
Hypogonadism in Patients with Prader Willi Syndrome: A Narrative Review


 , ,
, ,
Abstract
:1. Epidemiology
2. Molecular Genetics
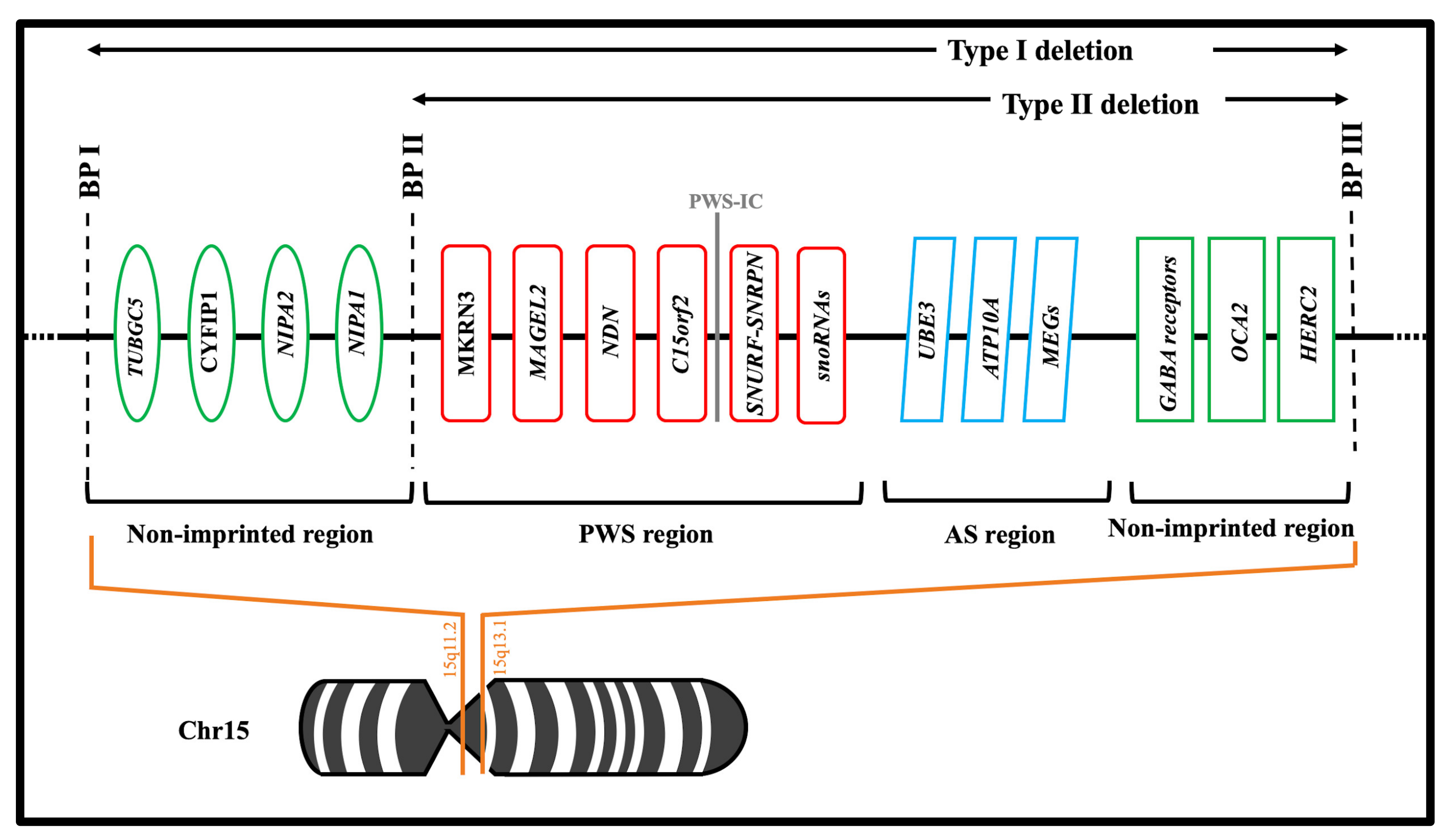
2.1. Chromosomal Abnormalities
2.2. Encoded Genes
3. Hypogonadism in PWS
3.1. Pathophysiology
3.2. Clinical Presentation of Hypogonadism in PWS
3.3. Laboratory Data and Hormones Levels
3.4. Therapy for Hypogonadism in PWS
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Butler, M.G. Prader-Willi syndrome: Current understanding of cause and diagnosis. Am. J. Med Genet. 1990, 35, 319–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurren, B.J.; Flack, N.A. Prader-Willi Syndrome: A spectrum of anatomical and clinical features. Clin. Anat. 2016, 29, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, S.B.; Dykens, E.; Williams, C.A. Prader-Willi and Angelman syndromes: Sister imprinted disorders. Am. J. Med Genet. 2000, 97, 136–146. [Google Scholar] [CrossRef]
- Butler, M.G. Genomic imprinting disorders in humans: A mini-review. J. Assist. Reprod. Genet. 2009, 26, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittington, J.E.; Holland, A.J.; Webb, T.; Butler, J.; Clarke, D.; Boer, H. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J. Med. Genet. 2001, 38, 792–798. [Google Scholar] [CrossRef] [Green Version]
- Einfeld, S.L.; Kavanagh, S.J.; Smith, A.; Evans, E.J.; Tonge, B.J.; Taffe, J. Mortality in Prader-Willi syndrome. Am. J. Ment. Retard. 2006, 111, 193–198. [Google Scholar] [CrossRef]
- Stevenson, D.A.; Anaya, T.M.; Clayton-Smith, J.; Hall, B.D.; Van Allen, M.I.; Zori, R.T.; Zackai, E.H.; Frank, G.; Clericuzio, C.L. Unexpected death and critical illness in Prader-Willi syndrome: Report of ten individuals. Am. J. Med Genet. 2004, 124A, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.M.; Stafford, D.E.J. Prader Willi syndrome: Endocrine updates and new medical therapies. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 56–62. [Google Scholar] [CrossRef]
- Kim, S.J.; Miller, J.L.; Kuipers, P.J.; German, J.R.; Beaudet, A.L.; Sahoo, T.; Driscoll, D.J. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur. J. Hum. Genet. 2012, 20, 283–290. [Google Scholar] [CrossRef]
- Ledbetter, D.H.; Riccardi, V.M.; Airhart, S.D.; Strobel, R.J.; Keenan, B.S.; Crawford, J.D. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N. Engl. J. Med. 1981, 304, 325–329. [Google Scholar] [CrossRef]
- Butler, M.G.; Miller, J.L.; Forster, J.L. Prader-Willi Syndrome—Clinical Genetics, Diagnosis and Treatment Approaches: An Update. Curr. Pediatr. Rev. 2019, 15, 207–244. [Google Scholar] [CrossRef]
- Yamazawa, K.; Ogata, T.; Ferguson-Smith, A.C. Uniparental disomy and human disease: An overview. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Christian, S.L.; Robinson, W.P.; Huang, B.; Mutirangura, A.; Line, M.R.; Nakao, M.; Surti, U.; Chakravarti, A.; Ledbetter, D.H. Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am. J. Hum. Genet. 1995, 57, 40–48. [Google Scholar]
- Irizarry, K.A.; Miller, M.; Freemark, M.; Haqq, A.M. Prader Willi Syndrome: Genetics, Metabolomics, Hormonal Function, and New Approaches to Therapy. Adv. Pediatr. 2016, 63, 47–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, C.K. Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 126–135. [Google Scholar] [CrossRef]
