, A.; Feixas, F.; et al. p38gamma is essential for cell cycle progression and liver tumorigenesis. Nature 2019, 568, 557–560. [Google Scholar] [CrossRef] [Green Version]
- Gupta, J.; del Barco Barrantes, I.; Igea, A.; Sakellariou, S.; Pateras, I.S.; Gorgoulis, V.G.; Nebreda, A.R. Dual function of p38alpha MAPK in colon cancer: Suppression of colitis-associated tumor initiation but requirement for cancer cell survival. Cancer Cell 2014, 25, 484–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Bambang, I.F.; Lu, D.; Li, H.; Chiu, L.L.; Lau, Q.C.; Koay, E.; Zhang, D. Cytokeratin 19 regulates endoplasmic reticulum stress and inhibits ERp29 expression via p38 MAPK/XBP-1 signaling in breast cancer cells. Exp. Cell Res. 2009, 315, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhu, H.; Sheng, F.; Tian, Y.; Zhou, J.; Chen, Y.; Li, S.; Lin, J. Activation of the MAPK11/12/13/14 (p38 MAPK) pathway regulates the transcription of autophagy genes in response to oxidative stress induced by a novel copper complex in HeLa cells. Autophagy 2014, 10, 1285–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Wu, J.; Silke, J. An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Thoms, H.C.; Dunlop, M.G.; Stark, L.A. p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res. 2007, 67, 1660–1669. [Google Scholar] [CrossRef] [Green Version]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Faust, D.; Schmitt, C.; Oesch, F.; Oesch-Bartlomowicz, B.; Schreck, I.; Weiss, C.; Dietrich, C. Differential p38-dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun. Signal. 2012, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adewumi, I.; Lopez, C.; Davie, J.R. Mitogen and stress- activated protein kinase regulated gene expression in cancer cells. Adv. Biol. Regul. 2019, 71, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, A.; Nakayama, K.; Williams, S.; Davis, R.J.; Mustelin, T.; Ronai, Z. Regulation of the ring finger E3 ligase Siah2 by p38 MAPK. J. Biol. Chem. 2006, 281, 35316–35326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Min, Y.; Wuest, T.; Lin, P.C. Vav1 is Essential for HIF-1alpha Activation via a Lysosomal VEGFR1-Mediated Degradation Mechanism in Endothelial Cells. Cancers 2020, 12, 1374. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Stender, J.D.; Joshi, S.; Wu, G.; Katzenellenbogen, B.S. OCT-4: A novel estrogen receptor-alpha collaborator that promotes tamoxifen resistance in breast cancer cells. Oncogene 2016, 35, 5722–5734. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, M.Y.; Ann, E.J.; Mo, J.S.; Yoon, J.H.; Park, H.S. Presenilin-2 regulates the degradation of RBP-Jk protein through p38 mitogen-activated protein kinase. J. Cell Sci. 2012, 125, 1296–1308. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.E.; Han, C.; Zhao, R.; Wani, G.; Zhu, Q.; Gong, L.; Battu, A.; Racoma, I.; Sharma, N.; Wani, A.A. p38 MAPK- and Akt-mediated p300 phosphorylation regulates its degradation to facilitate nucleotide excision repair. Nucleic Acids Res. 2013, 41, 1722–1733. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zhang, C.; Li, B.; Xie, W.; Zhang, J.; Nie, X.; Tan, P.; Zheng, L.; Wu, S.; Qin, Y.; et al. Mutual Stabilization between TRIM9 Short Isoform and MKK6 Potentiates p38 Signaling to Synergistically Suppress Glioblastoma Progression. Cell Rep. 2018, 23, 838–851. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Park, Y.; Yoon, S.K.; Yoon, J.B. Osmotic stress inhibits proteasome by p38 MAPK-dependent phosphorylation. J. Biol. Chem. 2010, 285, 41280–41289. [Google Scholar] [CrossRef] [Green Version]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome Activation by Small Molecules. Cell Chem. Biol. 2017, 24, 725–736.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Jones, S.; Minis, A.; Rodriguez, J.; Molina, H.; Steller, H. PI31 Is an Adaptor Protein for Proteasome Transport in Axons and Required for Synaptic Development. Dev. Cell 2019, 50, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Trempolec, N.; Dave-Coll, N.; Nebreda, A.R. SnapShot: p38 MAPK substrates. Cell 2013, 152, 924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, F.W.; Brown, B.; Palmenberg, A.C. Nucleoporin phosphorylation triggered by the encephalomyocarditis virus leader protein is mediated by mitogen-activated protein kinases. J. Virol. 2010, 84, 12538–12548. [Google Scholar] [CrossRef] [Green Version]
- Chahine, M.N.; Mioulane, M.; Sikkel, M.B.; O’Gara, P.; Dos Remedios, C.G.; Pierce, G.N.; Lyon, A.R.; Foldes, G.; Harding, S.E. Nuclear pore rearrangements and nuclear trafficking in cardiomyocytes from rat and human failing hearts. Cardiovasc. Res. 2015, 105, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Wu, C.; Boye, A.; Wu, J.; Wang, J.; Yang, X.; Yang, Y. MAPK inhibitors modulate Smad2/3/4 complex cyto-nuclear translocation in myofibroblasts via Imp7/8 mediation. Mol. Cell. Biochem. 2015, 406, 255–262. [Google Scholar] [CrossRef]
- Hayes, S.A.; Huang, X.; Kambhampati, S.; Platanias, L.C.; Bergan, R.C. p38 MAP kinase modulates Smad-dependent changes in human prostate cell adhesion. Oncogene 2003, 22, 4841–4850. [Google Scholar] [CrossRef] [Green Version]
- Ho, K.K.; McGuire, V.A.; Koo, C.Y.; Muir, K.W.; de Olano, N.; Maifoshie, E.; Kelly, D.J.; McGovern, U.B.; Monteiro, L.J.; Gomes, A.R.; et al. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. J. Biol. Chem. 2012, 287, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.Z.; Thuraisingam, T.; Morais, D.A.; Rola-Pleszczynski, M.; Radzioch, D. Nuclear translocation of beta-actin is involved in transcriptional regulation during macrophage differentiation of HL-60 cells. Mol. Biol. Cell 2010, 21, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, Y.; Xu, J. Factors influencing RhoA protein distribution in the nucleus. Mol. Med. Rep. 2011, 4, 1115–1119. [Google Scholar] [CrossRef] [Green Version]
