
Innate Receptor Activation Patterns Involving TLR and NLR Synergisms in COVID-19, ALI/ARDS and Sepsis Cytokine Storms: A Review and Model Making Novel Predictions and Therapeutic Suggestions


Abstract
:
1. Introduction: The Problem of What Causes Cytokine Overproduction Syndromes
2. Innate Immune System Receptor Activation in Cytokine Storms
2.1. The Presence of Multiple Concurrent Infections in Cytokine Release Syndromes
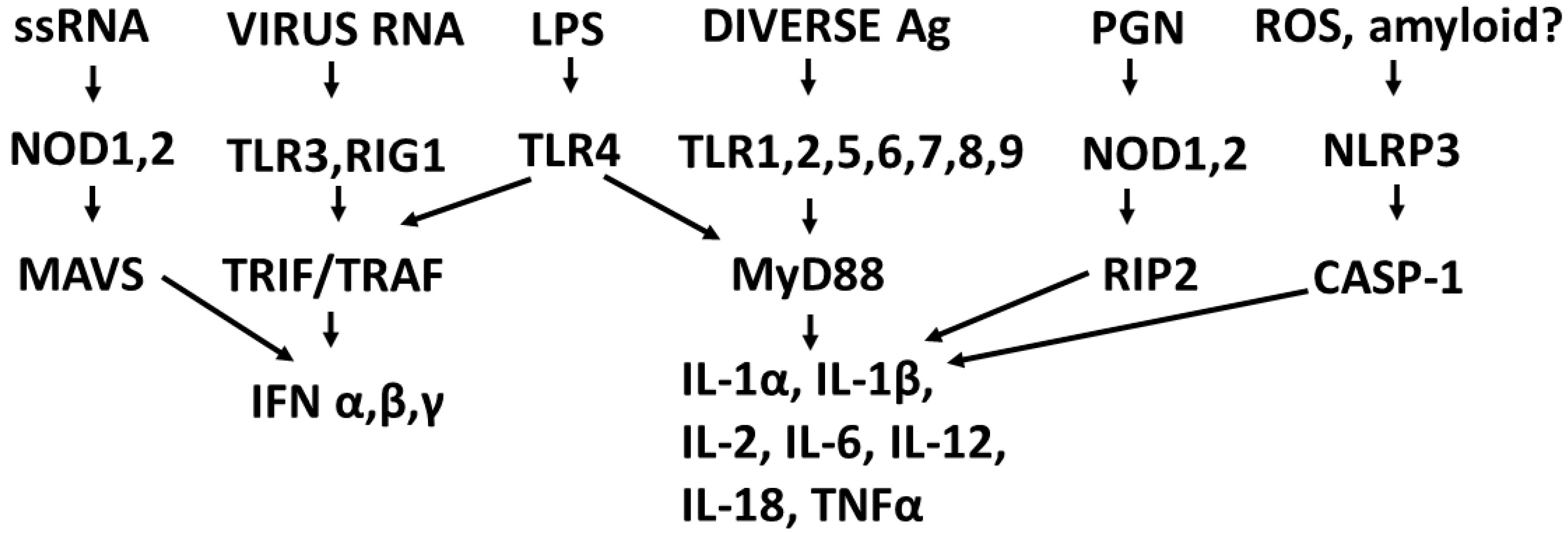
2.2. Overview of Receptor Regulation of Cytokine Production
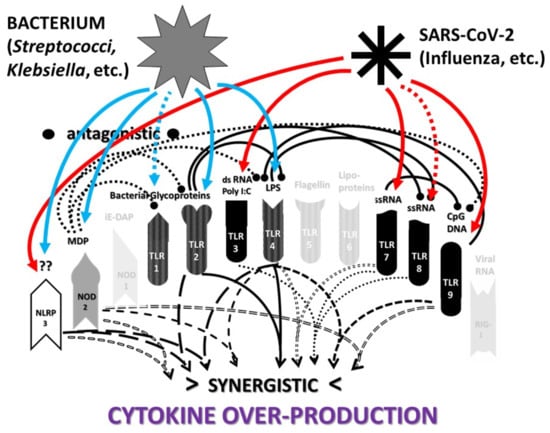
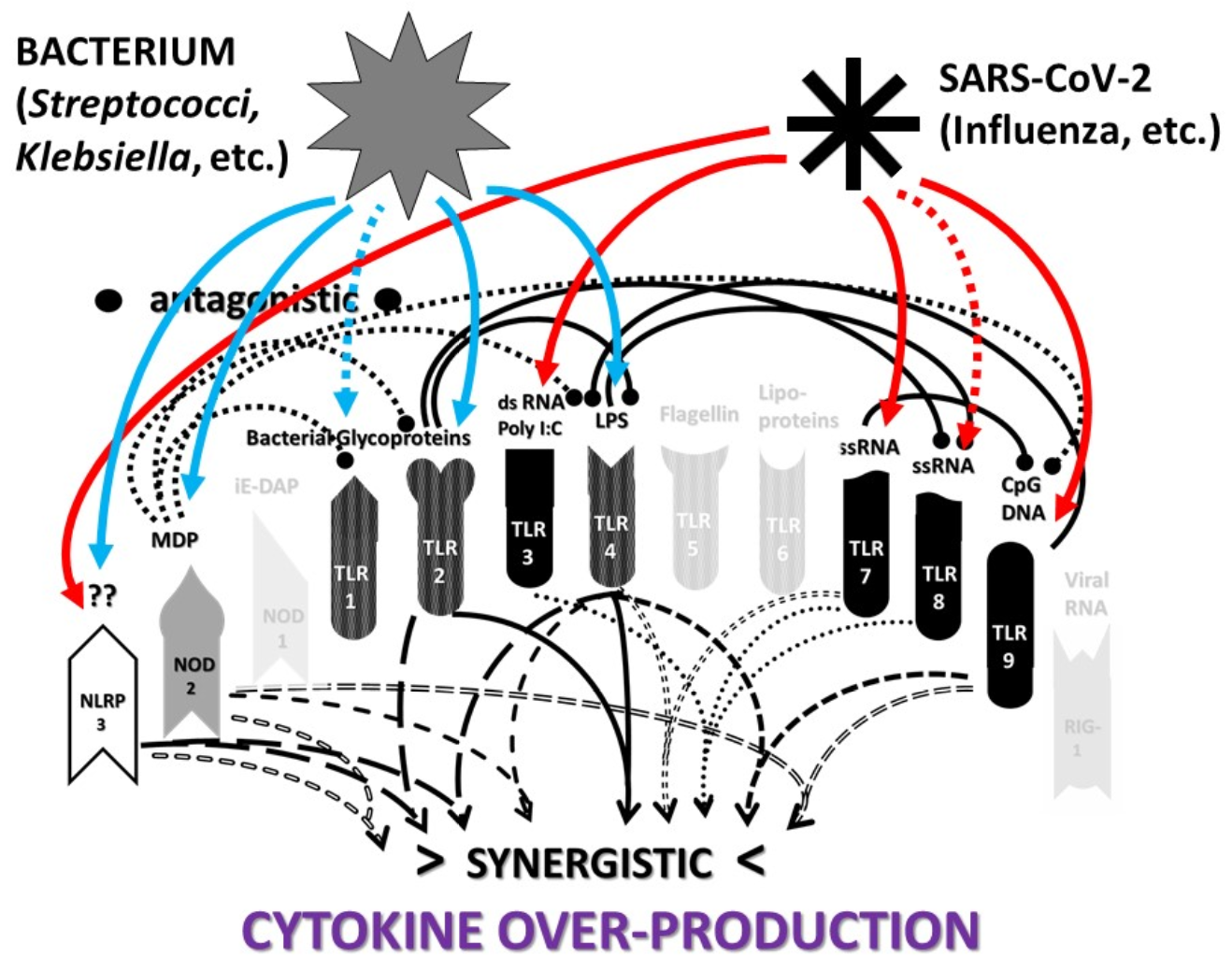
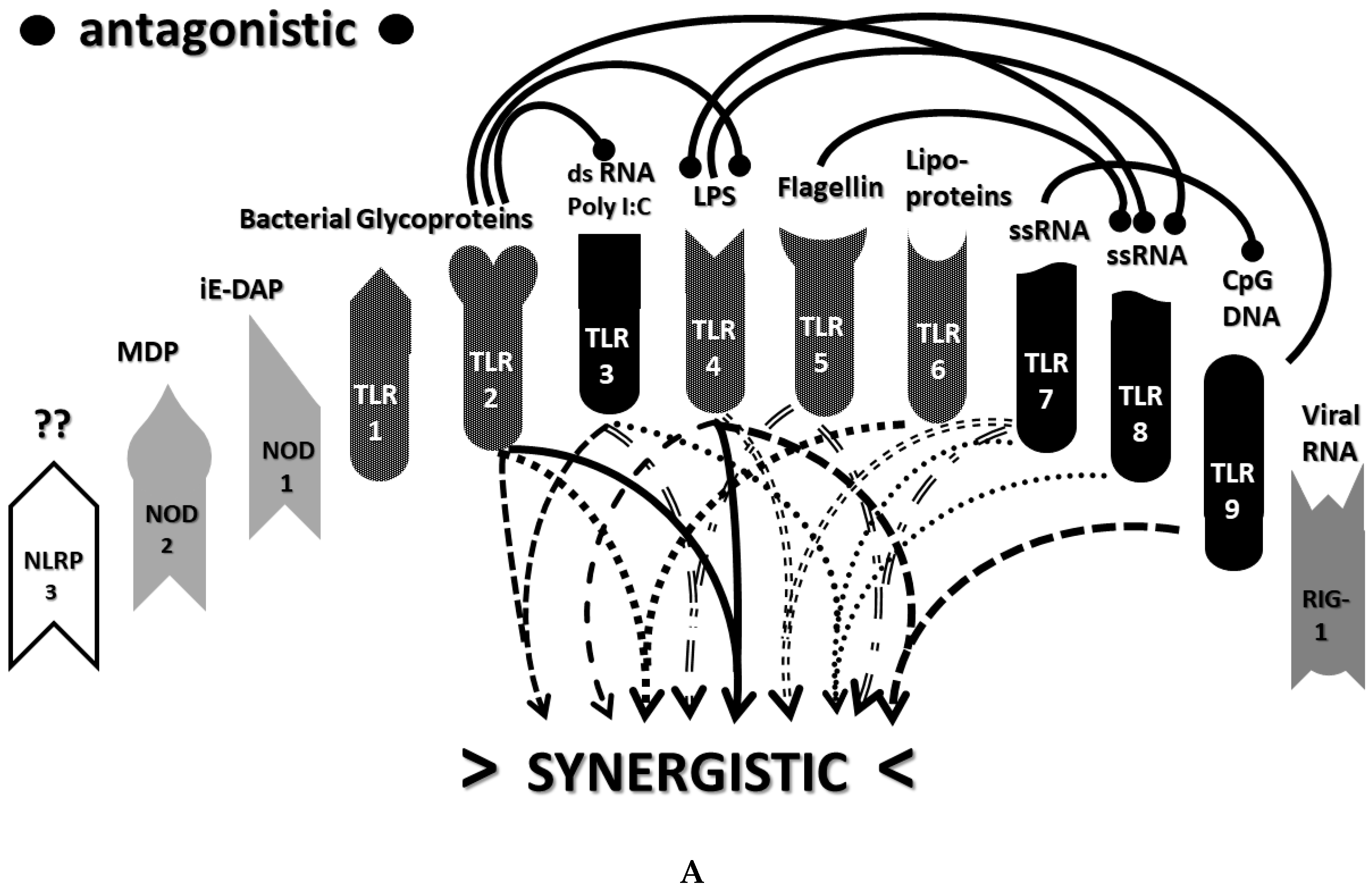
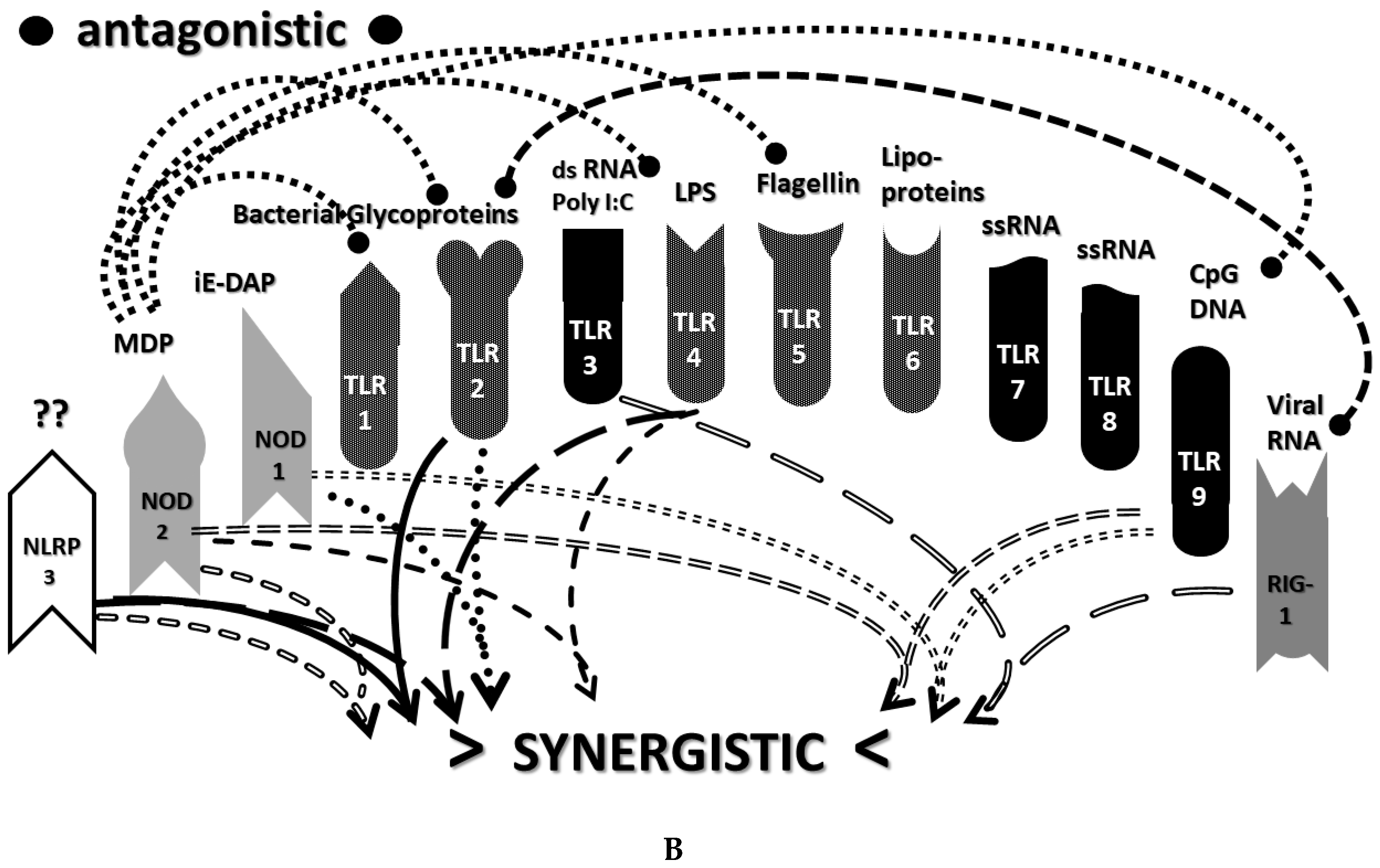
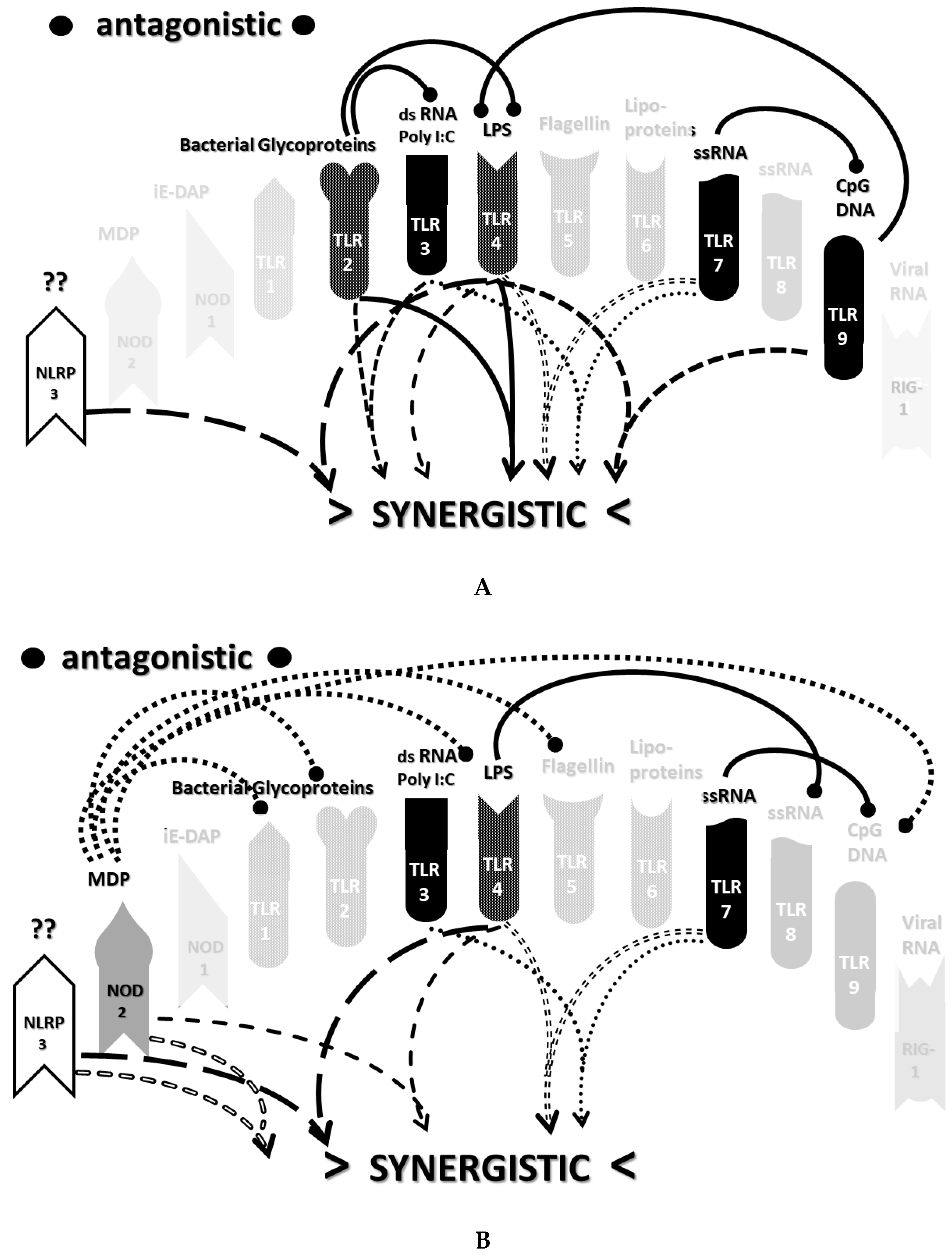
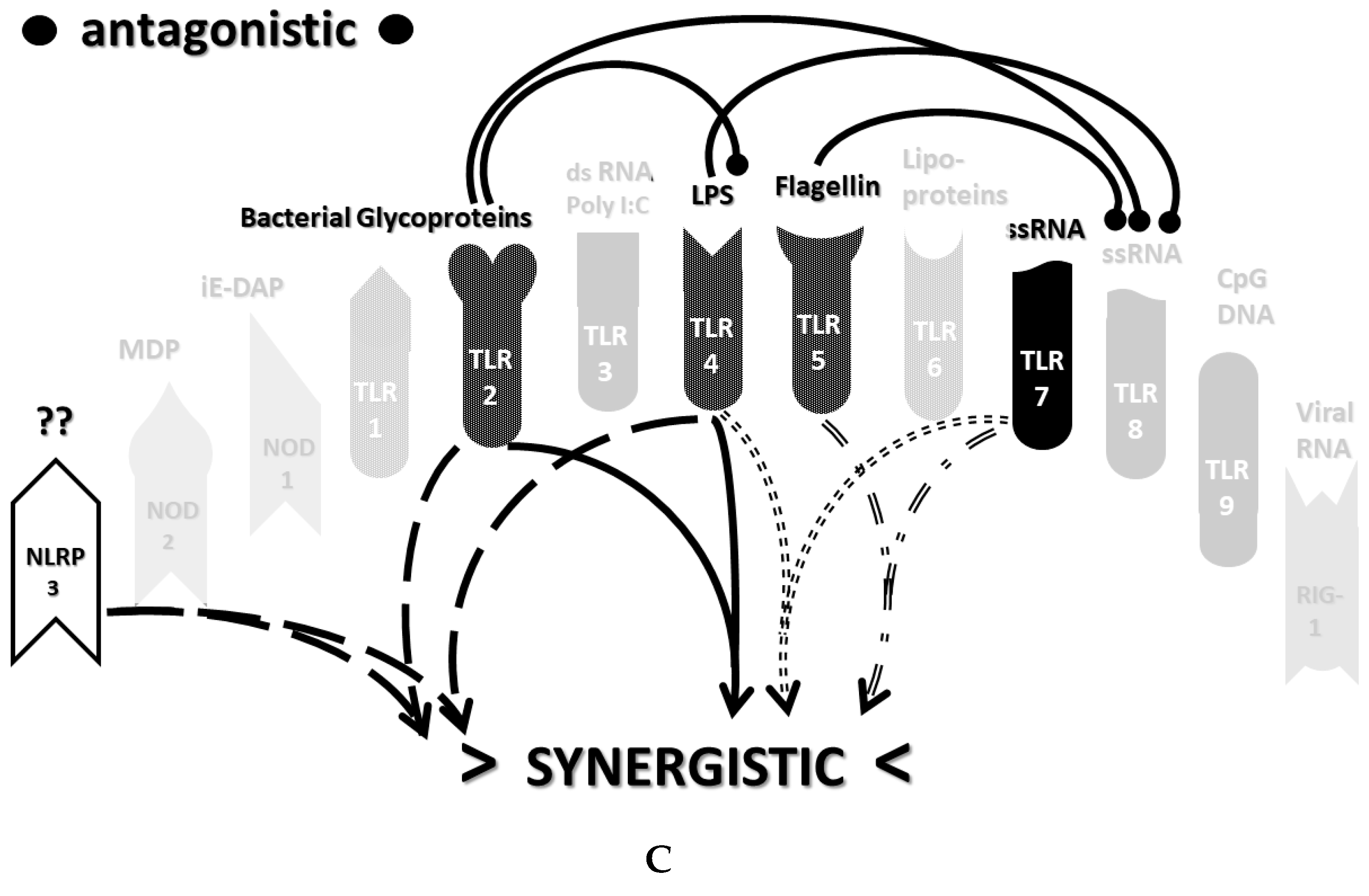
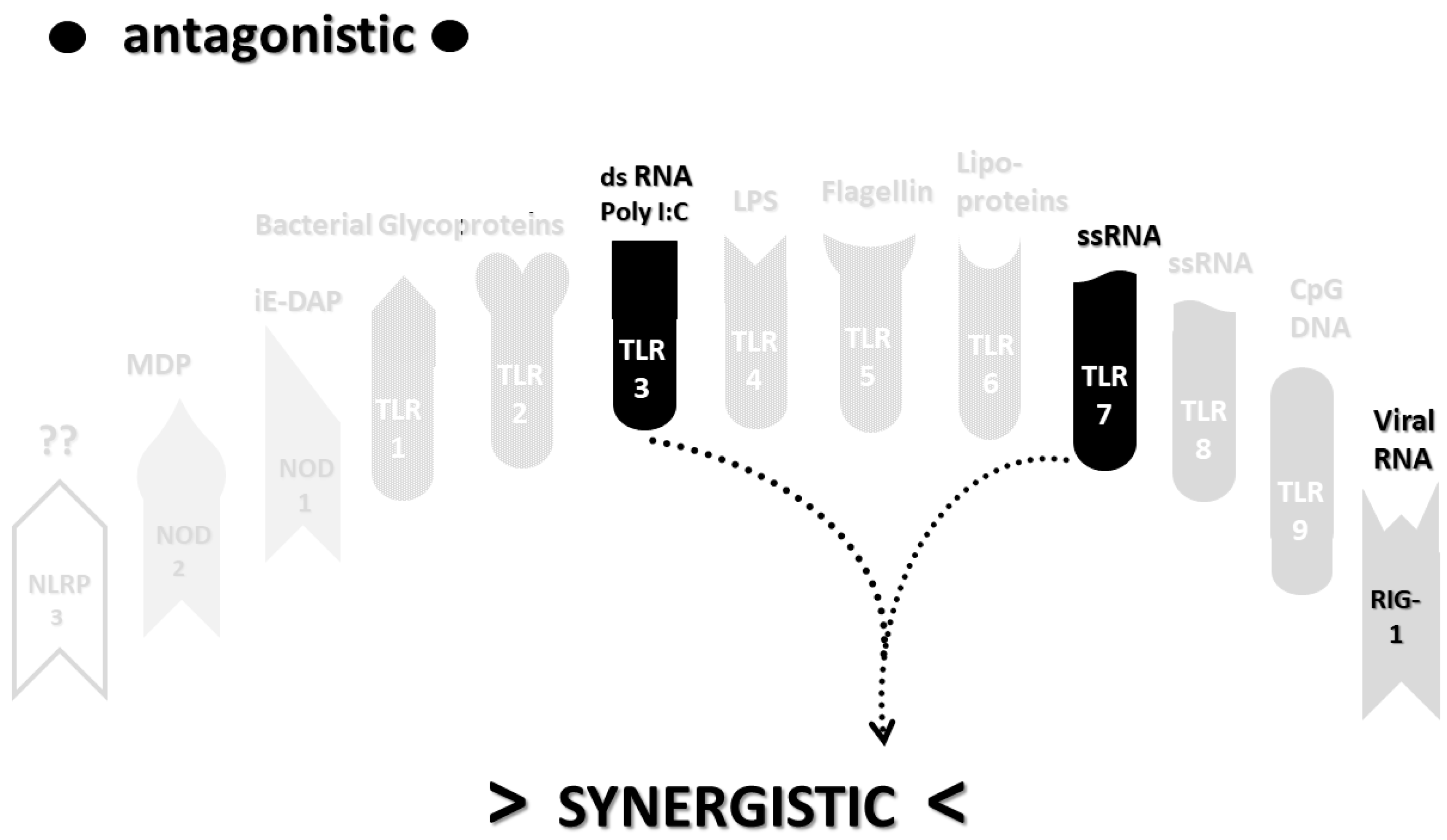
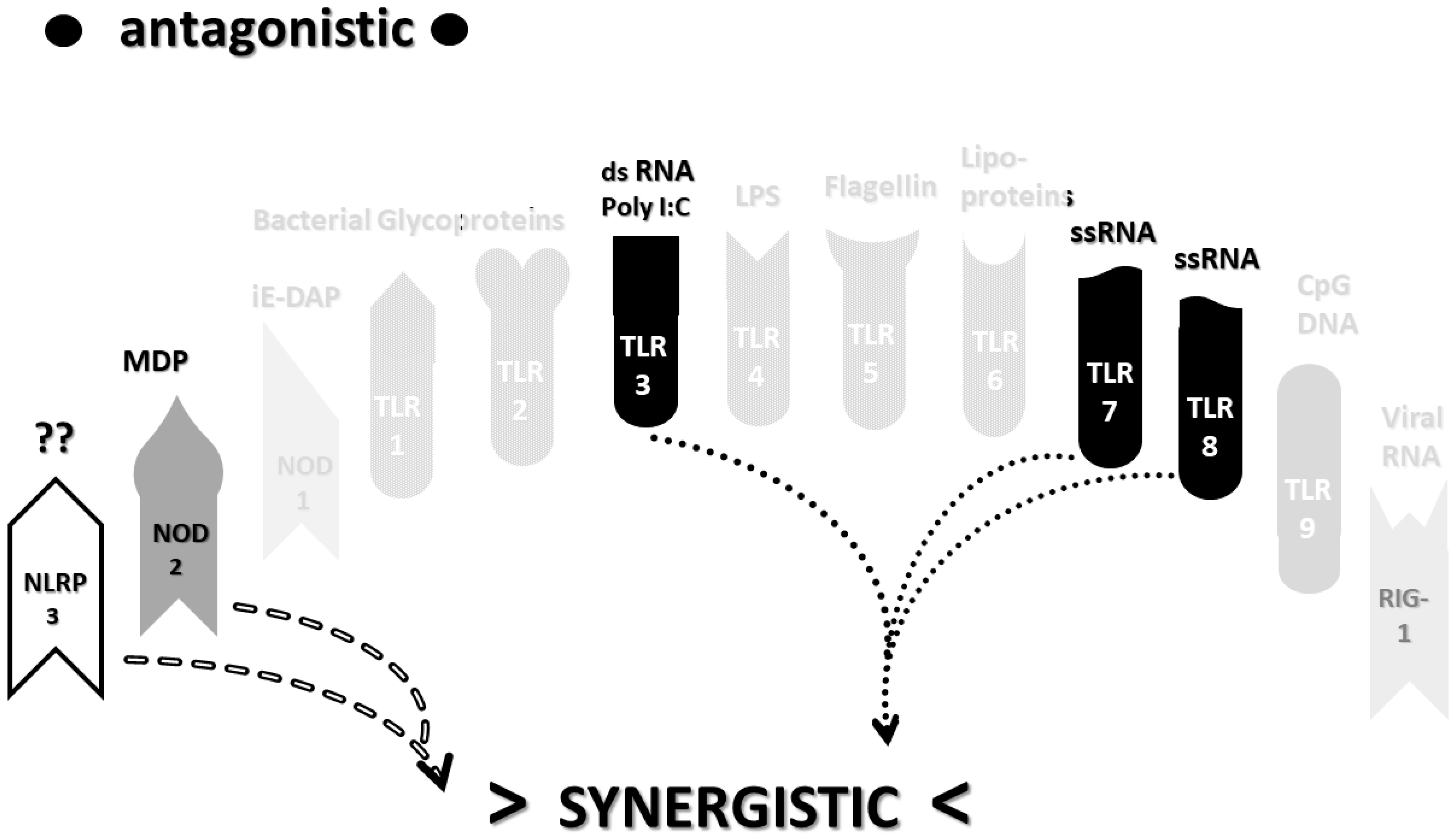
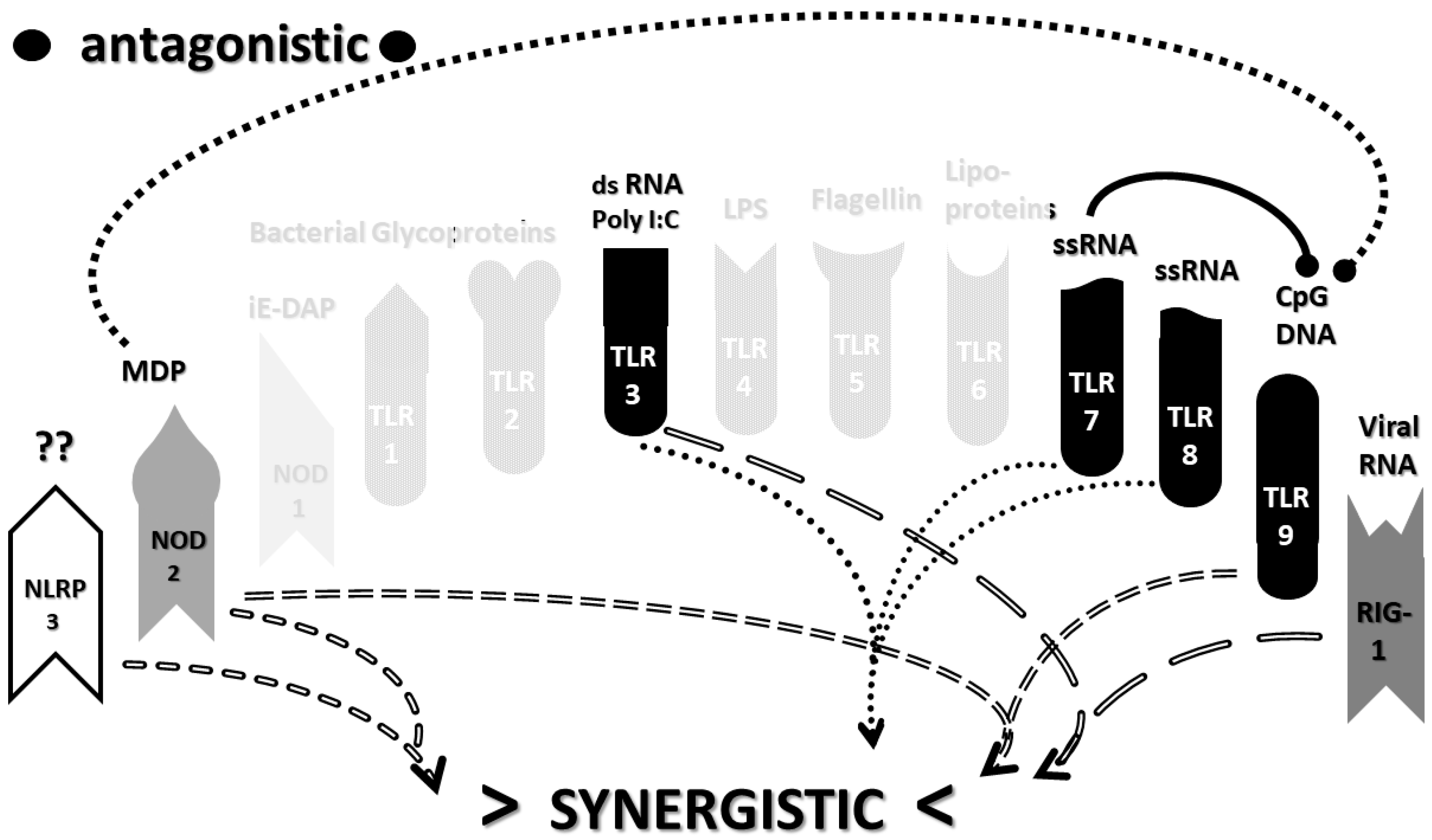
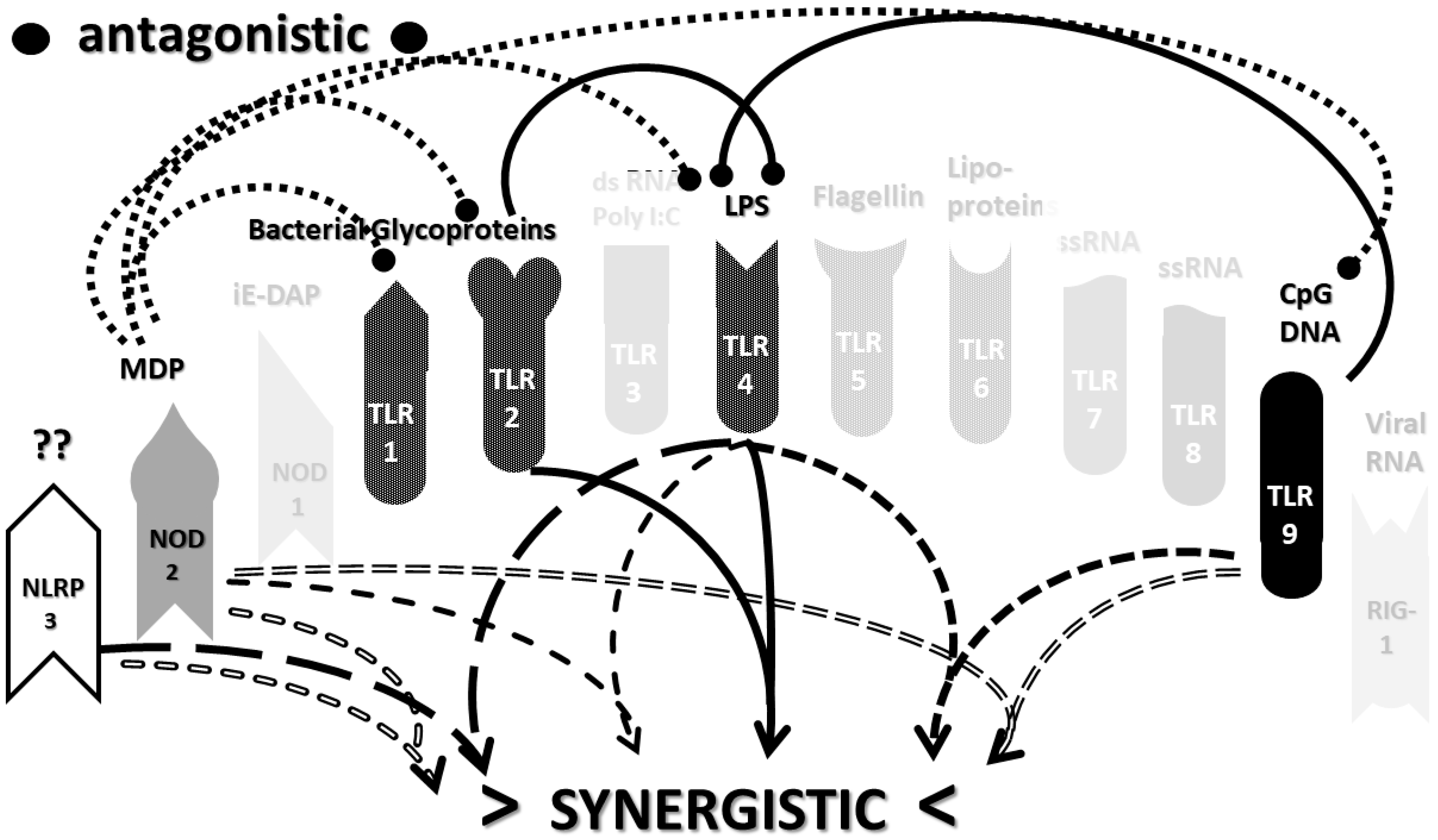
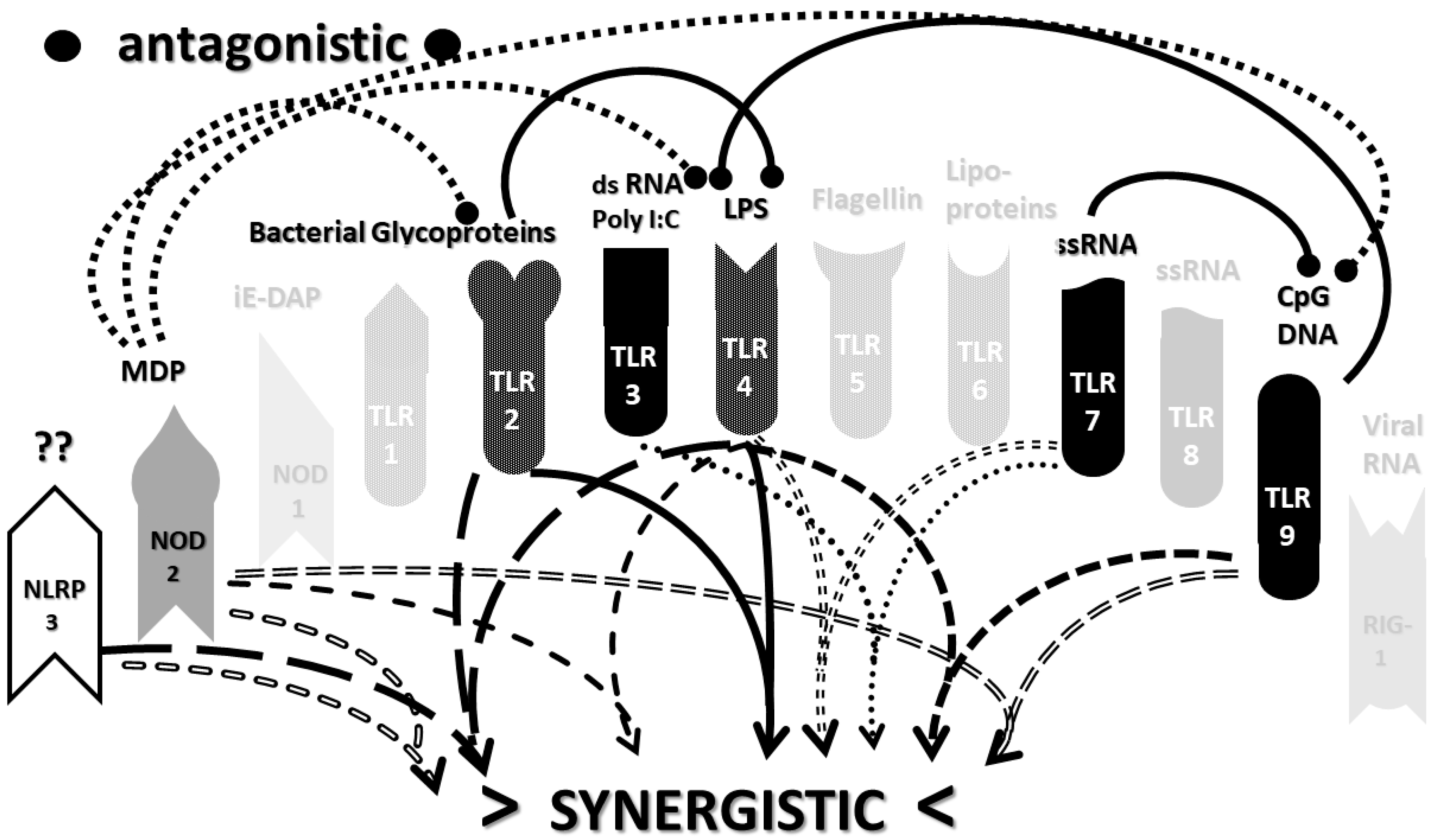
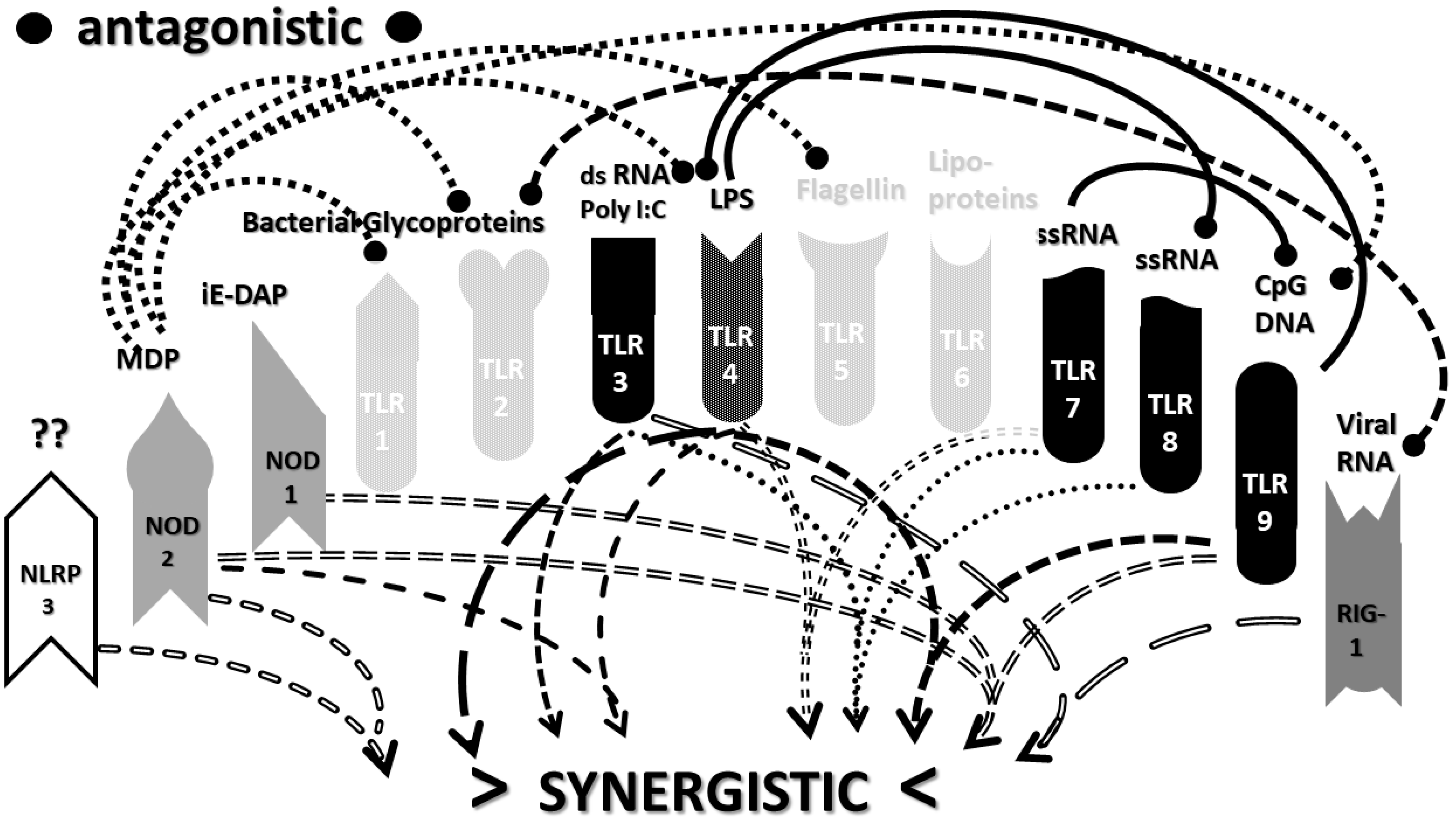
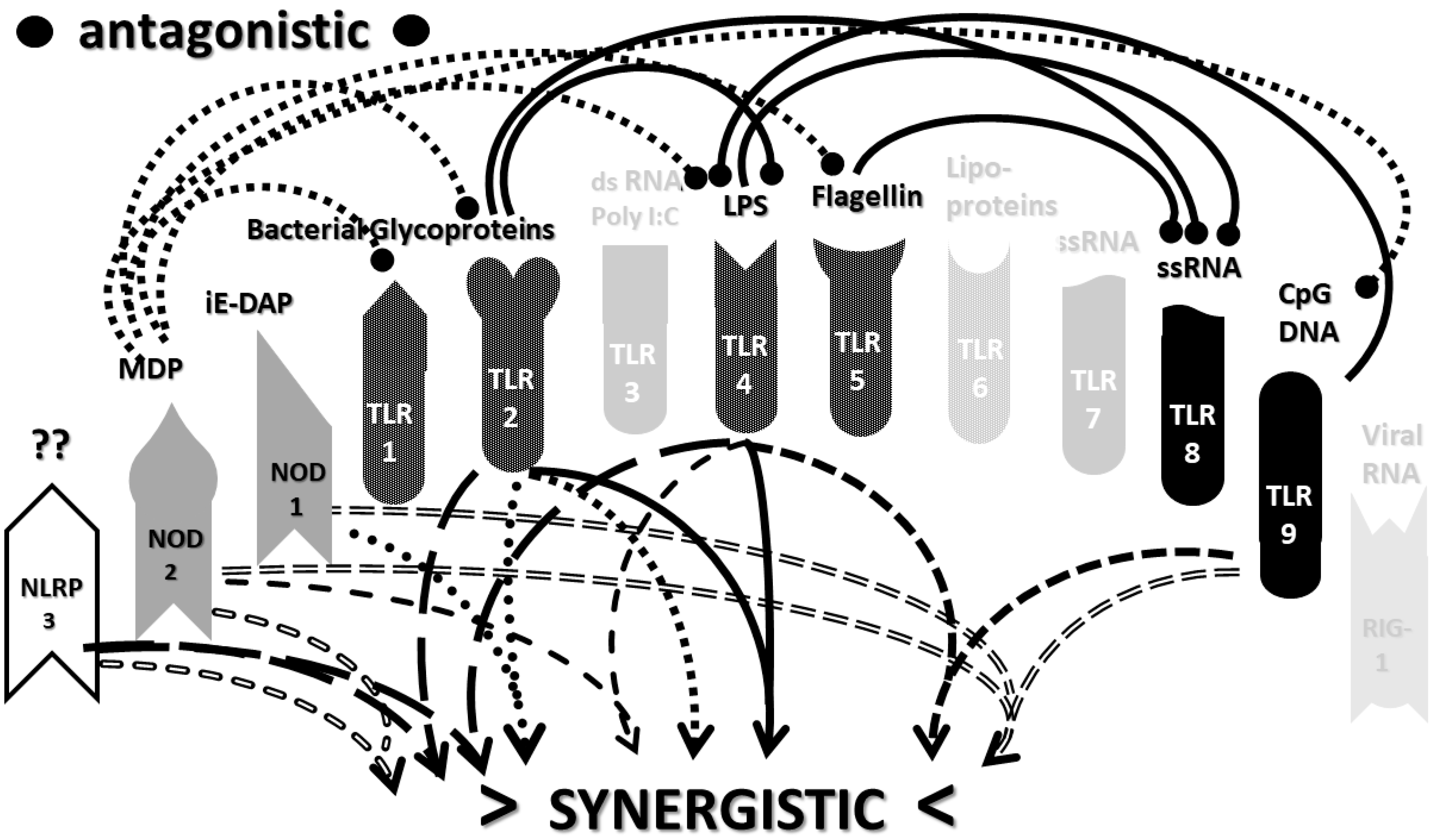
2.3. Synergistic and Antagonistic Receptor Interactions within the Innate Immune System
2.4. A Hypothesis Concerning the Mechanism Producing Cytokine Storms
2.5. Methods for Reviewing Literature Relevant for Comparing Alternative Hypotheses
2.6. Synergistic and Antagonistic Receptor Activation Networks in Severe COVID-19, ALI/ARDS and Sepsis
2.7. Varied Receptor Activation by PAMP Produced by Different Pathogens
2.8. Innate Receptor Activation by Bacterial and Fungal Infections Associated with Coronavirus, Influenza and Other ALI/ARDS Syndromes
2.9. Do Combinations of Viruses and Bacteria Explain Innate Receptor Activation Patterns in Severe COVID-19 and ALI/ARDS?
2.10. Role of DAMPs in Driving COVID-19 and Other Cytokine Release Syndromes
3. Discussion
3.1. Summary of the Synergistic Activation of TLR and NLR in Cytokine Storm Syndromes
3.2. The Role of Pathogen Synergisms in COVID-19
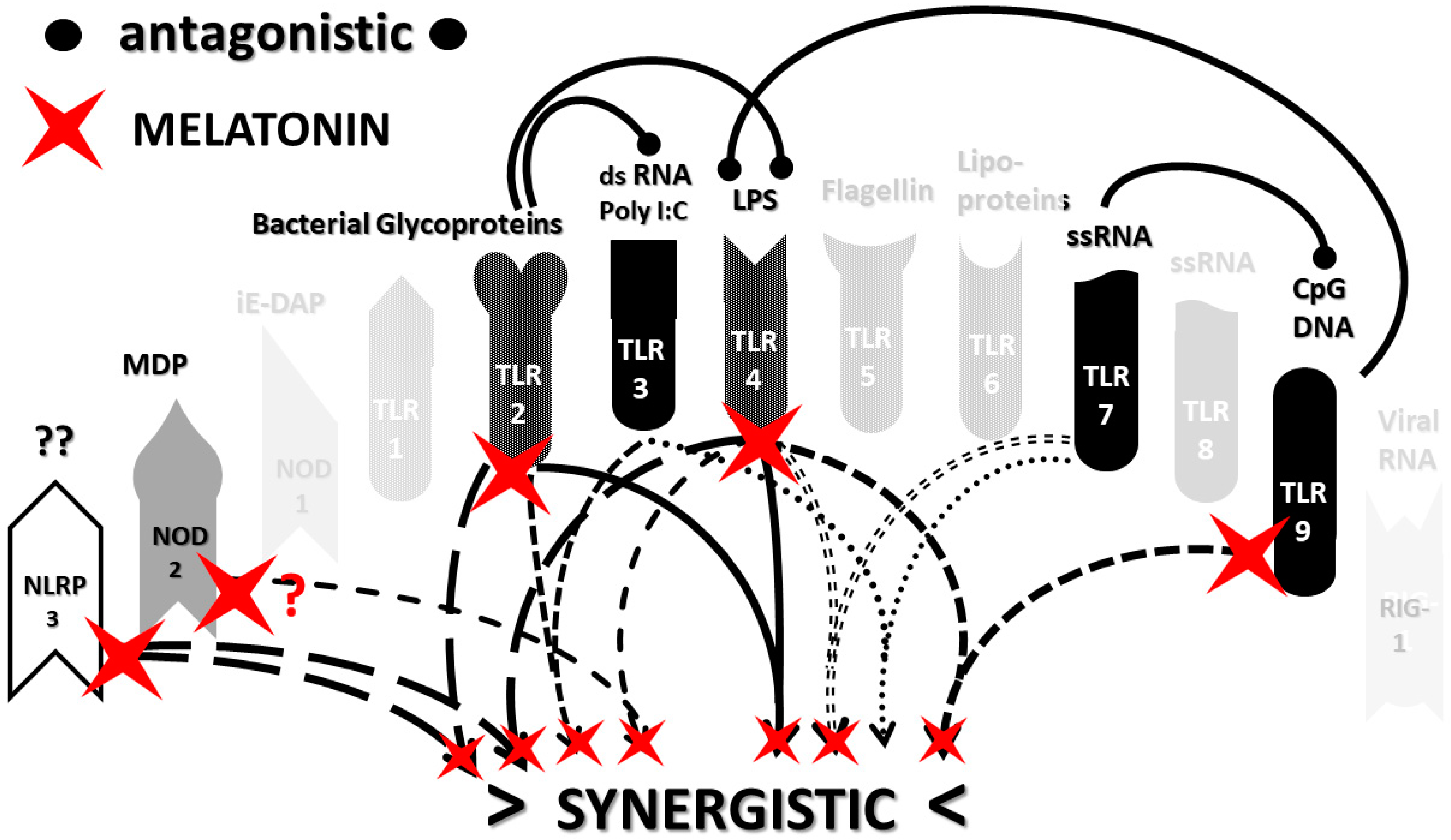
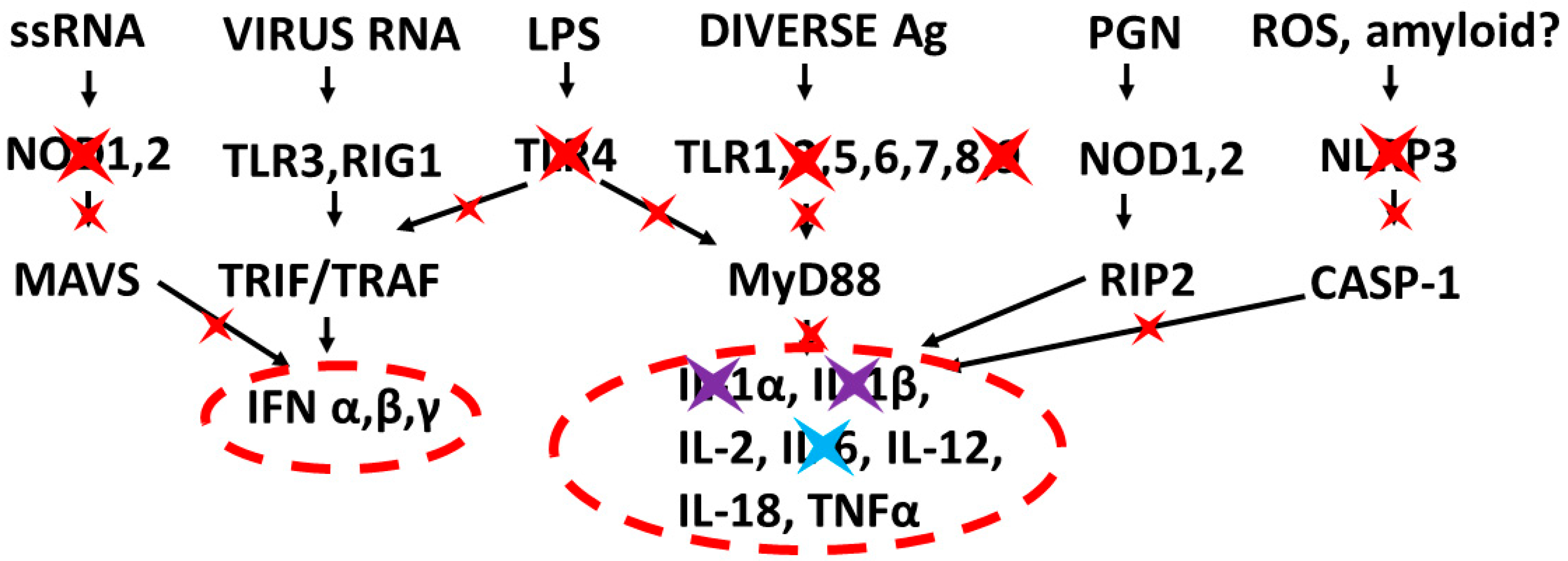
3.3. Implications of Innate Receptor Activation Profiles for Tretment of COVID-19 and Other Cytokine Release Syndromes
3.4. Directions for Future Research
