Cardiovascular Damage in COVID-19: Therapeutic Approaches Targeting the Renin-Angiotensin-Aldosterone System

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
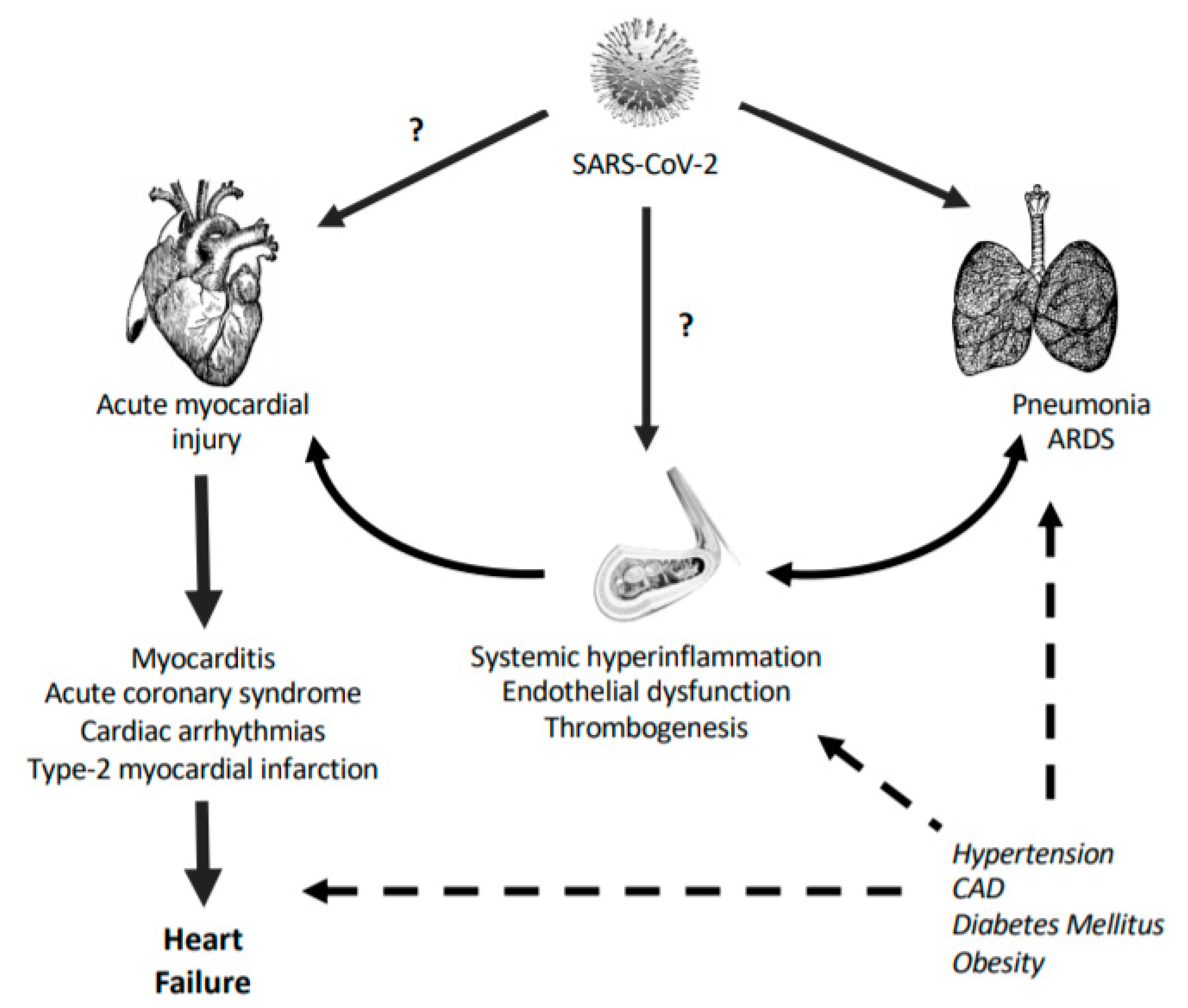
1.1. SARS-CoV-2 and the CV System
1.1.1. Endothelial Dysfunction
1.1.2. Acute Cardiac Injury
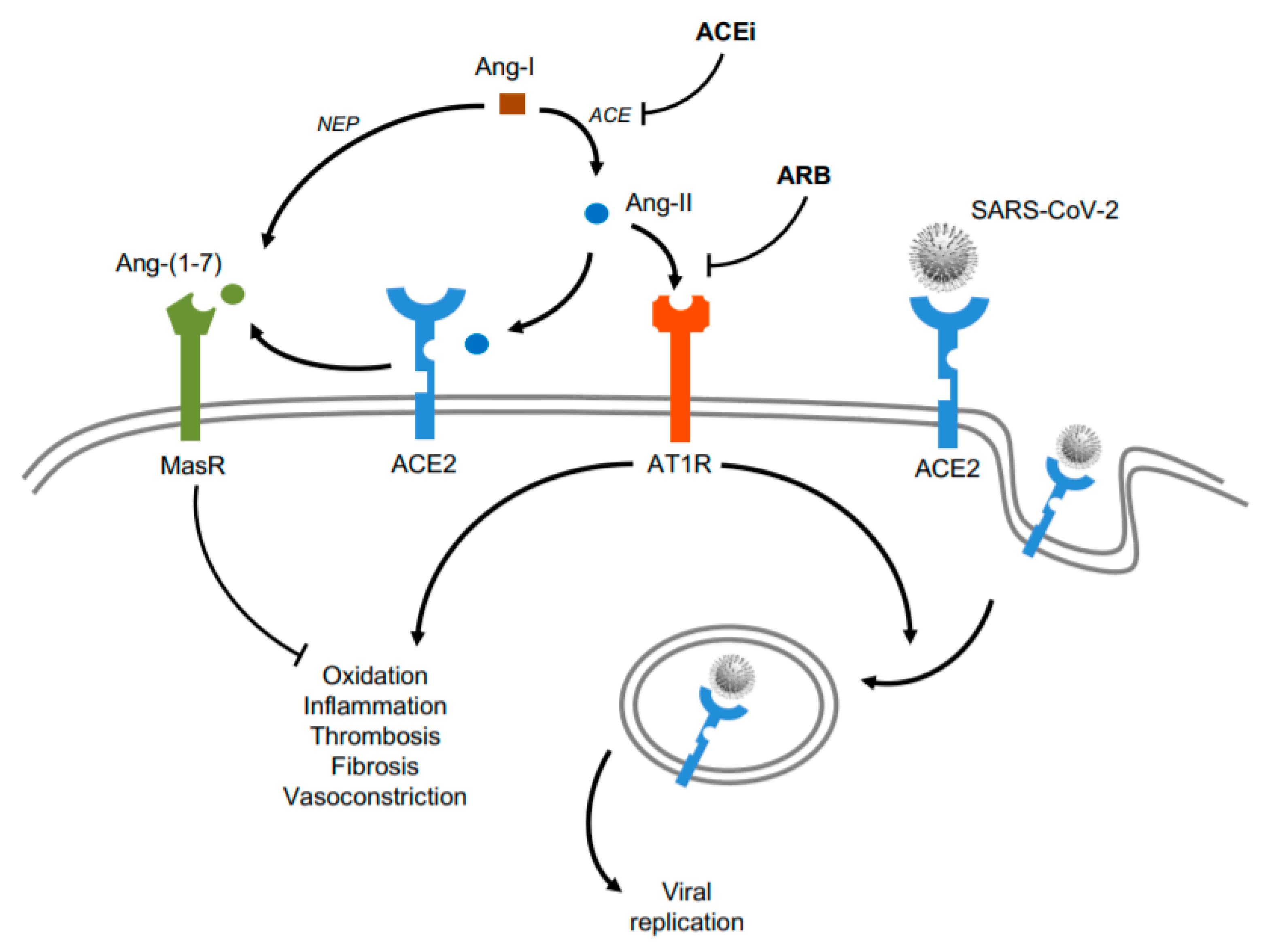
1.2. Renin-Angiotensin-Aldosterone System (RAAS) Modulators in CV Damage of COVID-19
1.3. Influence of ACEi and ARB on SARS-CoV-2 Infection
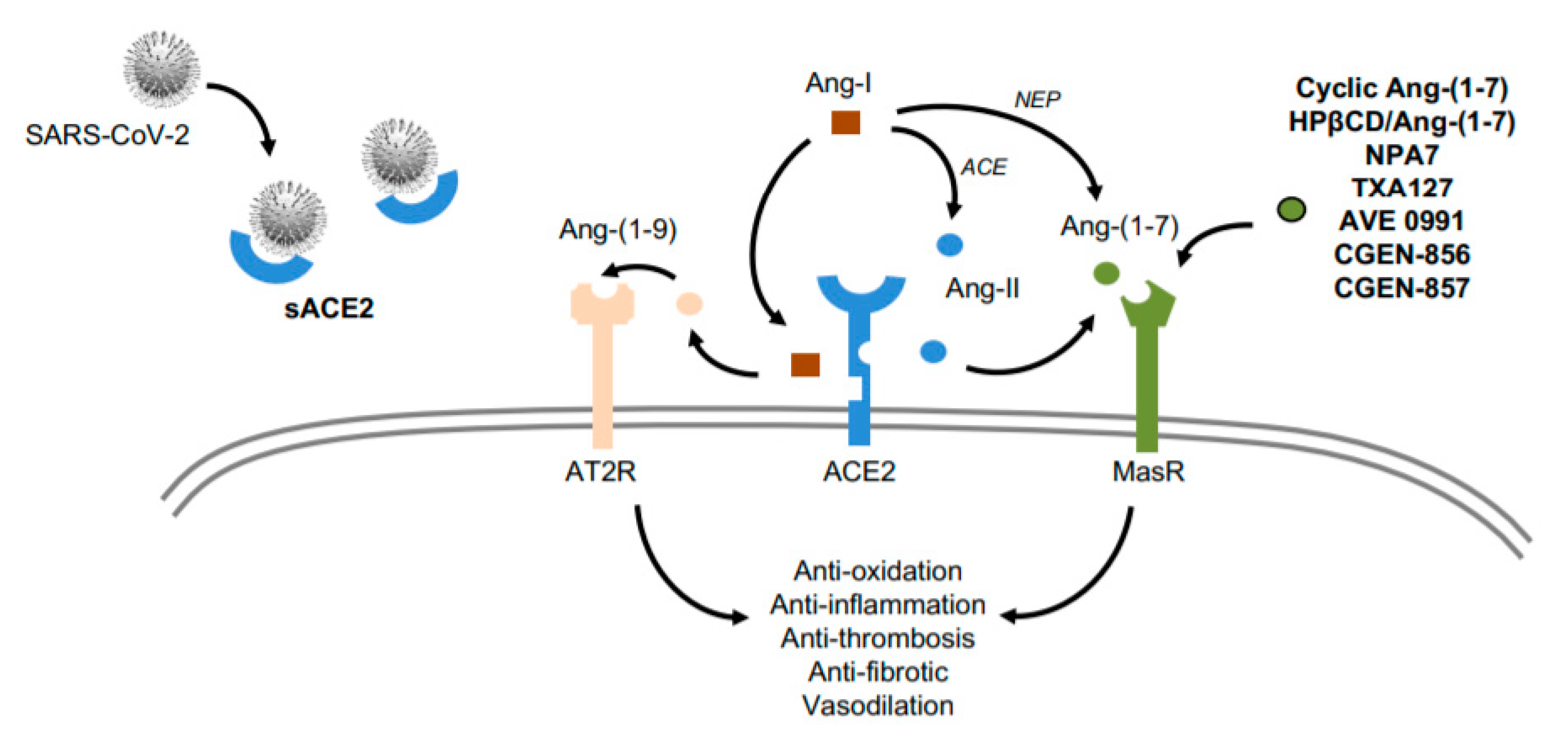
1.4. ACE2 as a Potential Target for CV Therapy During COVID-19
1.5. Ang-(1–7): A Cardioprotective Peptide for COVID-19
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | angiotensin convertase enzyme |
| ACE2 | angiotensin-converting enzyme-2 |
| ACEi | angiotensin-converting enzyme inhibitors |
| ADAM-17 | short for a disintegrin and metalloproteinase domain 17 |
| Ang- | angiotensin |
| ARB | angiotensin receptor blockers |
| ARDS | acute respiratory stress syndrome |
| AT1R | Angiotensin receptor type-I |
| AT2R | Angiotensin receptor type-II |
| ATCO | Angiotensin-(1,7) Treatment in COVID-19 |
| BH4 | coenzyme tetrahydrobiopterin |
| CAD | coronary artery disease |
| CK | creatine kinase |
| COVID-19 | Coronavirus disease 2019 |
| cTnI | cardiac troponin-I |
| DIC | disseminated intravascular coagulation |
| EMMPRIN-CD147 | extracellular matrix metalloproteinase inducer |
| HDL-C | high-density lipoprotein-cholesterol |
| HPβCD-Ang-(1-7) | hydroxypropyl β-cyclodextrin-Ang-(1-7) |
| IFN-γ | interferon gamma |
| IP-10 | IFN-γ-induced protein-10 |
| LDL-C | low-density lipoprotein-cholesterol |
| MasR | Mas receptor |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MERS | Middle East Respiratory Syndrome coronavirus |
| Mrg-D | Mas-related receptor |
| NEP | Neprilysin |
| NF-κB | nuclear factor-κB |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein-3 |
| NOD | Nucleotide-binding and oligomerization domain |
| NT-proBNP | N-terminal pro-B-type natriuretic peptide |
| PAI-1 | plasminogen activator inhibitor type-1 |
| RAAS | renin-angiotensin-aldosterone system |
| ROS | reactive oxidant species |
| sACE2 | Soluble ACE2 |
| SARS-CoV-1 | Severe acute respiratory syndrome coronavirus-1 |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus-2 |
| SNPs | single nucleotide polymorphisms |
| TC | total colesterol |
| TG | triglycerides |
| TMPRSS2 | serine protease transmembrane serine protease-2 |
| TNF-α | Tumor necrosis factor-alpha |
References
- Chan, J.W.M.; Ng, C.K.; Chan, Y.H.; Mok, T.Y.W.; Lee, S.; Chu, S.Y.Y.; Law, W.L.; Lee, M.P.; Li, P.C.K. Short Term Outcome and Risk Factors for Adverse Clinical Outcomes in Adults With Severe Acute Respiratory Syndrome (SARS). Thorax 2003, 58, 686–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawi, A.; Ryoo, S.G. Prevalence of Comorbidities in the Middle East Respiratory Syndrome Coronavirus (MERS-CoV): A Systematic Review and Meta-Analysis. Int. J. Infect. Dis. 2016, 49, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raoult, D.; Zumla, A.; Locatelli, F.; Ippolito, G.; Kroemer, G. Coronavirus Infections: Epidemiological, Clinical and Immunological Features and Hypotheses. Cell Stress 2020, 4, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Pei, S.; Chen, B.; Song, Y.; Zhang, T.; Yang, W.; Shaman, J. Substantial Undocumented Infection Facilitates the Rapid Dissemination of Novel Coronavirus (SARS-CoV-2). Science 2020, 368, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Letko, M.; Marzi, A.; Munster, V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Senapati, S.; Kumar, S.; Singh, A.K.; Banerjee, P.; Bhagavatula, S. Assessment of Risk Conferred by Coding and Regulatory Variations of TMPRSS2 and CD26 in Susceptibility to SARS-CoV-2 Infection in Human. J. Genet. 2020, 99, 1–5. [Google Scholar] [CrossRef]