- Gaestel, M. MAPKAP kinases-MKs-two’s company, three’s a crowd. Nat. Rev. Mol. Cell Biol. 2006, 7, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Seternes, O.M.; Mikalsen, T.; Johansen, B.; Michaelsen, E.; Armstrong, C.G.; Morrice, N.A.; Turgeon, B.; Meloche, S.; Moens, U.; Keyse, S.M. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 2004, 23, 4780–4791. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Zhou, H.; Kim, J.H.; Yan, T.D.; Lee, K.H.; Wu, H.; Lin, F.; Lu, N.; Liu, J.; Zeng, J.Z.; et al. A unique cytoplasmic localization of retinoic acid receptor-gamma and its regulations. J. Biol. Chem. 2009, 284, 18503–18514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gioeli, D.; Black, B.E.; Gordon, V.; Spencer, A.; Kesler, C.T.; Eblen, S.T.; Paschal, B.M.; Weber, M.J. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol. Endocrinol. 2006, 20, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Bai, W. Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol. Cell. Biol. 2002, 22, 5835–5845. [Google Scholar] [CrossRef] [Green Version]
- Flamand, N.; Luo, M.; Peters-Golden, M.; Brock, T.G. Phosphorylation of serine 271 on 5-lipoxygenase and its role in nuclear export. J. Biol. Chem. 2009, 284, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.C.; Moroishi, T.; Meng, Z.; Jeong, H.S.; Plouffe, S.W.; Sekido, Y.; Han, J.; Park, H.W.; Guan, K.L. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat. Cell Biol. 2017, 19, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Cotticelli, M.G.; Xia, S.; Kaur, A.; Lin, D.; Wang, Y.; Ruff, E.; Tobias, J.W.; Wilson, R.B. Identification of p38 MAPK as a novel therapeutic target for Friedreich’s ataxia. Sci. Rep. 2018, 8, 5007. [Google Scholar] [CrossRef] [Green Version]
- Bohush, A.; Niewiadomska, G.; Filipek, A. Role of Mitogen Activated Protein Kinase Signaling in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 2973. [Google Scholar] [CrossRef] [Green Version]
- Kheiri, G.; Dolatshahi, M.; Rahmani, F.; Rezaei, N. Role of p38/MAPKs in Alzheimer’s disease: Implications for amyloid beta toxicity targeted therapy. Rev. Neurosci. 2018, 30, 9–30. [Google Scholar] [CrossRef]
- Liu, R.; Molkentin, J.D. Regulation of cardiac hypertrophy and remodeling through the dual-specificity MAPK phosphatases (DUSPs). J. Mol. Cell. Cardiol. 2016, 101, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Shi, J.; Shi, Q.; Xu, X.; Xia, Y.; He, X. Mitogen-Activated Protein Kinases and Hypoxic/Ischemic Nephropathy. Cell. Physiol. Biochem. 2016, 39, 1051–1067. [Google Scholar] [CrossRef] [PubMed]
- Nandipati, K.C.; Subramanian, S.; Agrawal, D.K. Protein kinases: Mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol. Cell. Biochem. 2017, 426, 27–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossi, V.; Peserico, A.; Tezil, T.; Simone, C. p38alpha MAPK pathway: A key factor in colorectal cancer therapy and chemoresistance. World J. Gastroenterol. 2014, 20, 9744–9758. [Google Scholar] [CrossRef]
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [Green Version]
- Yong, H.Y.; Koh, M.S.; Moon, A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin. Investig. Drugs 2009, 18, 1893–1905. [Google Scholar] [CrossRef]
- Tiedje, C.; Holtmann, H.; Gaestel, M. The role of mammalian MAPK signaling in regulation of cytokine mRNA stability and translation. J. Interferon Cytokine Res. 2014, 34, 220–232. [Google Scholar] [CrossRef]
- Winzen, R.; Kracht, M.; Ritter, B.; Wilhelm, A.; Chen, C.Y.; Shyu, A.B.; Muller, M.; Gaestel, M.; Resch, K.; Holtmann, H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999, 18, 4969–4980. [Google Scholar] [CrossRef]
- Haller, V.; Nahidino, P.; Forster, M.; Laufer, S.A. An updated patent review of p38 MAP kinase inhibitors (2014–2019). Expert Opin. Ther. Pat. 2020, 30, 453–466. [Google Scholar] [CrossRef]
- Xing, L. Clinical candidates of small molecule p38 MAPK inhibitors for inflammatory diseases. MAP Kinase 2015, 4, 24–30. [Google Scholar] [CrossRef]
- Brancho, D.; Tanaka, N.; Jaeschke, A.; Ventura, J.J.; Kelkar, N.; Tanaka, Y.; Kyuuma, M.; Takeshita, T.; Flavell, R.A.; Davis, R.J. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003, 17, 1969–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacNeil, A.J.; Jiao, S.C.; McEachern, L.A.; Yang, Y.J.; Dennis, A.; Yu, H.; Xu, Z.; Marshall, J.S.; Lin, T.J. MAPK kinase 3 is a tumor suppressor with reduced copy number in breast cancer. Cancer Res. 2014, 74, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishi, H.; Nakagawa, K.; Matsumoto, M.; Suga, M.; Ando, M.; Taya, Y.; Yamaizumi, M. Osmotic shock induces G1 arrest through p53 phosphorylation at Ser33 by activated p38MAPK without phosphorylation at Ser15 and Ser20. J. Biol. Chem. 2001, 276, 39115–39122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFbetaRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef] [Green Version]
- Lalaoui, N.; Hanggi, K.; Brumatti, G.; Chau, D.; Nguyen, N.Y.; Vasilikos, L.; Spilgies, L.M.; Heckmann, D.A.; Ma, C.; Ghisi, M.; et al. Targeting p38 or MK2 Enhances the Anti-Leukemic Activity of Smac-Mimetics. Cancer Cell 2016, 29, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.L.; Wu, Z.; Zhang, P.; Wood, L.D.; Bhakta, K.S.; Han, J.; Feramisco, J.R.; Karin, M.; Wang, J.Y. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 2000, 14, 574–584. [Google Scholar]
- Bollaert, E.; de Rocca Serra, A.; Demoulin, J.B. The HMG box transcription factor HBP1: A cell cycle inhibitor at the crossroads of cancer signaling pathways. Cell. Mol. Life Sci. 2019, 76, 1529–1539. [Google Scholar] [CrossRef]