3.5. Limitations and Sources of Bias in This Study
4. Conclusions
Funding
Conflicts of Interest
References
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef]
- Moore, B.J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulchandani, R.; Lyngdoh, T.; Kakkar, A.K. Deciphering the COVID-19 cytokine storm: Systematic review and meta-analysis. Eur. J. Clin. Investig. 2021, 51, e13429. [Google Scholar] [CrossRef]
- Notz, Q.; Schmalzing, M.; Wedekink, F.; Schlesinger, T.; Gernert, M.; Herrmann, J.; Sorger, L.; Weismann, D.; Schmid, B.; Sitter, M.; et al. Pro- and Anti-Inflammatory Responses in Severe COVID-19-Induced Acute Respiratory Distress Syndrome—An Observational Pilot Study. Front. Immunol. 2020, 11, 581338. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, X. Acute respiratory failure in COVID-19: Is it “typical” ARDS? Crit. Care 2020, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ng, O.T.; Marimuthu, K.; Koh, V. SARS-CoV-2 seroprevalence and transmission risk factors among high-risk close contacts: A retrospective cohort study. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- King, J.A.; Whitten, T.A.; Bakal, J.A.; McAlister, F.A. Symptoms associated with a positive result for a swab for SARS-CoV-2 infection among children in Alberta. Can. Med. Assoc. J. 2021, 193, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Hue, S.; Beldi-Ferchiou, A.; Bendib, I.; Surenaud, M.; Fourati, S.; Frapard, T.; Rivoal, S.; Razazi, K.; Carteaux, G.; Delfau-Larue, M.-H.; et al. Uncontrolled Innate and Impaired Adaptive Immune Responses in Patients with COVID-19 Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 202, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-Y.; Hong, S.-B. Sepsis and Acute Respiratory Distress Syndrome: Recent Update. Tuberc. Respir. Dis. 2016, 79, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-Z.J.; Thomas, P.G. New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 2017, 39, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Matthay, M.A.; Calfee, C.S. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern. Med. 2020, 180, 1152. [Google Scholar] [CrossRef] [PubMed]