- Kim, C.-H. SARS-CoV-2 Evolutionary Adaptation Toward Host Entry and Recognition of Receptor O-Acetyl Sialylation in Virus–Host Interaction. Int. J. Mol. Sci. 2020, 21, 4549. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 Cleave ACE2 Differentially and Only Proteolysis by TMPRSS2 Augments Entry Driven by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2013, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Li, M.-Y.; Li, L.; Zhang, Y.; Wang, X. Expression of the SARS-CoV-2 Cell Receptor Gene ACE2 in a Wide Variety of Human Tissues. Infect. Dis. Poverty 2020, 9, 1–7. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.T.; Navis, G.; Van Goor, H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, S.; Herbert, J.A.; McNamara, P.S.; Hedrich, C.M. COVID-19: Immunology and Treatment Options. Clin. Immunol. 2020, 215, 108448. [Google Scholar] [CrossRef] [PubMed]
- Tufan, A.; Guler, A.A.; Matucci-Cerinic, M. COVID-19, Immune System Response, Hyperinflammation and Repurposing Antirheumatic Drugs. Turk. J. Med Sci. 2020, 50, 620–632. [Google Scholar] [CrossRef]
- Sallenave, J.-M.; Guillot, L. Innate Immune Signaling and Proteolytic Pathways in the Resolution or Exacerbation of SARS-CoV-2 in Covid-19: Key Therapeutic Targets? Front. Immunol. 2020, 11, 1229. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 Infection and Overactivation of Nlrp3 Inflammasome As a Trigger of Cytokine “storm” and Risk Factor for Damage of Hematopoietic Stem Cells. Leukemia 2020, 34, 1726–1729. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and Pyroptosis As Therapeutic Targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef]
- De Lucena, T.M.C.; Santos, A.F.D.S.; De Lima, B.R.; Borborema, M.E.D.A.; Silva, J.D.A.; Fabrício, B.R.D.L. Mechanism of Inflammatory Response in Associated Comorbidities in COVID-19. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 597–600. [Google Scholar] [CrossRef]
- Weekly, C.C. The Novel Coronavirus Pneumonia Emergency Response Epidemiology Team. The Epidemiological Characteristics of an Outbreak of 2019 Novel Coronavirus Diseases (COVID-19) in China. China CDC Wkly. 2020, 2, 113–122. [Google Scholar] [CrossRef]
- Porfidia, A.; Pola, R. Venous Thromboembolism in COVID-19 Patients. J. Thromb. Haemost. 2020, 18, 1516–1517. [Google Scholar] [CrossRef] [Green Version]
- Sriram, K.; Insel, P.A. A Hypothesis for Pathobiology and Treatment of COVID-19: The Centrality of ACE1 / ACE2 Imbalance. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Hu, Y.; Sun, J.; Dai, Z.; Deng, H.; Li, X.; Huang, Q.; Wu, Y.; Sun, L.; Xu, Y. Prevalence and Severity of Corona Virus Disease 2019 (COVID-19): A Systematic Review and Meta-Analysis. J. Clin. Virol. 2020, 127, 104371. [Google Scholar] [CrossRef]
- Guzik, T.; Mohiddin, S.A.; DiMarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the Cardiovascular System: Implications for Risk Assessment, Diagnosis, and Treatment Options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients With COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Hendren, N.S.; Drazner, M.H.; Bozkurt, B.; Cooper, L.T. Description and Proposed Management of the Acute COVID-19 Cardiovascular Syndrome. Circulation 2020, 141, 1903–1914. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 Expression in Human Heart Indicates New Potential Mechanism of Heart Injury Among Patients Infected With SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Endemann, D.H.; Schiffrin, E.L. Endothelial Dysfunction. J Am Soc Nephrol. 2004, 1515, 1983–1992. [Google Scholar]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Pons, S.; Fodil, S.; Azoulay, E.; Zafrani, L. The Vascular Endothelium: The Cornerstone of Organ Dysfunction in Severe SARS-CoV-2 Infection. Crit. Care 2020, 24, 1–8. [Google Scholar] [CrossRef]