- Wen, S.Y.; Cheng, S.Y.; Ng, S.C.; Aneja, R.; Chen, C.J.; Huang, C.Y.; Kuo, W.W. Roles of p38alpha and p38beta mitogenactivated protein kinase isoforms in human malignant melanoma A375 cells. Int. J. Mol. Med. 2019, 44, 2123–2132. [Google Scholar] [CrossRef]
- Maik-Rachline, G.; Zehorai, E.; Hanoch, T.; Blenis, J.; Seger, R. The nuclear translocation of the kinases p38 and JNK promotes inflammation-induced cancer. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Gupta, J.; Nebreda, A.R. Roles of p38alpha mitogen-activated protein kinase in mouse models of inflammatory diseases and cancer. FEBS J. 2015, 282, 1841–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergote, I.; Heitz, F.; Buderath, P.; Powell, M.; Sehouli, J.; Lee, C.M.; Hamilton, A.; Fiorica, J.; Moore, K.N.; Teneriello, M.; et al. A randomized, double-blind, placebo-controlled phase 1b/2 study of ralimetinib, a p38 MAPK inhibitor, plus gemcitabine and carboplatin versus gemcitabine and carboplatin for women with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 2020, 156, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousseau, S.; Dolado, I.; Beardmore, V.; Shpiro, N.; Marquez, R.; Nebreda, A.R.; Arthur, J.S.; Case, L.M.; Tessier-Lavigne, M.; Gaestel, M.; et al. CXCL12 and C5a trigger cell migration via a PAK1/2-p38alpha MAPK-MAPKAP-K2-HSP27 pathway. Cell. Signal. 2006, 18, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Arechederra, M.; Priego, N.; Vazquez-Carballo, A.; Sequera, C.; Gutierrez-Uzquiza, A.; Cerezo-Guisado, M.I.; Ortiz-Rivero, S.; Roncero, C.; Cuenda, A.; Guerrero, C.; et al. p38 MAPK down-regulates fibulin 3 expression through methylation of gene regulatory sequences: Role in migration and invasion. J. Biol. Chem. 2015, 290, 4383–4397. [Google Scholar] [CrossRef] [Green Version]
- Leelahavanichkul, K.; Amornphimoltham, P.; Molinolo, A.A.; Basile, J.R.; Koontongkaew, S.; Gutkind, J.S. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol. Oncol. 2014, 8, 105–118. [Google Scholar] [CrossRef]
- Trempolec, N.; Dave-Coll, N.; Nebreda, A.R. SnapShot: p38 MAPK signaling. Cell 2013, 152, 656. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Blenis, J. MAPK Signaling in Human Diseases. In Apoptosis, Cell Signaling, and Human Diseases; Srivastava, R., Ed.; Humana Press: Totowa, NJ, USA, 2006; pp. 135–149. [Google Scholar]
- Iida, N.; Fujita, M.; Miyazawa, K.; Kobayashi, M.; Hattori, S. Proteomic identification of p38 MAP kinase substrates using in vitro phosphorylation. Electrophoresis 2014, 35, 554–562. [Google Scholar] [CrossRef]
- Knight, J.D.; Tian, R.; Lee, R.E.; Wang, F.; Beauvais, A.; Zou, H.; Megeney, L.A.; Gingras, A.C.; Pawson, T.; Figeys, D.; et al. A novel whole-cell lysate kinase assay identifies substrates of the p38 MAPK in differentiating myoblasts. Skelet. Muscle 2012, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.; Crespo, P. Working without kinase activity: Phosphotransfer-independent functions of extracellular signal-regulated kinases. Sci. Signal. 2011, 4, re3. [Google Scholar] [CrossRef]
- Mayor, F., Jr.; Jurado-Pueyo, M.; Campos, P.M.; Murga, C. Interfering with MAP kinase docking interactions: Implications and perspective for the p38 route. Cell Cycle 2007, 6, 528–533. [Google Scholar] [CrossRef] [Green Version]
- Tanoue, T.; Adachi, M.; Moriguchi, T.; Nishida, E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2000, 2, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.I.; Xu, B.E.; Akella, R.; Cobb, M.H.; Goldsmith, E.J. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol. Cell 2002, 9, 1241–1249. [Google Scholar] [CrossRef]
- Yang, S.H.; Whitmarsh, A.J.; Davis, R.J.; Sharrocks, A.D. Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J. 1998, 17, 1740–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardwell, A.J.; Bardwell, L. Two hydrophobic residues can determine the specificity of mitogen-activated protein kinase docking interactions. J. Biol. Chem. 2015, 290, 26661–26674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzarum, N.; Komornik, N.; Ben Chetrit, D.; Engelberg, D.; Livnah, O. DEF pocket in p38alpha facilitates substrate selectivity and mediates autophosphorylation. J. Biol. Chem. 2013, 288, 19537–19547. [Google Scholar] [CrossRef] [Green Version]
- Beenstock, J.; Ben-Yehuda, S.; Melamed, D.; Admon, A.; Livnah, O.; Ahn, N.G.; Engelberg, D. The p38beta mitogen-activated protein kinase possesses an intrinsic autophosphorylation activity, generated by a short region composed of the alpha-G helix and MAPK insert. J. Biol. Chem. 2014, 289, 23546–23556. [Google Scholar] [CrossRef] [Green Version]
- Purvis, J.E.; Lahav, G. Encoding and decoding cellular information through signaling dynamics. Cell 2013, 152, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Haq, R.; Brenton, J.D.; Takahashi, M.; Finan, D.; Finkielsztein, A.; Damaraju, S.; Rottapel, R.; Zanke, B. Constitutive p38HOG mitogen-activated protein kinase activation induces permanent cell cycle arrest and senescence. Cancer Res. 2002, 62, 5076–5082. [Google Scholar]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Lau, E.; Ronai, Z.A. ATF2 at the crossroad of nuclear and cytosolic functions. J. Cell Sci. 2012, 125, 2815–2824. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Sun, C.; Zhou, Y.; Lee, J.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef] [Green Version]