- Butler, M.G.; Fischer, W.; Kibiryeva, N.; Bittel, D.C. Array comparative genomic hybridization (aCGH) analysis in Prader-Willi syndrome. Am. J. Med. Genet. A 2008, 146a, 854–860. [Google Scholar] [CrossRef] [Green Version]
- Anderlid, B.M.; Lundin, J.; Malmgren, H.; Lehtihet, M.; Nordgren, A. Small mosaic deletion encompassing the snoRNAs and SNURF-SNRPN results in an atypical Prader-Willi syndrome phenotype. Am. J. Med. Genet. A 2014, 164, 425–431. [Google Scholar] [CrossRef]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Invest. 2015, 38, 1249–1263. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G.; Bittel, D.C.; Kibiryeva, N.; Talebizadeh, Z.; Thompson, T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics 2004, 113, 565–573. [Google Scholar] [CrossRef]
- Goldstone, A.P. Prader-Willi syndrome: Advances in genetics, pathophysiology and treatment. Trends Endocrinol. Metab. 2004, 15, 12–20. [Google Scholar] [CrossRef]
- Kanber, D.; Giltay, J.; Wieczorek, D.; Zogel, C.; Hochstenbach, R.; Caliebe, A.; Kuechler, A.; Horsthemke, B.; Buiting, K. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader-Willi syndrome. Eur. J. Hum. Genet. 2009, 17, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Bittel, D.C.; Butler, M.G. Prader-Willi syndrome:Clinical genetics, cytogenetics and molecular biology. Expert Rev. Mol. Med. 2005, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Runte, M.; Hüttenhofer, A.; Gross, S.; Kiefmann, M.; Horsthemke, B.; Buiting, K. The IC-SNURF-SNRPN transcript serves as a Host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 2001, 10, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.P.; Gonzalez-Garay, M.L.; Xia, F.; Potocki, L.; Gripp, K.W.; Zhang, B.; Peters, B.A.; McElwain, M.A.; Drmanac, R.; Beaudet, A.L.; et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 2013, 45, 1405–1408. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Wevrick, R. Identification of novel imprinted transcripts in the Prader-Willi syndrome and Angelman syndrome deletion region: Further evidence for regional imprinting control. Am. J. Hum. Genet. 2000, 66, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kozlov, S.; Hernandez, L.; Chamberlain, S.J.; Brannan, C.I.; Stewart, C.L.; Wevrick, R. Expression and imprinting of MAGEL2 suggest a role in Prader-willi syndrome and the homologous murine imprinting phenotype. Hum. Mol. Genet. 2000, 9, 1813–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macedo, D.B.; Abreu, A.P.; Reis, A.C.; Montenegro, L.R.; Dauber, A.; Beneduzzi, D.; Cukier, P.; Silveira, L.F.; Teles, M.G.; Carroll, R.S.; et al. Central precocious puberty that appears to be sporadic caused by paternally inherited mutations in the imprinted gene makorin ring finger 3. J. Clin. Endocrinol. Metab. 2014, 99, E1097–E1103. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.R.; Lee, H.J.; Shim, Y.S.; Kang, M.J.; Yang, S.; Hwang, I.T. Serum Makorin ring finger protein 3 values for predicting Central precocious puberty in girls. Gynecol. Endocrinol. 2019, 35, 732–736. [Google Scholar] [CrossRef]
- Färber, C.; Gross, S.; Neesen, J.; Buiting, K.; Horsthemke, B. Identification of a testis-specific gene (C15orf2) in the Prader-Willi syndrome region on chromosome 15. Genomics 2000, 65, 174–183. [Google Scholar]