- Aimes, R.; Zijlstra, A.; Hooper, J.; Ogbourne, S.; Sit, M.-L.; Fuchs, S.; Gotley, D.; Quigley, J.P.; Antalis, T. Endothelial Cell Serine Proteases Expressed During Vascular Morphogenesis and Angiogenesis. Thromb. Haemost. 2003, 89, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Vanarsdall, A.L.; Pritchard, S.R.; Wisner, T.W.; Liu, J.; Jardetzky, T.S.; Johnson, D.C. CD147 Promotes Entry of Pentamer-Expressing Human Cytomegalovirus into Epithelial and Endothelial Cells. mBio 2018, 9, e00781-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef] [PubMed]
- Whyte, C.; Morrow, G.B.; Mitchell, J.L.; Chowdary, P.; Mutch, N.J. Fibrinolytic Abnormalities in Acute Respiratory Distress Syndrome (ARDS) and Versatility of Thrombolytic Drugs to Treat COVID-19. J. Thromb. Haemost. 2020, 18, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Menter, T.; Haslbauer, J.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. A Post-Mortem Examination of COVID19 Patients Reveals Diffuse Alveolar Damage With Severe Capillary Congestion and Variegated Findings of Lungs and Other Organs Suggesting Vascular Dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef]
- Pouletty, M.; Borocco, C.; Ouldali, N.; Caseris, M.; Basmaci, R.; Lachaume, N.; Bensaid, P.; Pichard, S.; Kouider, H.; Morelle, G.; et al. Paediatric Multisystem Inflammatory Syndrome Temporally Associated With SARS-CoV-2 Mimicking Kawasaki Disease (Kawa-COVID-19): A Multicentre Cohort. Ann. Rheum. Dis. 2020, 79, 999–1006. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial Cell Control of Thrombosis. BMC Cardiovasc. Disord. 2015, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Wichmann, D.; Sperhake, J.-P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef]
- Mackman, N.; Antoniak, S.; Wolberg, A.; Kasthuri, R.; Key, N. Coagulation Abnormalities and Thrombosis in Patients Infected With SARS-CoV-2 and Other Pandemic Viruses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2033. [Google Scholar] [CrossRef]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-Vessel Stroke As a Presenting Feature of Covid-19 in the Young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant Treatment Is Associated With Decreased Mortality in Severe Coronavirus Disease 2019 Patients With Coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Paranjpe, I.; Fuster, V.; Lala, A.; Russak, A.J.; Glicksberg, B.S.; Levin, M.A.; Charney, A.W.; Narula, J.; Fayad, Z.A.; Bagiella, E.; et al. Association of Treatment Dose Anticoagulation With In-Hospital Survival Among Hospitalized Patients With COVID-19. J. Am. Coll. Cardiol. 2020, 76, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Santamarina, M.G.; Boisier, D.; Contreras, R.; Baque, M.; Volpacchio, M.; Beddings, I. COVID-19: A Hypothesis Regarding the Ventilation-Perfusion Mismatch. Crit. Care 2020, 24, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary Post-Mortem Findings in a Series of COVID-19 Cases from Northern Italy: A Two-Centre Descriptive Study. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Sardu, C.; Gargiulo, G.; Esposito, G.; Paolisso, G.; Marfella, R. Impact of Diabetes Mellitus on Clinical Outcomes in Patients Affected by Covid-19. Cardiovasc. Diabetol. 2020, 19, 1–4. [Google Scholar] [CrossRef]