- Rauch, C.; Loughna, P.T. Static stretch promotes MEF2A nuclear translocation and expression of neonatal myosin heavy chain in C2Cl2 myocytes in a calcineurin- and p38-dependent manner. Am. J. Physiol. Cell Physiol. 2005, 288, C593–C605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.H.; Sarnecki, C.; Blenis, J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol. Cell. Biol. 1992, 12, 915–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, C.D.; Thornton, T.M.; Sabio, G.; Davis, R.A.; Rincon, M. Nuclear localization of p38 MAPK in response to DNA damage. Int. J. Biol. Sci. 2009, 5, 428–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Aparicio, C.; Torres, J.; Pulido, R. A novel regulatory mechanism of MAP kinases activation and nuclear translocation mediated by PKA and the PTP-SL tyrosine phosphatase. J. Cell Biol. 1999, 147, 1129–1136. [Google Scholar] [CrossRef] [Green Version]
- Posen, Y.; Kalchenko, V.; Seger, R.; Brandis, A.; Scherz, A.; Salomon, Y. Manipulation of redox signaling in mammalian cells enabled by controlled photogeneration of reactive oxygen species. J. Cell Sci. 2005, 118, 1957–1969. [Google Scholar] [CrossRef] [Green Version]
- Ben-Levy, R.; Hooper, S.; Wilson, R.; Paterson, H.F.; Marshall, C.J. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr. Biol. 1998, 8, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Wainstein, E.; Seger, R. The dynamic subcellular localization of ERK: Mechanisms of translocation and role in various organelles. Curr. Opin. Cell Biol. 2016, 39, 15–20. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, S.; Lim, Y.; Yoon, H.N.; Ku, N.O. Keratins regulate Hsp70-mediated nuclear localization of p38 mitogen-activated protein kinase. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K.; Davis, R.J. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 2003, 19, 91–118. [Google Scholar] [CrossRef]
- Gong, X.; Ming, X.; Deng, P.; Jiang, Y. Mechanisms regulating the nuclear translocation of p38 MAP kinase. J. Cell. Biochem. 2010, 110, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Zehorai, E.; Seger, R. Beta-Like Importins Mediate the Nuclear Translocation of MAPKs. Cell. Physiol. Biochem. 2019, 52, 802–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreiro, I.; Barragan, M.; Gubern, A.; Ballestar, E.; Joaquin, M.; Posas, F. The p38 SAPK is recruited to chromatin via its interaction with transcription factors. J. Biol. Chem. 2010, 285, 31819–31828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, K.; Seger, R. Stimulated nuclear import by β-like importins. F1000Prime Rep. 2013, 5. [Google Scholar] [CrossRef]
- Chuderland, D.; Konson, A.; Seger, R. Identification and characterization of a general nuclear translocation signal in signaling proteins. Mol. Cell 2008, 31, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Chuderland, D.; Karamansha, Y.; Livnah, O.; Seger, R. Nuclear ERK Translocation is Mediated by Protein Kinase CK2 and Accelerated by Autophosphorylation. Cell. Physiol. Biochem. 2019, 53, 366–387. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Luo, T.; Deng, P.; Liu, Z.; Xiu, J.; Shi, H.; Jiang, Y. Stress-induced interaction between p38 MAPK and HSP70. Biochem. Biophys. Res. Commun. 2012, 425, 357–362. [Google Scholar] [CrossRef]
- Wang, P.Y.; Hsu, P.I.; Wu, D.C.; Chen, T.C.; Jarman, A.P.; Powell, L.M.; Chen, A. SUMOs Mediate the Nuclear Transfer of p38 and p-p38 during Helicobacter Pylori Infection. Int. J. Mol. Sci. 2018, 19, 2482. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, A.; Flores, K.; Maik-Rachline, G.; Zehorai, E.; Kapri-Pardes, E.; Berti, D.A.; Hanoch, T.; Besser, M.J.; Seger, R. The nuclear translocation of ERK1/2 as an anticancer target. Nat. Commun. 2015, 6, 6685. [Google Scholar] [CrossRef]
- Maik-Rachline, G.; Hacohen-Lev-Ran, A.; Seger, R. Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. Int. J. Mol. Sci. 2019, 20, 1194. [Google Scholar] [CrossRef] [Green Version]
- Jivan, A.; Ranganathan, A.; Cobb, M.H. Reconstitution of the nuclear transport of the MAP kinase ERK2. Methods Mol. Biol. 2010, 661, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Arafeh, R.; Flores, K.; Karen-Paz, A.; Maik-Rachline, G.; Gutkind, N.; Rosenberg, S.; Seger, R.; Samuels, Y. Combinede inhibition of MEK and nuclear ERK translocation has synergistic antitumor activity in mutated NRAS, BRAF and NF1 melanoma cells. Sci. Rep. 2017, 7, 16345. [Google Scholar] [CrossRef] [Green Version]
- Casanovas, O.; Jaumot, M.; Paules, A.B.; Agell, N.; Bachs, O. P38SAPK2 phosphorylates cyclin D3 at Thr-283 and targets it for proteasomal degradation. Oncogene 2004, 23, 7537–7544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Nicolay, B.N.; Chick, J.M.; Gao, X.; Geng, Y.; Ren, H.; Gao, H.; Yang, G.; Williams, J.A.; Suski, J.M.; et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 2017, 546, 426–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buss, H.; Handschick, K.; Jurrmann, N.; Pekkonen, P.; Beuerlein, K.; Muller, H.; Wait, R.; Saklatvala, J.; Ojala, P.M.; Schmitz, M.L.; et al. Cyclin-dependent kinase 6 phosphorylates NF-kappaB P65 at serine 536 and contributes to the regulation of inflammatory gene expression. PLoS ONE 2012, 7, e51847. [Google Scholar] [CrossRef] [Green Version]
- Lluis, F.; Ballestar, E.; Suelves, M.; Esteller, M.; Munoz-Canoves, P. E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific gene transcription. EMBO J. 2005, 24, 974–984. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Li, X. The roles of HLH transcription factors in epithelial mesenchymal transition and multiple molecular mechanisms. Clin. Exp. Metastasis 2014, 31, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, M.C.; Wierer, M.; Schachtrup, K.; Downes, M.; Hubner, N.; Evans, R.M.; Uhlenhaut, N.H. E47 modulates hepatic glucocorticoid action. Nat. Commun. 2019, 10, 306. [Google Scholar] [CrossRef] [Green Version]