- Buiting, K.; Nazlican, H.; Galetzka, D.; Wawrzik, M.; Gross, S.; Horsthemke, B. C15orf2 and a novel noncoding transcript from the Prader-Willi/Angelman syndrome region show monoallelic expression in fetal brain. Genomics 2007, 89, 588–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, N.; Taniura, H.; Niinobe, M.; Takayama, C.; Tominaga-Yoshino, K.; Ogura, A.; Yoshikawa, K. The postmitotic growth suppressor necdin interacts with a calcium-binding protein (NEFA) in neuronal cytoplasm. J. Biol. Chem. 2000, 275, 31674–31681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanella, S.; Barthelemy, M.; Muscatelli, F.; Hilaire, G. Necdin gene, respiratory disturbances and Prader-Willi syndrome. Adv. Exp. Med. Biol. 2008, 605, 159–164. [Google Scholar]
- Rieusset, A.; Schaller, F.; Unmehopa, U.; Matarazzo, V.; Watrin, F.; Linke, M.; Georges, B.; Bischof, J.; Dijkstra, F.; Bloemsma, M.; et al. Stochastic loss of silencing of the imprinted Ndn/NDN allele, in a mouse model and humans with prader-willi syndrome, has functional consequences. PLoS Genet. 2013, 9, e1003752. [Google Scholar] [CrossRef]
- Miller, N.L.; Wevrick, R.; Mellon, P.L. Necdin, a Prader-Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Hum. Mol. Genet. 2009, 18, 248–260. [Google Scholar] [CrossRef] [Green Version]
- Burnett, L.C.; LeDuc, C.A.; Sulsona, C.R.; Paull, D.; Rausch, R.; Eddiry, S.; Carli, J.F.; Morabito, M.V.; Skowronski, A.A.; Hubner, G.; et al. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome. J. Clin. Invest. 2017, 127, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Jay, P.; Rougeulle, C.; Massacrier, A.; Moncla, A.; Mattei, M.G.; Malzac, P.; Roëckel, N.; Taviaux, S.; Lefranc, J.L.; Cau, P.; et al. The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat. Genet. 1997, 17, 357–361. [Google Scholar] [CrossRef]
- Kuslich, C.D.; Kobori, J.A.; Mohapatra, G.; Gregorio-King, C.; Donlon, T.A. Prader-Willi syndrome is caused by disruption of the SNRPN gene. Ame. J. Hum. Genet. 1999, 64, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccaccio, I.; Glatt-Deeley, H.; Watrin, F.; Roëckel, N.; Lalande, M.; Muscatelli, F. The human MAGEL2 gene and its mouse homologue are paternally expressed and mapped to the Prader-Willi region. Hum. Mol. Genet. 1999, 8, 2497–2505. [Google Scholar] [CrossRef] [Green Version]
- Siemensma, E.P.; Van Alfen-van der Velden, A.A.; Otten, B.J.; Laven, J.S.; Hokken-Koelega, A.C. Ovarian function and reproductive hormone levels in girls with Prader-Willi syndrome: A longitudinal study. J. Clin. Endocrinol. Metab. 2012, 97, E1766–E1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogels, A.; Moerman, P.; Frijns, J.P.; Bogaert, G.A. Testicular histology in boys with Prader-Willi syndrome: Fertile or infertile? J. Urol. 2008, 180, 1800–1804. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Matsui, F.; Matsuoka, K.; Iijima, M.; Takeuchi, M.; Ida, S.; Matsumoto, F.; Mizokami, A. Gonadal function and testicular histology in males with Prader-Willi syndrome. Endocrinol. Diabetes Metab. 2019, 2, e00049. [Google Scholar] [CrossRef]