- Mudd, P.A.; Crawford, J.C.; Turner, J.S.; Souquette, A.; Reynolds, D.; Bender, D.; Bosanquet, J.P.; Anand, N.J.; Striker, D.A.; Martin, R.S.; et al. Distinct inflammatory profiles distinguish COVID-19 from influenza with limited contributions from cytokine storm. Sci. Adv. 2020, 6, eabe3024. [Google Scholar] [CrossRef] [PubMed]
- Blot, M.; Bour, J.-B.; Quenot, J.P.; Bourredjem, A.; Nguyen, M.; Guy, J.; Monier, S.; Georges, M.; Large, A.; Dargent, A.; et al. The dysregulated innate immune response in severe COVID-19 pneumonia that could drive poorer outcome. J. Transl. Med. 2020, 18, 1–14. [Google Scholar] [CrossRef]
- Kox, M.; Waalders, N.J.B.; Kooistra, E.J.; Gerretsen, J.; Pickkers, P. Cytokine Levels in Critically Ill Patients With COVID-19 and Other Conditions. JAMA 2020, 324, 1565. [Google Scholar] [CrossRef] [PubMed]
- Kox, M.; Frenzel, T.; Schouten, J.; Van De Veerdonk, F.L.; Koenen, H.J.P.M.; Pickkers, P. COVID-19 patients exhibit less pronounced immune suppression compared with bacterial septic shock patients. Crit. Care 2020, 24, 263. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef] [PubMed]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Balamayooran, T.; Balamayooran, G.; Jeyaseelan, S. Review: Toll-like receptors and NOD-like receptors in pulmonary antibacterial immunity. Innate Immun. 2010, 16, 201–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Predominant role of bacterial pneumonia as a cause of death in pan-demic influenza: Implications for pandemic influenza preparedness. J. Infect. Dis. 2008, 198, 962. [Google Scholar] [CrossRef] [PubMed]
- Stefani, C. Prevention of Cervical Cancer in Women: Human Papillomavirus DNA Testing in Atypical Pap Smears. J. Virol. Antivir. Res. 2013, 2. [Google Scholar] [CrossRef]
- Buttenschoen, K.; Kornmann, M.; Berger, D.; Leder, G.; Beger, H.G.; Vasilescu, C. Endotoxemia and endotoxin tolerance in patients with ARDS. Langenbeck’s Arch. Surg. 2008, 393, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Bai, C.; Wang, X. The value of the lipopolysaccharide-induced acute lung injury model in respiratory medicine. Expert Rev. Respir. Med. 2010, 4, 773–783. [Google Scholar] [CrossRef]