- Briata, P.; Forcales, S.V.; Ponassi, M.; Corte, G.; Chen, C.Y.; Karin, M.; Puri, P.L.; Gherzi, R. p38-dependent phosphorylation of the mRNA decay-promoting factor KSRP controls the stability of select myogenic transcripts. Mol. Cell 2005, 20, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Degese, M.S.; Tanos, T.; Naipauer, J.; Gingerich, T.; Chiappe, D.; Echeverria, P.; LaMarre, J.; Gutkind, J.S.; Coso, O.A. An interplay between the p38 MAPK pathway and AUBPs regulates c-fos mRNA stability during mitogenic stimulation. Biochem. J. 2015, 467, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Bollmann, F.; Art, J.; Henke, J.; Schrick, K.; Besche, V.; Bros, M.; Li, H.; Siuda, D.; Handler, N.; Bauer, F.; et al. Resveratrol post-transcriptionally regulates pro-inflammatory gene expression via regulation of KSRP RNA binding activity. Nucleic Acids Res. 2014, 42, 12555–12569. [Google Scholar] [CrossRef]
- Danckwardt, S.; Gantzert, A.S.; Macher-Goeppinger, S.; Probst, H.C.; Gentzel, M.; Wilm, M.; Grone, H.J.; Schirmacher, P.; Hentze, M.W.; Kulozik, A.E. p38 MAPK controls prothrombin expression by regulated RNA 3’ end processing. Mol. Cell 2011, 41, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Kristiansen, I.; Johannsen, M.; Oelrich, B.; Scholmann, K.; Gunia, S.; May, M.; Meyer, H.A.; Behnke, S.; Moch, H.; et al. The FUSE binding proteins FBP1 and FBP3 are potential c-myc regulators in renal, but not in prostate and bladder cancer. BMC Cancer 2008, 8, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dmitrieva, N.I.; Bulavin, D.V.; Fornace, A.J., Jr.; Burg, M.B. Rapid activation of G2/M checkpoint after hypertonic stress in renal inner medullary epithelial (IME) cells is protective and requires p38 kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, F.; Zykova, T.A.; Peng, C.; Zhang, J.; Cho, Y.Y.; Zheng, D.; Yao, K.; Ma, W.Y.; Lau, A.T.; Bode, A.M.; et al. Phosphorylation of H2AX at Ser139 and a new phosphorylation site Ser16 by RSK2 decreases H2AX ubiquitination and inhibits cell transformation. Cancer Res. 2011, 71, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risques, R.A.; Lai, L.A.; Brentnall, T.A.; Li, L.; Feng, Z.; Gallaher, J.; Mandelson, M.T.; Potter, J.D.; Bronner, M.P.; Rabinovitch, P.S. Ulcerative colitis is a disease of accelerated colon aging: Evidence from telomere attrition and DNA damage. Gastroenterology 2008, 135, 410–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.P.; Ma, W.Y.; Dong, Z. ERKs and p38 kinases mediate ultraviolet B-induced phosphorylation of histone H3 at serine 10. J. Biol. Chem. 2000, 275, 20980–20984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.A.; Amnekar, R.; Khade, B.; Barreto, S.G.; Ramadwar, M.; Shrikhande, S.V.; Gupta, S. p38-MAPK/MSK1-mediated overexpression of histone H3 serine 10 phosphorylation defines distance-dependent prognostic value of negative resection margin in gastric cancer. Clin. Epigenetics 2016, 8, 88. [Google Scholar] [CrossRef] [Green Version]
- Saccani, S.; Pantano, S.; Natoli, G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat. Immunol. 2002, 3, 69–75. [Google Scholar] [CrossRef]
- Xiu, M.; Kim, J.; Sampson, E.; Huang, C.Y.; Davis, R.J.; Paulson, K.E.; Yee, A.S. The transcriptional repressor HBP1 is a target of the p38 mitogen-activated protein kinase pathway in cell cycle regulation. Mol. Cell. Biol. 2003, 23, 8890–8901. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Li, X.; Liu, S.; Gu, L.; Zhou, X. MircroRNA-19a promotes vascular inflammation and foam cell formation by targeting HBP-1 in atherogenesis. Sci. Rep. 2017, 7, 12089. [Google Scholar] [CrossRef]
- Roschger, C.; Cabrele, C. The Id-protein family in developmental and cancer-associated pathways. Cell Commun. Signal. 2017, 15, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.B.; Frattini, V.; Bansal, M.; Castano, A.M.; Sherman, D.; Hutchinson, K.; Bruce, J.N.; Califano, A.; Liu, G.; Cardozo, T.; et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature 2016, 529, 172–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, M.; Miyazaki, K.; Chen, S.; Itoi, M.; Miller, M.; Lu, L.F.; Varki, N.; Chang, A.N.; Broide, D.H.; Murre, C. Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat. Immunol. 2014, 15, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Oqani, R.K.; Lin, T.; Lee, J.E.; Kang, J.W.; Shin, H.Y.; Il Jin, D. Iws1 and Spt6 Regulate Trimethylation of Histone H3 on Lysine 36 through Akt Signaling and are Essential for Mouse Embryonic Genome Activation. Sci. Rep. 2019, 9, 3831. [Google Scholar] [CrossRef] [Green Version]
- Katz, S.; Aronheim, A. Differential targeting of the stress mitogen-activated protein kinases to the c-Jun dimerization protein 2. Biochem. J. 2002, 368, 939–945. [Google Scholar] [CrossRef]
- Tsai, M.H.; Wuputra, K.; Lin, Y.C.; Lin, C.S.; Yokoyama, K.K. Multiple functions of the histone chaperone Jun dimerization protein 2. Gene 2016, 590, 193–200. [Google Scholar] [CrossRef]
- Bitton-Worms, K.; Pikarsky, E.; Aronheim, A. The AP-1 repressor protein, JDP2, potentiates hepatocellular carcinoma in mice. Mol. Cancer 2010, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Rampalli, S.; Li, L.; Mak, E.; Ge, K.; Brand, M.; Tapscott, S.J.; Dilworth, F.J. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat. Struct. Mol. Biol. 2007, 14, 1150–1156. [Google Scholar] [CrossRef]
- Song, Z.; Feng, C.; Lu, Y.; Gao, Y.; Lin, Y.; Dong, C. Overexpression and biological function of MEF2D in human pancreatic cancer. Am. J. Transl. Res. 2017, 9, 4836–4847. [Google Scholar]