- Ryan, P.M.; Caplice, N.M. Is Adipose Tissue a Reservoir for Viral Spread, Immune Activation, and Cytokine Amplification in Coronavirus Disease 2019? Obesity 2020, 28, 1191–1194. [Google Scholar] [CrossRef] [Green Version]
- Rali, A.S.; Ranka, S.; Shah, Z.; Sauer, A.J. Mechanisms of Myocardial Injury in Coronavirus Disease 2019. Card. Fail. Rev. 2020, 6. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 19. [Google Scholar] [CrossRef] [Green Version]
- Clerkin, K.J.; Fried, J.A.; Raikhelkar, J.; Sayer, G.; Griffin, J.M.; Masoumi, A.; Jain, S.S.; Burkhoff, D.; Kumaraiah, D.; Rabbani, L.; et al. COVID-19 and Cardiovascular Disease. Circular 2020, 141, 1648–1655. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.-H.; Hung, C.-S.; Liao, C.-W.; Wei, L.-H.; Chen, C.-W.; Shun, C.-T.; Wen, W.-F.; Wan, C.-H.; Wu, X.-M.; Chang, Y.-Y.; et al. IL-6 Trans-Signalling Contributes to Aldosterone-Induced Cardiac Fibrosis. Cardiovasc. Res. 2018, 114, 690–702. [Google Scholar] [CrossRef]
- Gao, L.; Jiang, D.; Wen, X.-S.; Cheng, X.-C.; Sun, M.; He, B.; You, L.-N.; Lei, P.; Tan, X.-W.; Qin, S.; et al. Prognostic Value of NT-ProBNP in Patients With Severe COVID-19. Respir. Res. 2020, 21, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune Response in COVID-19: Addressing a Pharmacological Challenge by Targeting Pathways Triggered by SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bangalore, S.; Sharma, A.; Slotwiner, A.; Yatskar, L.; Harari, R.; Shah, B.; Ibrahim, H.; Friedman, G.H.; Thompson, C.; Alviar, C.L.; et al. ST-Segment Elevation in Patients With Covid-19: A Case Series. N. Engl. J. Med. 2020, 382, 2478–2480. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.J.; Hiscox, J.A.; Hooper, N.M. ACE2: From Vasopeptidase to SARS Virus Receptor. Trends Pharmacol. Sci. 2004, 2525, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.-Q.; Wang, H.; Shen, H.; Li, Z.; Geng, J.; Han, H.; Cai, J.; Li, X.; Kang, W.; Weng, D.; et al. The Clinical Pathology of Severe Acute Respiratory Syndrome (SARS): A Report from China. J. Pathol. 2003, 200, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Ma, F.; Wei, X.; Fang, Y. Coronavirus Fulminant Myocarditis Treated With Glucocorticoid and Human Immunoglobulin. Eur. Hear. J. 2020, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, P.; Li, W.; Xie, J.; Hou, Y.; You, C. Cytokine Storm Induced by SARS-CoV-2. Clin. Chim. Acta 2020, 509, 280–287. [Google Scholar] [CrossRef]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and Cardiovascular Disease: From Basic Mechanisms to Clinical Perspectives. Nat. Rev. Cardiol. 2020, 1–16. [Google Scholar] [CrossRef]
- Park, M.D. Macrophages: A Trojan Horse in COVID-19? Nat. Rev. Immunol. 2020, 20, 351. [Google Scholar] [CrossRef]
- Kochav, S.M.; Coromilas, E.; Nalbandian, A.; Ranard, L.S.; Gupta, A.; Chung, M.K.; Gopinathannair, R.; Biviano, A.; Garan, H.; Wan, E.Y. Cardiac Arrhythmias in COVID-19 Infection. Circ. Arrhythmia Electrophysiol. 2020, 13, e008719. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Bhatla, A.; Mayer, M.M.; Adusumalli, S.; Hyman, M.C.; Oh, E.; Tierney, A.; Moss, J.; Chahal, A.A.; Anesi, G.; Denduluri, S.; et al. COVID-19 and Cardiac Arrhythmias. Hear. Rhythm. 2020, 17, 1439–1444. [Google Scholar] [CrossRef]
- Goyal, P.; Choi, J.J.; Pinheiro, L.C.; Schenck, E.J.; Chen, R.; Jabri, A.; Satlin, M.J.; Campion, T.R.; Nahid, M.; Ringel, J.B.; et al. Clinical Characteristics of Covid-19 in New York City. N. Engl. J. Med. 2020, 382, 2372–2374. [Google Scholar] [CrossRef]