- Sirivongrangson, P.; Kulvichit, W.; Payungporn, S.; Pisitkun, T.; Chindamporn, A.; Peerapornratana, S.; Pisitkun, P.; Chitcharoen, S.; Sawaswong, V.; Worasilchai, N.; et al. Endotoxemia and circulating bacteriome in severe COVID-19 patients. Intensiv. Care Med. Exp. 2020, 8, 72. [Google Scholar] [CrossRef]
- Gonçalves, J.M.F.; Pérez, J.M.H.; Sorensen, M.A.; Pérez, A.L.W.; De La Rosa, E.M.R.; Castilla, J.L.T.; Pérez, D.D.; Ramallo-Fariña, Y. Biomarkers of acute respiratory distress syndrome in adults hospitalised for severe SARS-CoV-2 infection in Tenerife Island, Spain. BMC Res. Notes 2020, 13, 555. [Google Scholar] [CrossRef]
- Vargas-Vargas, M.; Cortés-Rojo, C. Ferritin levels and COVID-19. Rev. Panam. Salud Públ. 2020, 44, e72. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y.; Li, B.; Song, X.; Zhou, X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020, 127, 104370. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Huang, Y.; Shi, F.; Tan, K.; Ma, Q.; Chen, Y.; Jiang, X.; Li, X. C-reactive protein correlates with computed tomographic findings and predicts severe COVID-19 early. J. Med. Virol. 2020, 92, 856–862. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan; China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Zhu, X.; Ge, Y.; Wu, T.; Zhao, K.; Chen, Y.; Wu, B.; Zhu, F.; Zhu, B.; Cui, L. Co-infection with respiratory pathogens among COVID-2019 cases. Virus Res. 2020, 285, 198005. [Google Scholar] [CrossRef]
- Lavoignet, C.-E.; Network, A.T.C.; Le Borgne, P.; Chabrier, S.; Bidoire, J.; Slimani, H.; Chevrolet-Lavoignet, J.; Lefebvre, F.; Jebri, R.; Sengler, L.; et al. White blood cell count and eosinopenia as valuable tools for the diagnosis of bacterial infections in the ED. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Borges, L.; Pithon-Curi, T.C.; Curi, R.; Hatanaka, E. COVID-19 and Neutrophils: The Relationship between Hyperinflammation and Neutrophil Extracellular Traps. Mediat. Inflamm. 2020, 2020, 8829674. [Google Scholar] [CrossRef]
- Arcanjo, A.; Logullo, J.; Menezes, C.C.B.; Giangiarulo, T.C.D.S.C.; Dos Reis, M.C.; De Castro, G.M.M.; Fontes, Y.D.S.; Todeschini, A.R.; Freire-De-Lima, L.; Decoté-Ricardo, D.; et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci. Rep. 2020, 10, 19630. [Google Scholar] [CrossRef] [PubMed]