- Yang, S.; Gao, L.; Lu, F.; Wang, B.; Gao, F.; Zhu, G.; Cai, Z.; Lai, J.; Yang, Q. Transcription factor myocyte enhancer factor 2D regulates interleukin-10 production in microglia to protect neuronal cells from inflammation-induced death. J. Neuroinflamm. 2015, 12, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waskiewicz, A.J.; Flynn, A.; Proud, C.G.; Cooper, J.A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997, 16, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Maimon, A.; Mogilevsky, M.; Shilo, A.; Golan-Gerstl, R.; Obiedat, A.; Ben-Hur, V.; Lebenthal-Loinger, I.; Stein, I.; Reich, R.; Beenstock, J.; et al. Mnk2 alternative splicing modulates the p38-MAPK pathway and impacts Ras-induced transformation. Cell Rep. 2014, 7, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.E.; Pickford, J.; Cagampang, F.R.; Stead, R.L.; Tian, S.; Zhao, X.; Tang, X.; Byrne, C.D.; Proud, C.G. MNK1 and MNK2 mediate adverse effects of high-fat feeding in distinct ways. Sci. Rep. 2016, 6, 23476. [Google Scholar] [CrossRef] [Green Version]
- Deak, M.; Clifton, A.D.; Lucocq, L.M.; Alessi, D.R. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998, 17, 4426–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan-Ganga, M.; Garcia-Yague, A.J.; Lastres-Becker, I. Role of MSK1 in the Induction of NF-kappaB by the Chemokine CX3CL1 in Microglial Cells. Cell. Mol. Neurobiol. 2019, 39, 331–340. [Google Scholar] [CrossRef]
- Caivano, M.; Cohen, P. Role of mitogen-activated protein kinase cascades in mediating lipopolysaccharide-stimulated induction of cyclooxygenase-2 and IL-1 beta in RAW264 macrophages. J. Immunol. 2000, 164, 3018–3025. [Google Scholar] [CrossRef] [Green Version]
- Ananieva, O.; Darragh, J.; Johansen, C.; Carr, J.M.; McIlrath, J.; Park, J.M.; Wingate, A.; Monk, C.E.; Toth, R.; Santos, S.G.; et al. The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat. Immunol. 2008, 9, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Lafarga, V.; Cheung, P.C.; Dolado, I.; Llanos, S.; Cohen, P.; Nebreda, A.R. A new p38 MAP kinase-regulated transcriptional coactivator that stimulates p53-dependent apoptosis. EMBO J. 2007, 26, 2115–2126. [Google Scholar] [CrossRef]
- Uehara, I.; Tanaka, N. Role of p53 in the Regulation of the Inflammatory Tumor Microenvironment and Tumor Suppression. Cancers 2018, 10, 219. [Google Scholar] [CrossRef] [Green Version]
- Bulavin, D.V.; Saito, S.; Hollander, M.C.; Sakaguchi, K.; Anderson, C.W.; Appella, E.; Fornace, A.J., Jr. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999, 18, 6845–6854. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1alpha in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar] [PubMed]
- Rius-Perez, S.; Torres-Cuevas, I.; Millan, I.; Ortega, A.L.; Perez, S. PGC-1alpha, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Barger, P.M.; Browning, A.C.; Garner, A.N.; Kelly, D.P. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: A potential role in the cardiac metabolic stress response. J. Biol. Chem. 2001, 276, 44495–44501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Aboud, O.; Donohoe, D.; Bultman, S.; Fitch, M.; Riiff, T.; Hellerstein, M.; Weiss, R.H. PPARalpha inhibition modulates multiple reprogrammed metabolic pathways in kidney cancer and attenuates tumor growth. Am. J. Physiol. Cell Physiol. 2015, 308, C890–C898. [Google Scholar] [CrossRef] [Green Version]
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.; van Rooijen, N.; Staels, B.; Kersten, S.; Muller, M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 2010, 51, 511–522. [Google Scholar] [CrossRef]
- Li, J.; Yin, W.H.; Takeuchi, K.; Guan, H.; Huang, Y.H.; Chan, J.K. Inflammatory myofibroblastic tumor with RANBP2 and ALK gene rearrangement: A report of two cases and literature review. Diagn. Pathol. 2013, 8, 147. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Nath, N.; Minden, A.; Chellappan, S. Regulation of Rb and E2F by signal transduction cascades: Divergent effects of JNK1 and p38 kinases. EMBO J. 1999, 18, 1559–1570. [Google Scholar] [CrossRef] [Green Version]
- Indovina, P.; Pentimalli, F.; Casini, N.; Vocca, I.; Giordano, A. RB1 dual role in proliferation and apoptosis: Cell fate control and implications for cancer therapy. Oncotarget 2015, 6, 17873–17890. [Google Scholar] [CrossRef]
- Kitajima, S.; Takahashi, C. Intersection of retinoblastoma tumor suppressor function, stem cells, metabolism, and inflammation. Cancer Sci. 2017, 108, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.S.; Satelli, A.; Zhang, S.; Srivastava, S.K.; Srivenugopal, K.S.; Rao, U.S. RNF2 is the target for phosphorylation by the p38 MAPK and ERK signaling pathways. Proteomics 2009, 9, 2776–2787. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.; Akdemir, K.C.; Kwong, L.N.; Fiziev, P.; Wu, C.J.; Keung, E.Z.; Sharma, S.; Samant, N.S.; Williams, M.; Axelrad, J.B.; et al. Dual Roles of RNF2 in Melanoma Progression. Cancer Discov. 2015, 5, 1314–1327. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Jiang, M.; Wang, W.; Liu, W.; Song, X.; Ma, Z.; Zhang, S.; Liu, L.; Liu, Y.; Cao, X. Nuclear RNF2 inhibits interferon function by promoting K33-linked STAT1 disassociation from DNA. Nat. Immunol. 2018, 19, 41–52. [Google Scholar] [CrossRef]
- Huang, L.; Jian, Z.; Gao, Y.; Zhou, P.; Zhang, G.; Jiang, B.; Lv, Y. RPN2 promotes metastasis of hepatocellular carcinoma cell and inhibits autophagy via STAT3 and NF-kappaB pathways. Aging 2019, 11, 6674–6690. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Yu, J.; Gui, X.; Lu, Z.; Wang, X.; Guo, H.; Zhou, Y. Overexpression of RPN2 promotes osteogenic differentiation of hBMSCs through the JAK/STAT3 pathway. FEBS Open Bio 2020, 10, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Greenblatt, M.B.; Shim, J.H.; Zou, W.; Sitara, D.; Schweitzer, M.; Hu, D.; Lotinun, S.; Sano, Y.; Baron, R.; Park, J.M.; et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Investig. 2010, 120, 2457–2473. [Google Scholar] [CrossRef] [Green Version]