- Atri, D.; Siddiqi, H.K.; Lang, J.P.; Nauffal, V.; Morrow, D.A.; Bohula, E.A. COVID-19 for the Cardiologist. JACC Basic Transl. Sci. 2020, 5, 518–536. [Google Scholar] [CrossRef]
- Libby, P.; Loscalzo, J.; Ridker, P.M.; Farkouh, M.E.; Hsue, P.Y.; Fuster, V.; Hasan, A.A.; Amar, S. Inflammation, Immunity, and Infection in Atherothrombosis. J. Am. Coll. Cardiol. 2018, 72, 2071–2081. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Executive Group on Behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth Universal Definition of Myocardial Infarction (2018). J. Am. Coll Cardiol. 2018, 7272, 2231–2264. [Google Scholar]
- Wu, C.-H.; Mohammadmoradi, S.; Chen, J.Z.; Sawada, H.; Daugherty, A.; Lu, H.S. Renin-Angiotensin System and Cardiovascular Functions. Arter. Thromb. Vasc. Boil. 2018, 38, e108–e116. [Google Scholar] [CrossRef] [Green Version]
- Dijkman, R.; Jebbink, M.F.; Deijs, M.; Milewska, A.; Pyrc, K.; Buelow, E.; Van Der Bijl, A.; Van Der Hoek, L. Replication-Dependent Downregulation of Cellular Angiotensin-Converting Enzyme 2 Protein Expression by Human Coronavirus NL63. J. Gen. Virol. 2012, 93, 1924–1929. [Google Scholar] [CrossRef]
- Dielis, A.W.; Smid, M.; Spronk, H.M.; Hamulyak, K.A.; Kroon, A.; Cate, H.T.; De Leeuw, P.W. The Prothrombotic Paradox of Hypertension. Hypertension 2005, 46, 1236–1242. [Google Scholar] [CrossRef] [Green Version]
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-Regulatory renin–angiotensin System in Cardiovascular Disease. Nat. Rev. Cardiol. 2019, 17, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Povlsen, A.L.; Grimm, D.; Wehland, M.; Infanger, M.; Krüger, M. The Vasoactive Mas Receptor in Essential Hypertension. J. Clin. Med. 2020, 9, 267. [Google Scholar] [CrossRef] [Green Version]
- Romero, A.; San Hipólito-Luengo, Á.; Villalobos, L.A.; Vallejo, S.; Valencia, I.; Michalska, P.; Pajuelo-Lozano, N.; Sánchez-Pérez, I.; León, R.; Bartha, J.L.; et al. The Angiotensin-(1-7)/Mas Receptor Axis Protects from Endothelial Cell Senescence via Klotho and Nrf2 Activation. Aging Cell 2019, 18. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, W.O.; De Castro, C.H.; Santos, R.A.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1-7) Counterregulates Angiotensin II Signaling in Human Endothelial Cells. Hypertension 2007, 50, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Karnik, S.S.; Singh, K.D.; Tirupula, K.; Unal, H. Significance of Angiotensin 1-7 Coupling With MAS1 Receptor and Other GPCRs to the Renin-Angiotensin System: IUPHAR Review 22. J. Pharmacol. 2017, 174, 737–753. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Baessler, A.; Schunkert, H. Renin Angiotensin System and Gender Differences in the Cardiovascular System. Cardiovasc. Res. 2002, 53, 672–677. [Google Scholar] [CrossRef] [Green Version]
- Komukai, K.; Mochizuki, S.; Yoshimura, M. Gender and the Renin-Angiotensin-Aldosterone System. Fundam. Clin. Pharmacol. 2010, 24, 687–698. [Google Scholar] [CrossRef]
- La Vignera, S.; Cannarella, R.; Condorelli, R.A.; Torre, F.; Aversa, A.; Calogero, A.E. Sex-Specific SARS-CoV-2 Mortality: Among Hormone-Modulated ACE2 Expression, Risk of Venous Thromboembolism and Hypovitaminosis D. Int. J. Mol. Sci. 2020, 21, 2948. [Google Scholar] [CrossRef]
- Nascimento, I.J.B.D.; Cacic, N.; Abdulazeem, H.M.; Von Groote, T.C.; Jayarajah, U.; Weerasekara, I.; Esfahani, M.A.; Civile, V.T.; Marušić, A.; Jerončić, A.; et al. Novel Coronavirus Infection (COVID-19) in Humans: A Scoping Review and Meta-Analysis. J. Clin. Med. 2020, 9, 941. [Google Scholar] [CrossRef] [Green Version]