- Schönrich, G.; Raftery, M.J. Neutrophil Extracellular Traps Go Viral. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Clancy, C.J.; Nguyen, M.H. Coronavirus Disease 2019, Superinfections, and Antimicrobial Development: What Can We Expect? Clin. Infect. Dis. 2020, 71, 2736–2743. [Google Scholar] [CrossRef]
- Rawson, T.M.; Moore, L.S.P.; Zhu, N.; Ranganathan, N.; Skolimowska, K.; Gilchrist, M.; Satta, G.; Cooke, G.; Holmes, A. Bacterial and Fungal Coinfection in Individuals with Coronavirus: A Rapid Review to Support COVID-19 Antimicrobial Prescribing. Clin. Infect. Dis. 2020, 530. [Google Scholar] [CrossRef]
- Xia, W.; Shao, J.; Guo, Y.; Peng, X.; Li, Z.; Hu, D. Clinical and CT features in pediatric patients with COVID-19 infection: Different points from adults. Pediatr. Pulmonol. 2020, 55, 1169–1174. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; He, W.; Yu, X.; Hu, D.; Bao, M.; Liu, H.; Zhou, J.; Jiang, H. Coronavirus disease 2019 in elderly patients: Characteristics and prognostic factors based on 4-week follow-up. J. Infect. 2020, 80, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-J.; Dong, X.; Cao, Y.-Y.; Yuan, Y.-D.; Yang, Y.-B.; Yan, Y.-Q.; Akdis, C.A.; Gao, Y.-D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.S.; Li, H.; Zhao, S.C.; Lu, R.J.; Niu, P.H.; Tan, W.J. Viral and bacterial etiology of acute febrile respiratory syn-drome among patients in Qinghai; China. Biomed. Environ. Sci. 2019, 32, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V. Toll-like receptors in sepsis-associated cytokines storm and their endogenous negative regulators as future immunomodulatory targets. Int. Immunopharmacol. 2020, 89, 107087. [Google Scholar] [CrossRef] [PubMed]
- Kuss-Duerkop, S.K.; Keestra-Gounder, A.M. NOD1 and NOD2 Activation by Diverse Stimuli: A Possible Role for Sensing Pathogen-Induced Endoplasmic Reticulum Stress. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef] [PubMed]
- Tolle, L.B.; Standiford, T.J. Danger-associated molecular patterns (DAMPs) in acute lung injury. J. Pathol. 2013, 229, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 493–518. [Google Scholar] [CrossRef] [Green Version]