- Ge, C.; Zhao, G.; Li, Y.; Li, H.; Zhao, X.; Pannone, G.; Bufo, P.; Santoro, A.; Sanguedolce, F.; Tortorella, S.; et al. Role of Runx2 phosphorylation in prostate cancer and association with metastatic disease. Oncogene 2016, 35, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Raaz, U.; Schellinger, I.N.; Chernogubova, E.; Warnecke, C.; Kayama, Y.; Penov, K.; Hennigs, J.K.; Salomons, F.; Eken, S.; Emrich, F.C.; et al. Transcription Factor Runx2 Promotes Aortic Fibrosis and Stiffness in Type 2 Diabetes Mellitus. Circ. Res. 2015, 117, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Al-Ayoubi, A.M.; Zheng, H.; Liu, Y.; Bai, T.; Eblen, S.T. Mitogen-activated protein kinase phosphorylation of splicing factor 45 (SPF45) regulates SPF45 alternative splicing site utilization, proliferation, and cell adhesion. Mol. Cell. Biol. 2012, 32, 2880–2893. [Google Scholar] [CrossRef] [Green Version]
- Sampath, J.; Long, P.R.; Shepard, R.L.; Xia, X.; Devanarayan, V.; Sandusky, G.E.; Perry, W.L., 3rd; Dantzig, A.H.; Williamson, M.; Rolfe, M.; et al. Human SPF45, a splicing factor, has limited expression in normal tissues, is overexpressed in many tumors, and can confer a multidrug-resistant phenotype to cells. Am. J. Pathol. 2003, 163, 1781–1790. [Google Scholar] [CrossRef] [Green Version]
- Gianni, M.; Parrella, E.; Raska, I., Jr.; Gaillard, E.; Nigro, E.A.; Gaudon, C.; Garattini, E.; Rochette-Egly, C. P38MAPK-dependent phosphorylation and degradation of SRC-3/AIB1 and RARalpha-mediated transcription. EMBO J. 2006, 25, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, R.C.; O’Malley, B.W. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 2009, 9, 615–630. [Google Scholar] [CrossRef] [Green Version]
- Al-Otaiby, M.; Tassi, E.; Schmidt, M.O.; Chien, C.D.; Baker, T.; Salas, A.G.; Xu, J.; Furlong, M.; Schlegel, R.; Riegel, A.T.; et al. Role of the nuclear receptor coactivator AIB1/SRC-3 in angiogenesis and wound healing. Am. J. Pathol. 2012, 180, 1474–1484. [Google Scholar] [CrossRef] [Green Version]
- Sohn, M.; Shin, S.; Yoo, J.Y.; Goh, Y.; Lee, I.H.; Bae, Y.S. Ahnak promotes tumor metastasis through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Sci. Rep. 2018, 8, 14379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Fukuda, T.; Nagayasu, S.; Nakanishi, J.; Yoshida, K.; Hirata-Tsuchiya, S.; Nakao, Y.; Sano, T.; Yamashita, A.; Yamada, S.; et al. Dental pulp cell-derived powerful inducer of TNF-alpha comprises PKR containing stress granule rich microvesicles. Sci. Rep. 2019, 9, 3825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geest, C.R.; Buitenhuis, M.; Laarhoven, A.G.; Bierings, M.B.; Bruin, M.C.; Vellenga, E.; Coffer, P.J. p38 MAP kinase inhibits neutrophil development through phosphorylation of C/EBPalpha on serine 21. Stem Cells 2009, 27, 2271–2282. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.B.; Wang, Y.W.; Xing, A.Y.; Gao, J.W.; Zhang, H.; Guo, X.Y.; Gao, P. C/EBPalpha-induced miR-100 expression suppresses tumor metastasis and growth by targeting ZBTB7A in gastric cancer. Cancer Lett. 2015, 369, 376–385. [Google Scholar] [CrossRef]
- Friedman, A.D. C/EBPalpha induces PU.1 and interacts with AP-1 and NF-kappaB to regulate myeloid development. Blood Cells Mol. Dis. 2007, 39, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Horie, R.; Ishida, T.; Maruyama-Nagai, M.; Ito, K.; Watanabe, M.; Yoneyama, A.; Higashihara, M.; Kimura, S.; Watanabe, T. TRAF activation of C/EBPbeta (NF-IL6) via p38 MAPK induces HIV-1 gene expression in monocytes/macrophages. Microbes Infect. 2007, 9, 721–728. [Google Scholar] [CrossRef]
- Yeh, H.W.; Lee, S.S.; Chang, C.Y.; Lang, Y.D.; Jou, Y.S. A New Switch for TGFbeta in Cancer. Cancer Res. 2019, 79, 3797–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahid, M.D.K.; Rogowski, M.; Ponce, C.; Choudhury, M.; Moustaid-Moussa, N.; Rahman, S.M. CCAAT/enhancer-binding protein beta (C/EBPbeta) knockdown reduces inflammation, ER stress, and apoptosis, and promotes autophagy in oxLDL-treated RAW264.7 macrophage cells. Mol. Cell. Biochem. 2020, 463, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Anbalagan, M.; Rowan, B.G. Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol. Cell. Endocrinol. 2015, 418, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J.A. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Rouse, J.; Cohen, P.; Trigon, S.; Morange, M.; Alonso-Llamazares, A.; Zamanillo, D.; Hunt, T.; Nebreda, A.R. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 1994, 78, 1027–1037. [Google Scholar] [CrossRef]
- Singh, R.K.; Najmi, A.K. Novel Therapeutic Potential of Mitogen-Activated Protein Kinase Activated Protein Kinase 2 (MK2) in Chronic Airway Inflammatory Disorders. Curr. Drug Targets 2019, 20, 367–379. [Google Scholar] [CrossRef]
- Manke, I.A.; Nguyen, A.; Lim, D.; Stewart, M.Q.; Elia, A.E.; Yaffe, M.B. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 2005, 17, 37–48. [Google Scholar] [CrossRef]
- McLaughlin, M.M.; Kumar, S.; McDonnell, P.C.; Van Horn, S.; Lee, J.C.; Livi, G.P.; Young, P.R. Identification of mitogen-activated protein (MAP) kinase-activated protein kinase-3, a novel substrate of CSBP p38 MAP kinase. J. Biol. Chem. 1996, 271, 8488–8492. [Google Scholar] [CrossRef] [Green Version]
- Reynoird, N.; Mazur, P.K.; Stellfeld, T.; Flores, N.M.; Lofgren, S.M.; Carlson, S.M.; Brambilla, E.; Hainaut, P.; Kaznowska, E.B.; Arrowsmith, C.H.; et al. Coordination of stress signals by the lysine methyltransferase SMYD2 promotes pancreatic cancer. Genes Dev. 2016, 30, 772–785. [Google Scholar] [CrossRef]
- Ronkina, N.; Menon, M.B.; Schwermann, J.; Tiedje, C.; Hitti, E.; Kotlyarov, A.; Gaestel, M. MAPKAP kinases MK2 and MK3 in inflammation: Complex regulation of TNF biosynthesis via expression and phosphorylation of tristetraprolin. Biochem. Pharmacol. 2010, 80, 1915–1920. [Google Scholar] [CrossRef]