- Chanana, N.; Palmo, T.; Sharma, K.; Kumar, R.; Graham, B.B.; Pasha, Q. Sex-Derived Attributes Contributing to SARS-CoV-2 Mortality. Am. J. Physiol. Metab. 2020. [Google Scholar] [CrossRef]
- Bukowska, A.; Spiller, L.; Wolke, C.; Lendeckel, U.; Weinert, S.; Hoffmann, J.; Bornfleth, P.; Kutschka, I.; Gardemann, A.; Isermann, B.; et al. Protective Regulation of the ACE2/ACE Gene Expression by Estrogen in Human Atrial Tissue from Elderly Men. Exp Biol Med. 2017, 242, 1412–1423. [Google Scholar] [CrossRef]
- Stelzig, K.E.; Canepa-Escaro, F.; Schiliro, M.; Berdnikovs, S.; Prakash, Y.S.; Chiarella, S.E. Estrogen Regulates the Expression of SARS-CoV-2 Receptor ACE2 in Differentiated Airway Epithelial Cells. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L1280–L1281. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A Crucial Role of Angiotensin Converting Enzyme 2 (ACE2) in SARS coronavirus–induced Lung Injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex Differences in Immune Responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Straub, R.H. The Complex Role of Estrogens in Inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef] [Green Version]
- Groß, S.; Jahn, C.; Cushman, S.; Bär, C.; Thum, T. SARS-CoV-2 Receptor ACE2-Dependent Implications on the Cardiovascular System: From Basic Science to Clinical Implications. J. Mol. Cell. Cardiol. 2020, 144, 47–53. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Mullick, A.E. Renin Angiotensin Aldosterone Inhibition in the Treatment of Cardiovascular Disease. Pharmacol. Res. 2017, 125, 57–71. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A Human Homolog of Angiotensin-Converting Enzyme. J. Boil. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Basu, R.; Poglitsch, M.; Yogasundaram, H.; Thomas, J.; Rowe, B.H.; Oudit, G.Y. Roles of Angiotensin Peptides and Recombinant Human ACE2 in Heart Failure. J. Am. Coll. Cardiol. 2017, 69, 805–819. [Google Scholar] [CrossRef]
- Sukumaran, V.; Veeraveedu, P.T.; Gurusamy, N.; Yamaguchi, K.; Lakshmanan, A.P.; Ma, M.; Ma, M.; Suzuki, K.; Kodama, M.; Watanabe, K. Cardioprotective Effects of Telmisartan Against Heart Failure in Rats Induced By Experimental Autoimmune Myocarditis through the Modulation of Angiotensin-Converting Enzyme-2/Angiotensin 1-7/Mas Receptor Axis. Int. J. Biol. Sci. 2011, 7, 1077–1092. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary Angiotensin-Converting Enzyme 2 in Hypertensive Patients May Be Increased by Olmesartan, an Angiotensin II Receptor Blocker. Am. J. Hypertens. 2014, 28, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Karakiulakis, G.; Roth, M. Are Patients With Hypertension and Diabetes Mellitus at Increased Risk for COVID-19 Infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Khan, M.S.; Abraham, W.T.; Bauersachs, J.; Bocchi, E.; Bozkurt, B.; Braunwald, E.; Chopra, V.K.; Cleland, J.G.; et al. Conducting Clinical Trials in Heart Failure During (and After) the COVID-19 Pandemic: An Expert Consensus Position Paper from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. Hear. J. 2020, 41, 2109–2117. [Google Scholar] [CrossRef]
- Deshotels, M.R.; Xia, H.; Sriramula, S.; Lazartigues, E.; Filipeanu, C.M. Angiotensin II Mediates Angiotensin Converting Enzyme Type 2 Internalization and Degradation through an Angiotensin II Type I Receptor-Dependent Mechanism. Hypertension 2014, 64, 1368–1375. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04355429 (accessed on 3 September 2020).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04366050 (accessed on 3 September 2020).