- Root-Bernstein, R. Synergistic Activation of Toll-Like and NOD Receptors by Complementary Antigens as Facilitators of Autoimmune Disease: Review, Model and Novel Predictions. Int. J. Mol. Sci. 2020, 21, 4645. [Google Scholar] [CrossRef]
- Hosseini, A.M.; Majidi, J.; Baradaran, B.; Yousefi, M. Toll-Like Receptors in the Pathogenesis of Autoimmune Diseases. Adv. Pharm. Bull. 2015, 5, 605–614. [Google Scholar] [CrossRef]
- Dabbagh, K.; Lewis, D.B. Toll-like receptors and T-helper-1/T-helper-2 responses. Curr. Opin. Infect. Dis. 2003, 16, 199–204. [Google Scholar] [CrossRef]
- Tukhvatulin, A.I.; Gitlin, I.I.; Shcheblyakov, D.V.; Artemicheva, N.M.; Burdelya, L.G.; Shmarov, M.M.; Naroditsky, B.S.; Gudkov, A.V.; Gintsburg, A.L.; Logunov, D.Y. Combined Stimulation of Toll-Like Receptor 5 and NOD1 Strongly Potentiates Activity of NF-κB, Resulting in Enhanced Innate Immune Reactions and Resistance to Salmonella enterica Serovar Typhimurium Infection. Infect. Immun. 2013, 81, 3855–3864. [Google Scholar] [CrossRef] [Green Version]
- Moreira, L.O.; Zamboni, D.S. NOD1 and NOD2 Signaling in Infection and Inflammation. Front. Immunol. 2012, 3, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pashenkov, M.V.; Murugina, N.E.; Budikhina, A.S.; Pinegin, B.V. Synergistic interactions between NOD receptors and TLRs: Mechanisms and clinical implications. J. Leukoc. Biol. 2019, 105, 669–680. [Google Scholar] [CrossRef]
- Caron, G.; Duluc, D.; Frémaux, I.; Jeannin, P.; David, C.; Gascan, H.; Delneste, Y. Direct Stimulation of Human T Cells via TLR5 and TLR7/8: Flagellin and R-848 Up-Regulate Proliferation and IFN-γ Production by Memory CD4+T Cells. J. Immunol. 2005, 175, 1551–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lantier, L.; Drouet, F.; Guesdon, W.; Mancassola, R.; Metton, C.; Lo-Man, R.; Werts, C.; Laurent, F.; Lacroix-Lamandé, S. Poly(I:C)-Induced Protection of Neonatal Mice Against Intestinal Cryptosporidium parvum Infection Requires an Additional TLR5 Signal Provided by the Gut Flora. J. Infect. Dis. 2013, 209, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, C.E.; O’Neill, L.A.J. Inflammasomes in inflammatory disorders: The role of TLRs and their interactions with NLRs. Semin. Immunopathol. 