- New, L.; Jiang, Y.; Zhao, M.; Liu, K.; Zhu, W.; Flood, L.J.; Kato, Y.; Parry, G.C.; Han, J. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 1998, 17, 3372–3384. [Google Scholar] [CrossRef] [Green Version]
- Al-Mahdi, R.; Babteen, N.; Thillai, K.; Holt, M.; Johansen, B.; Wetting, H.L.; Seternes, O.M.; Wells, C.M. A novel role for atypical MAPK kinase ERK3 in regulating breast cancer cell morphology and migration. Cell Adh. Migr. 2015, 9, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moens, U.; Kostenko, S.; Sveinbjornsson, B. The Role of Mitogen-Activated Protein Kinase-Activated Protein Kinases (MAPKAPKs) in Inflammation. Genes 2013, 4, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Suelves, M.; Lluis, F.; Ruiz, V.; Nebreda, A.R.; Munoz-Canoves, P. Phosphorylation of MRF4 transactivation domain by p38 mediates repression of specific myogenic genes. EMBO J. 2004, 23, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arons, E.; Zhou, H.; Sokolsky, M.; Gorelik, D.; Potocka, K.; Davies, S.; Fykes, E.; Still, K.; Edelman, D.C.; Wang, Y.; et al. Expression of the muscle-associated gene MYF6 in hairy cell leukemia. PLoS ONE 2020, 15, e0227586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.T.; Xiong, Q.; Enslen, H.; Davis, R.J.; Chow, C.W. Phosphorylation of NFATc4 by p38 mitogen-activated protein kinases. Mol. Cell. Biol. 2002, 22, 3892–3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.J.; Iyengar, M.; Panesso-Gomez, S.; O’Hayer, P.; Chan, D.; Delgoffe, G.M.; Aird, K.M.; Yoon, E.; Bai, S.; Buckanovich, R.J. NFATC4 promotes quiescence and chemotherapy resistance in ovarian cancer. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.B.; Kumar, A.; Wang, L.; Liu, G.H.; Keller, S.R.; Lawrence, J.C., Jr.; Finck, B.N.; Harris, T.E. Lipin 1 represses NFATc4 transcriptional activity in adipocytes to inhibit secretion of inflammatory factors. Mol. Cell. Biol. 2010, 30, 3126–3139. [Google Scholar] [CrossRef] [Green Version]
- Sekine, Y.; Takagahara, S.; Hatanaka, R.; Watanabe, T.; Oguchi, H.; Noguchi, T.; Naguro, I.; Kobayashi, K.; Tsunoda, M.; Funatsu, T.; et al. p38 MAPKs regulate the expression of genes in the dopamine synthesis pathway through phosphorylation of NR4A nuclear receptors. J. Cell Sci. 2011, 124, 3006–3016. [Google Scholar] [CrossRef] [Green Version]
- Mohan, H.M.; Aherne, C.M.; Rogers, A.C.; Baird, A.W.; Winter, D.C.; Murphy, E.P. Molecular pathways: The role of NR4A orphan nuclear receptors in cancer. Clin. Cancer Res. 2012, 18, 3223–3228. [Google Scholar] [CrossRef] [Green Version]
- McMorrow, J.P.; Murphy, E.P. Inflammation: A role for NR4A orphan nuclear receptors? Biochem. Soc. Trans. 2011, 39, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, I.; Bruun, J.A.; Bjorkoy, G.; Holm, T.; Johansen, T. Phosphorylation of the transactivation domain of Pax6 by extracellular signal-regulated kinase and p38 mitogen-activated protein kinase. J. Biol. Chem. 1999, 274, 15115–15126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Yue, W.; Zhang, L.; Ma, L.; Jia, W.; Qian, Z.; Zhang, C.; Wang, Y. Downregulation of PAX6 by shRNA inhibits proliferation and cell cycle progression of human non-small cell lung cancer cell lines. PLoS ONE 2014, 9, e85738. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Scheme showing the mechanism of nuclear p38α/β translocation and its inhibition by the PERY peptide. A. p38α/β are localized in the cytoplasm of resting cells and some of the molecules are phosphorylated upon stimulation. B. Phosphorylated or non-phosphorylated p38α/β bind to a dimer of Imp7/3 or Imp9/3, which escort them to the nuclear pores. C. Imp3 stays outside, while Imp7 or Imp9 escort the p38α/β through the nuclear pores, to the nucleus, where they dissociate from the importins, and are free to phosphorylate their substrates. D. Translocation of the kinases to the nucleus can be inhibited using the inhibitory PERY peptide that was synthesized according to the sequence of the p38α–Imp binding site as described in the text.
Figure 1.
Scheme showing the mechanism of nuclear p38α/β translocation and its inhibition by the PERY peptide. A. p38α/β are localized in the cytoplasm of resting cells and some of the molecules are phosphorylated upon stimulation. B. Phosphorylated or non-phosphorylated p38α/β bind to a dimer of Imp7/3 or Imp9/3, which escort them to the nuclear pores. C. Imp3 stays outside, while Imp7 or Imp9 escort the p38α/β through the nuclear pores, to the nucleus, where they dissociate from the importins, and are free to phosphorylate their substrates. D. Translocation of the kinases to the nucleus can be inhibited using the inhibitory PERY peptide that was synthesized according to the sequence of the p38α–Imp binding site as described in the text.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Maik-Rachline, G.; Lifshits, L.; Seger, R. Nuclear P38: Roles in Physiological and Pathological Processes and Regulation of Nuclear Translocation. Int. J. Mol. Sci. 2020, 21, 6102. https://doi.org/10.3390/ijms21176102
AMA Style
Maik-Rachline G, Lifshits L, Seger R. Nuclear P38: Roles in Physiological and Pathological Processes and Regulation of Nuclear Translocation. International Journal of Molecular Sciences. 2020; 21(17):6102. https://doi.org/10.3390/ijms21176102
Chicago/Turabian StyleMaik-Rachline, Galia, Lucia Lifshits, and Rony Seger. 2020. "Nuclear P38: Roles in Physiological and Pathological Processes and Regulation of Nuclear Translocation" International Journal of Molecular Sciences 21, no. 17: 6102. https://doi.org/10.3390/ijms21176102
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.