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-Angiotensin System Inhibitors Improve the Clinical Outcomes of COVID-19 Patients With Hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef]
- Klimas, J.; Olvedy, M.; Ochodnicka-Mackovicova, K.; Kruzliak, P.; Cacanyiova, S.; Kristek, F.; Krenek, P.; Ochodnický, P. Perinatally Administered Losartan Augments Renal ACE2 Expression But Not Cardiac or Renal Mas Receptor in Spontaneously Hypertensive Rats. J. Cell. Mol. Med. 2015, 19, 1965–1974. [Google Scholar] [CrossRef]
- Wysocki, J.; Ye, M.; Rodriguez, E.; González-Pacheco, F.R.; Barrios, C.; Evora, K.; Schuster, M.; Loibner, H.; Brosnihan, K.B.; Ferrario, C.M.; et al. Targeting the Degradation of Angiotensin II With Recombinant Angiotensin-Converting Enzyme 2: Prevention of Angiotensin II-Dependent Hypertension. Hypertension 2009, 55, 90–98. [Google Scholar] [CrossRef]
- Batlle, D.; Wysocki, J.; Satchell, K. Soluble Angiotensin-Converting Enzyme 2: A Potential Approach for Coronavirus Infection Therapy? Clin. Sci. 2020, 134, 543–545. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01597635 (accessed on 3 September 2020).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04287686 (accessed on 3 September 2020).
- Haschke, M.; Schuster, M.; Poglitsch, M.; Loibner, H.; Salzberg, M.; Bruggisser, M.; Penninger, J.M.; Krähenbühl, S. Pharmacokinetics and Pharmacodynamics of Recombinant Human Angiotensin-Converting Enzyme 2 in Healthy Human Subjects. Clin. Pharmacokinet. 2013, 52, 783–792. [Google Scholar] [CrossRef]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A Pilot Clinical Trial of Recombinant Human Angiotensin-Converting Enzyme 2 in Acute Respiratory Distress Syndrome. Crit. Care 2017, 21, 234. [Google Scholar] [CrossRef] [Green Version]
- Ocaranza, M.P.; Jalil, J.E. Protective Role of the ACE2/Ang-(1–9) Axis in Cardiovascular Remodeling. Int. J. Hypertens. 2012, e594361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif-Askari, S.N.; Sharif-Askari, S.F.; Alabed, M.; Temsah, M.-H.; Heialy, A.S.; Hamid, Q.; Halwani, R. Airways Expression of SARS-CoV-2 Receptor, ACE2, and TMPRSS2 Is Lower in Children Than Adults and Increases With Smoking and COPD. Mol. Ther. Methods Clin. Dev. 2020, 18, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation 2020, 142, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, C.G.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Jiang, S. A Suspicious Role of Interferon in the Pathogenesis of SARS-CoV-2 by Enhancing Expression of ACE2. Signal Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef]
- Hussain, M.; Jabeen, N.; Raza, F.; Shabbir, S.; Baig, A.A.; Amanullah, A.; Aziz, B. Structural Variations in Human ACE2 May Influence Its Binding With SARS-CoV-2 Spike Protein. J. Med. Virol. 2020, 92, 1580–1586. [Google Scholar] [CrossRef] [Green Version]
- Devaux, C.A.; Rolain, J.-M.; Raoult, D. ACE2 Receptor Polymorphism: Susceptibility to SARS-CoV-2, Hypertension, Multi-Organ Failure, and COVID-19 Disease Outcome. J. Microbiol. Immunol. Infect. 2020, 53, 425–435. [Google Scholar] [CrossRef]
- Benetti, E.; GEN-COVID Multicenter Study; Tita, R.; Spiga, O.; Ciolfi, A.; Birolo, G.; Bruselles, A.; Doddato, G.; Giliberti, A.; Marconi, C.; et al. ACE2 Gene Variants May Underlie Interindividual Variability and Susceptibility to COVID-19 in the Italian Population. Eur. J. Hum. Genet. 2020, 1–13. [Google Scholar] [CrossRef]
- Darbani, B. The Expression and Polymorphism of Entry Machinery for COVID-19 in Human: Juxtaposing Population Groups, Gender, and Different Tissues. Int. J. Environ. Res. Public Health 2020, 17, 3433. [Google Scholar] [CrossRef]
- Wang, J.; Xu, X.; Zhou, X.; Chen, P.; Liang, H.; Li, X.; Zhong, W.; Hao, P. Molecular Simulation of SARS-CoV-2 Spike Protein Binding to Pangolin ACE2 or Human ACE2 Natural Variants Reveals Altered Susceptibility to Infection. J. Gen. Virol. 2020, jgv0014. [Google Scholar] [CrossRef]
- Stawiski, E.W.; Diwanji, D.; Suryamohan, K.; Gupta, R.A.; Fellouse, F.; Sathirapongsasuti, F.; Liu, J.; Jiang, Y.-P.; Ratan, A.; Mis, M.; et al. Human ACE2 Receptor Polymorphisms Predict SARS-CoV-2 Susceptibility. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, P.; Santos, J.; Oliveira, E.; Miglioli, N.; Assis, A.; Monteleone-Cassiano, A.; Ribeiro, V.; Duarte, M.; Machado, M.; Mascarenhas, R.; et al. A Crispr-Cas9 System Designed to Introduce Point Mutations into the Human ACE2 Gene to Weaken the Interaction of the ACE2 Receptor With the SARS-CoV-2 S Protein. Preprints 2020. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, T.; Li, Y.; Guan, T.; Lai, Y.; Shen, Y.; Zeyaweiding, A.; Maimaiti, T.; Li, F.; Zhao, H.; et al. Association of ACE2 Polymorphisms With Susceptibility to Essential Hypertension and Dyslipidemia in Xinjiang, China. Lipids Health Dis. 2018, 17, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Liu, C.; Guan, T.; Li, Y.; Lai, Y.; Li, F.; Zhao, H.; Maimaiti, T.; Zeyaweiding, A. Correction: Association of ACE2 Genetic Polymorphisms With Hypertension-Related Target Organ Damages in South Xinjiang. Hypertens. Res. 2019, 42, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Niu, W.; Qi, Y.; Hou, S.; Zhou, W.; Qiu, C. Correlation of Angiotensin-Converting Enzyme 2 Gene Polymorphisms With Stage 2 Hypertension in Han Chinese. Transl. Res. 2007, 150, 374–380. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Guan, T.; Lai, Y.; Shen, Y.; Zeyaweiding, A.; Zhao, H.; Li, F.; Maimaiti, T. ACE2 Polymorphisms Associated With Cardiovascular Risk in Uygurs With Type 2 Diabetes Mellitus. Cardiovasc. Diabetol. 2018, 17, 127. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Wu, G.; Yue, M.; Ye, J.; Chen, Y.; Xu, B.; Shu, Z.; Zhu, J.; Lu, N.; Tan, X. Hypertension and Hypertensive Left Ventricular Hypertrophy Are Associated With ACE2 Genetic Polymorphism. Life Sci. 2019, 225, 39–45. [Google Scholar] [CrossRef]