- Eldar-Geva, T.; Hirsch, H.J.; Benarroch, F.; Rubinstein, O.; Gross-Tsur, V. Hypogonadism in females with Prader-Willi syndrome from infancy to adulthood: Variable combinations of a primary gonadal defect and hypothalamic dysfunction. Eur. J. Endocrinol. 2010, 162, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Siemensma, E.P.; de Lind van Wijngaarden, R.F.; Otten, B.J.; de Jong, F.H.; Hokken-Koelega, A.C. Testicular failure in boys with Prader-Willi syndrome: Longitudinal studies of reproductive hormones. J. Clin. Endocrinol. Metab. 2012, 97, E452–E459. [Google Scholar] [CrossRef] [Green Version]
- Eldar-Geva, T.; Hirsch, H.J.; Rabinowitz, R.; Benarroch, F.; Rubinstein, O.; Gross-Tsur, V. Primary ovarian dysfunction contributes to the hypogonadism in women with Prader-Willi Syndrome. Horm. Res. 2009, 72, 153–159. [Google Scholar] [CrossRef]
- Hirsch, H.J.; Eldar-Geva, T.; Bennaroch, F.; Pollak, Y.; Gross-Tsur, V. Sexual dichotomy of gonadal function in Prader-Willi syndrome from early infancy through the fourth decade. Hum. Reprod. 2015, 30, 2587–2596. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G.; Manzardo, A.M.; Forster, J.L. Prader-Willi Syndrome: Clinical Genetics and Diagnostic Aspects with Treatment Approaches. Curr. Pediatr. Rev. 2016, 12, 136–166. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.J.; Eldar-Geva, T.; Erlichman, M.; Pollak, Y.; Gross-Tsur, V. Characterization of minipuberty in infants with Prader-Willi syndrome. Horm. Res. Paediatr. 2014, 82, 230–237. [Google Scholar] [CrossRef]
- Gross-Tsur, V.; Hirsch, H.J.; Benarroch, F.; Eldar-Geva, T. The FSH-inhibin axis in prader-willi syndrome: Heterogeneity of gonadal dysfunction. Reprod. Biol. Endocrinol. 2012, 10, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elena, G.; Bruna, C.; Benedetta, M.; Stefania, D.C.; Giuseppe, C. Prader-Willi syndrome: Clinical aspects. J. Obes. 2012, 2012, 473941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillion, M.; Deal, C.L.; Van Vliet, G. Normal minipuberty of infancy in boys with Prader-Willi syndrome. J. Pediatr. 2006, 149, 874–876. [Google Scholar] [CrossRef]
- Pacilli, M.; Heloury, Y.; O’Brien, M.; Lionti, T.; Rowell, M.; Hutson, J. Orchidopexy in children with Prader-Willi syndrome: Results of a long-term follow-up study. J. Pediatr. Urol. 2018, 14, 63.e1–63.e6. [Google Scholar] [CrossRef] [PubMed]
- Emerick, J.E.; Vogt, K.S. Endocrine manifestations and management of Prader-Willi syndrome. Int. J. Pediatr. Endocrinol. 2013, 2013, 14. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.G.; Radaeli, R.F.; Silva, M.M.; Romero, C.M.; Carrilho, A.J.; Bessa, D.; Macedo, D.B.; Oliveira, M.L.; Latronico, A.C.; Mazzuco, T.L. A boy with Prader-Willi syndrome: Unmasking precocious puberty during growth hormone replacement therapy. Arch. Endocrinol. Metabol. 2016, 60, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Monai, E.; Johansen, A.; Clasen-Linde, E.; Rajpert-De Meyts, E.; Skakkebæk, N.E.; Main, K.M.; Jørgensen, A.; Jensen, R.B. Central Precocious Puberty in Two Boys with Prader-Willi Syndrome on Growth Hormone Treatment. AACE Clin. Case Rep. 2019, 5, e352–e356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crinò, A.; Schiaffini, R.; Ciampalini, P.; Spera, S.; Beccaria, L.; Benzi, F.; Bosio, L.; Corrias, A.; Gargantini, L.; Salvatoni, A.; et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur. J. Pediatr. 2003, 162, 327–333. [Google Scholar]