2007, 29, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Oviedo-Boyso, J.; Bravo-Patiño, A.; Baizabal-Aguirre, V.M. Collaborative Action of Toll-Like and Nod-Like Receptors as Modulators of the Inflammatory Response to Pathogenic Bacteria. Mediat. Inflamm. 2014, 2014, 432785. [Google Scholar] [CrossRef]
- Pandey, S.; Gruenbaum, A.; Kanashova, T.; Mertins, P.; Cluzel, P.; Chevrier, N. Pairwise Stimulations of Pathogen-Sensing Pathways Predict Immune Responses to Multi-adjuvant Combinations. Cell Syst. 2020, 11, 495–508.e10. [Google Scholar] [CrossRef] [PubMed]
- Nasirudeen, A.M.A.; Wong, H.H.; Thien, P.; Xu, S.; Lam, K.-P.; Liu, D.X. RIG-I, MDA5 and TLR3 Synergistically Play an Important Role in Restriction of Dengue Virus Infection. PLoS Negl. Trop. Dis. 2011, 5, e926. [Google Scholar] [CrossRef] [PubMed]
- Hotz, C.; Roetzer, L.C.; Huber, T.; Sailer, A.; Oberson, A.; Treinies, M.; Heidegger, S.; Herbst, T.; Endres, S.; Bourquin, C. TLR and RLR Signaling Are Reprogrammed in Opposite Directions after Detection of Viral Infection. J. Immunol. 2015, 195, 4387–4395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Asano, N.; Murray, P.J.; Ozato, K.; Tailor, P.; Fuss, I.J.; Kitani, A.; Strober, W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J. Clin. Investig. 2008, 118, 545–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Coveney, A.P.; Wu, M.; Huang, J.; Blankson, S.; Zhao, H.; O’Leary, D.P.; Bai, Z.; Li, Y.; Redmond, H.P.; et al. Activation of Both TLR and NOD Signaling Confers Host Innate Immunity-Mediated Protection Against Microbial Infection. Front. Immunol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, Y.; Pandey, R.K.; Sodhi, A. NOD2 downregulates TLR2/1 mediated IL1 beta gene expression in mouse peritoneal macrophages. PLoS ONE 2011, 6, e27828. [Google Scholar] [CrossRef]
- Watanabe, T.; Kitani, A.; Murray, P.J.; Strober, W. NOD2 is a negative regulator of Toll-like receptor 2–mediated T helper type 1 responses. Nat. Immunol. 2004, 5, 800–808. [Google Scholar] [CrossRef]