- Wang, S.-X.; Tao, T.; Fu, Z.-Q.; Xie, X.-Z.; Wang, H.; Wang, Y.-T. Polymorphisms of Angiotensin-Converting Enzyme 2 Gene Confer a Risk to Lone Atrial Fibrillation in Chinese Male Patients. Chin. Med. J. 2013, 126, 4608–4611. [Google Scholar]
- Chen, Y.; Yu, H. A3216 Relationship Between Genetic Variants of ACE2 Gene and Circulating Levels of ACE2 and Its Metabolites. J. Hypertens. 2018, 36. [Google Scholar] [CrossRef]
- Peiró, C.; Moncada, S. Substituting Angiotensin-(1-7) to Prevent Lung Damage in SARS-CoV-2 Infection? Circular 2020, 141, 1665–1666. [Google Scholar] [CrossRef] [Green Version]
- Shete, A. Urgent Need for Evaluating Agonists of Angiotensin-(1-7)/Mas Receptor Axis for Treating Patients With COVID-19. Int. J. Infect. Dis. 2020, 96, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhong, J.-C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1–7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, L.K.; Totou, N.L.; Moura, S.; Kangussu, L.; Millán, R.D.S.; Campagnole-Santos, M.J.; Coelho, D.; Motta-Santos, D.; Santos, R.A. Eccentric Overload Muscle Damage Is Attenuated By a Novel Angiotensin- (1-7) Treatment. Int. J. Sports Med. 2018, 39, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Meems, L.M.; Andersen, I.A.; Pan, S.; Harty, G.; Chen, Y.; Zheng, Y.; Harders, G.E.; Ichiki, T.; Heublein, D.M.; Iyer, S.R.; et al. Design, Synthesis, and Actions of an Innovative Bispecific Designer Peptide. Hypertension 2019, 73, 900–909. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04401423 (accessed on 3 September 2020).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04375124 (accessed on 3 September 2020).
- ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04332666 (accessed on 3 September 2020).




{kind=link}
{kind=link}
{kind=link}
| ID SNPs | Nucleotide Change | Genotype | Related Pathology | Sex | Reference |
|---|---|---|---|---|---|
| rs4646188 | A > G | rs4646188 (TT + CT) | Increased TC Decreased HDL-C Hypertension | M/F | [117] |
| rs4646188 (CC) | Increased TG Increased LDL-C | M/F | [117] | ||
| rs4646188 (TT) | T2DM | M/F | [120] | ||
| rs4646188 (CC + CT) | Ischemic stroke | M/F | [117] | ||
| rs2106809 | A > G | rs2106809 (TT) | Increased LDL-C Hypertension Reduced Ang-(1–7) Ventricular Hypertrophy | M/F F | [117] [121,123] |
| rs2106809 (CC + CT) | Increased TG Decreased HDL-C | M/F | [117] | ||
| rs2106809 (T) | Atrial Fibrillation | M | [122] | ||
| rs2074192 | C > T | rs2074192 (TT + CT) | Increased TC Hypertension | M/F | [117] |
| rs2074192 (TT) | Reduced Ang-(1–7) Ventricular Hypertrophy | F | [121,123] | ||
| rs2074192 (CC) | T2DM | M/F | [120] | ||
| rs4240157 | C > G, T | rs4240157 (CC + CT) | Increased TC Increased TG Decreased HDL-C Hypertension Ischemic stroke T2DM | M/F | [117,120] |
| rs1978124 | T > A, C, G | rs1978124 (TT + CT) | Increased LDL-C T2DM | M/F | [117,120] |
| rs1978124 (G) | Hypertension | M | [119] | ||
| rs233575 | G > A, C | rs233575 (CC + CT) | Increased LDL-C Increased SBP T2DM | M/F | [117,120] |
| rs4830542 | C > A, G, T | rs4830542 (CC + CT) | Ischemic stroke | M/F | [117] |
| rs879922 | C > G | rs879922 (CC + CG) | Increased LDL-C Increased TC Increased TG Hypertension T2DM | M/F | [117,118,120] |
| rs4646156 | A > C, G, T | rs4646156 (AA + AT) | Increased SBP T2DM | M/F | [120] |
| rs2048683 | T > A, G | rs2048683 (TT + GT) | Increased SBP T2DM | M/F | [120] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lumpuy-Castillo, J.; Lorenzo-Almorós, A.; Pello-Lázaro, A.M.; Sánchez-Ferrer, C.; Egido, J.; Tuñón, J.; Peiró, C.; Lorenzo, Ó. Cardiovascular Damage in COVID-19: Therapeutic Approaches Targeting the Renin-Angiotensin-Aldosterone System. Int. J. Mol. Sci. 2020, 21, 6471. https://doi.org/10.3390/ijms21186471
Lumpuy-Castillo J, Lorenzo-Almorós A, Pello-Lázaro AM, Sánchez-Ferrer C, Egido J, Tuñón J, Peiró C, Lorenzo Ó. Cardiovascular Damage in COVID-19: Therapeutic Approaches Targeting the Renin-Angiotensin-Aldosterone System. International Journal of Molecular Sciences. 2020; 21(18):6471. https://doi.org/10.3390/ijms21186471
Chicago/Turabian StyleLumpuy-Castillo, Jairo, Ana Lorenzo-Almorós, Ana María Pello-Lázaro, Carlos Sánchez-Ferrer, Jesús Egido, José Tuñón, Concepción Peiró, and Óscar Lorenzo. 2020. "Cardiovascular Damage in COVID-19: Therapeutic Approaches Targeting the Renin-Angiotensin-Aldosterone System" International Journal of Molecular Sciences 21, no. 18: 6471. https://doi.org/10.3390/ijms21186471