- Lee, H.S.; Hwang, J.S. Central precocious puberty in a girl with Prader-Willi syndrome. J. Pediatr. Endocrinol. Metab. 2013, 26, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Pusz, E.R.; Rotenstein, D. Treatment of precocious puberty in a female with Prader-Willi syndrome. J. Pediatr. Endocrinol. Metab. 2008, 21, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; Formoso, G.; Pugliese, G.; Ruggeri, R.M.; Scarano, E.; Colao, A. Prader- Willi syndrome: An uptodate on endocrine and metabolic complications. Rev. Endocr. Metab. Disord. 2019, 20, 239–250. [Google Scholar] [CrossRef]
- Sinnema, M.; Maaskant, M.A.; van Schrojenstein Lantman-de Valk, H.M.; van Nieuwpoort, I.C.; Drent, M.L.; Curfs, L.M.; Schrander-Stumpel, C.T. Physical health problems in adults with Prader-Willi syndrome. Am. J. Med. Genet. A 2011, 155, 2112–2124. [Google Scholar] [CrossRef]
- Valdes-Socin, H.; Rubio Almanza, M.; Tomé Fernández-Ladreda, M.; Debray, F.G.; Bours, V.; Beckers, A. Reproduction, smell, and neurodevelopmental disorders: Genetic defects in different hypogonadotropic hypogonadal syndromes. Front. Endocrinol. 2014, 5, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heksch, R.; Kamboj, M.; Anglin, K.; Obrynba, K. Review of Prader-Willi syndrome: The endocrine approach. Transl. Pediatr. 2017, 6, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Eldar-Geva, T.; Hirsch, H.J.; Pollak, Y.; Benarroch, F.; Gross-Tsur, V. Management of hypogonadism in adolescent girls and adult women with Prader-Willi syndrome. Am. J. Med. Genet. A 2013, 161, 3030–3034. [Google Scholar] [CrossRef] [PubMed]
- Unanue, N.; Bazaes, R.; Iñiguez, G.; Cortés, F.; Avila, A.; Mericq, V. Adrenarche in Prader-Willi syndrome appears not related to insulin sensitivity and serum adiponectin. Horm. Res. 2007, 67, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Prasad, H.K.; Khadilkar, V.V.; Jahagirdar, R.; Khadilkar, A.V.; Lalwani, S.K. Evaluation of GnRH analogue testing in diagnosis and management of children with pubertal disorders. Indian J. Endocrinol. Metab. 2012, 16, 400–405. [Google Scholar] [PubMed]
- Kimonis, V.E.; Tamura, R.; Gold, J.A.; Patel, N.; Surampalli, A.; Manazir, J.; Miller, J.L.; Roof, E.; Dykens, E.; Butler, M.G.; et al. Early Diagnosis in Prader-Willi Syndrome Reduces Obesity and Associated Co-Morbidities. Genes 2019, 10, 898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, C.; Diene, G.; Molinas, C.; Bieth, E.; Casper, C.; Tauber, M. Early diagnosis and care is achieved but should be improved in infants with Prader-Willi syndrome. Orphanet J. Rare Dis. 2017, 12, 118. [Google Scholar] [CrossRef] [Green Version]
- Bakker, N.E.; Wolffenbuttel, K.P.; Looijenga, L.H.; Hokken-Koelega, A.C. Testes in infants with Prader-Willi syndrome: Human chorionic gonadotropin treatment, surgery and histology. J. Urol. 2015, 193, 291–298. [Google Scholar] [CrossRef]
- Eiholzer, U.; Grieser, J.; Schlumpf, M.; l’Allemand, D. Clinical effects of treatment for hypogonadism in male adolescents with Prader-Labhart-Willi syndrome. Horm. Res. Paediatr. 2007, 68, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Carrel, A.L.; Myers, S.E.; Whitman, B.Y.; Allen, D.B. Benefits of long-term GH therapy in Prader-Willi syndrome: A 4-year study. J. Clin. Endocrinol. Metab. 2002, 87, 1581–1585. [Google Scholar] [CrossRef]
- Goldstone, A.P.; Holland, A.J.; Hauffa, B.P.; Hokken-Koelega, A.C.; Tauber, M. Recommendations for the diagnosis and management of Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4183–4197. [Google Scholar] [CrossRef] [PubMed]
- Gross-Tsur, V.; Eldar-Geva, T.; Benarroch, F.; Rubinstein, O.; Hirsch, H.J. Body image and sexual interests in adolescents and young adults with Prader-Willi syndrome. J. Pediatr. Endocrinol. Metab. 2011, 24, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Kido, Y.; Sakazume, S.; Abe, Y.; Oto, Y.; Itabashi, H.; Shiraishi, M.; Yoshino, A.; Tanaka, Y.; Obata, K.; Murakami, N.; et al. Testosterone replacement therapy to improve secondary sexual characteristics and body composition without adverse behavioral problems in adult male patients with Prader-Willi syndrome: An observational study. Am. J. Med. Genet. A 2013, 161, 2167–2173. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Function | Potential Role in PWS |
|---|---|---|
| MKRN3 | Hormonal regulation, familial central precocious puberty (CPP) [28,29] | ↓ Hypothalamic GnRH secretion [29] |
| MAGEL2 | Brain structure development, human reproduction, fertility [25] | Eating disorder (hyperphagia) [26,27] |
| NDN | GnRH neurons development [34] | Hypogonadotropic hypogonadal phenotype [2,32] |
| SNORD116 | ProConvertase 1 (PC) activation and regulation of some hormonal pathways [36] | hormonal changes: -↓GHRH → short stature [36] -↓ProGnRH → Hypogonadism [36] -↓ProGrelin → Hypergrelinemia [36] -↓Proinsulin → Hypoinsulinemia and DM2 [36] -↓ Proopiomelanocortin → Hypocortisolism [36] -↓ProTRH → Hypothyroidism [36] |
| C15orf2 | Regulation of several spermatid-specific genes [31] | Impaired spermatogenesis and male fertility [31] |
| Male | Female |
|---|---|
| Scrotal Hypoplasia [56] | Hypoplasia of Labia Minora and/or clitoris [48] |
| Cryptorchidism [51] | Delayed spontaneous puberal development with menarche [48,59] |
| Low penile length [59] | Amenorrhea or oligomenorrhea [60,61] |
| Low serum levels of Testosterone and Inhibin B [59] | Low serum levels of Estradiol and Inhibin B [53] |
| Tanner stage 3–4 [48] | Tanner stage 3–4 [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napolitano, L.; Barone, B.; Morra, S.; Celentano, G.; La Rocca, R.; Capece, M.; Morgera, V.; Turco, C.; Caputo, V.F.; Spena, G.; et al. Hypogonadism in Patients with Prader Willi Syndrome: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 1993. https://doi.org/10.3390/ijms22041993
Napolitano L, Barone B, Morra S, Celentano G, La Rocca R, Capece M, Morgera V, Turco C, Caputo VF, Spena G, et al. Hypogonadism in Patients with Prader Willi Syndrome: A Narrative Review. International Journal of Molecular Sciences. 2021; 22(4):1993. https://doi.org/10.3390/ijms22041993
Chicago/Turabian StyleNapolitano, Luigi, Biagio Barone, Simone Morra, Giuseppe Celentano, Roberto La Rocca, Marco Capece, Vincenzo Morgera, Carmine Turco, Vincenzo Francesco Caputo, Gianluca Spena, and et al. 2021. "Hypogonadism in Patients with Prader Willi Syndrome: A Narrative Review" International Journal of Molecular Sciences 22, no. 4: 1993. https://doi.org/10.3390/ijms22041993