- Moen, S.H.; Ehrnström, B.; Kojen, J.F.; Yurchenko, M.; Beckwith, K.S.; Afset, J.E.; Damås, J.K.; Hu, Z.; Yin, H.; Espevik, T.; et al. Human Toll-like Receptor 8 (TLR8) Is an Important Sensor of Pyogenic Bacteria, and Is Attenuated by Cell Surface TLR Signaling. Front. Immunol. 2019, 10, 1209. [Google Scholar] [CrossRef] [Green Version]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. TYPE I INTERFERONS (α/β) IN IMMUNITY AND AUTOIMMUNITY. Annu. Rev. Immunol. 2005, 23, 307–335. [Google Scholar] [CrossRef]
- Køllgaard, T.; Enevold, C.; Bendtzen, K.; Hansen, P.R.; Givskov, M.; Holmstrup, P.; Nielsen, C.H. Cholesterol crystals enhance TLR2- and TLR4-mediated pro-inflammatory cytokine responses of monocytes to the proatherogenic oral bacterium Porphyromonas gingivalis. PLoS ONE 2017, 12, e0172773. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Mouktaroudi, M.; Bodar, E.; Van Der Ven, J.; Kullberg, B.-J.; Netea, M.G.; Van Der Meer, J.W.M. Crystals of monosodium urate monohydrate enhance lipopolysaccharide-induced release of interleukin 1β by mononuclear cells through a caspase 1-mediated process. Ann. Rheum. Dis. 2008, 68, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Seydoux, E.; Liang, H.; Cauwelaert, N.D.; Archer, M.; Rintala, N.D.; Kramer, R.; Carter, D.; Fox, C.B.; Orr, M.T. Effective Combination Adjuvants Engage Both TLR and Inflammasome Pathways to Promote Potent Adaptive Immune Responses. J. Immunol. 2018, 201, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Lacroix-Lamandé, S.; D’Andon, M.F.; Michel, E.; Ratet, G.; Philpott, D.J.; Girardin, S.E.; Boneca, I.G.; Vandewalle, A.; Werts, C. Downregulation of the Na/K-ATPase Pump by Leptospiral Glycolipoprotein Activates the NLRP3 Inflammasome. J. Immunol. 2012, 188, 2805–2814. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, M.-C.; Yang, J.; Wang, J.-F.; Zhu, Y.-H. Lactobacillus rhamnosus GR-1 Ameliorates Escherichia coli-Induced Inflammation and Cell Damage via Attenuation of ASC-Independent NLRP3 Inflammasome Activation. Appl. Environ. Microbiol. 2015, 82, 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conforti-Andreoni, C.; Beretta, O.; Licandro, G.; Qian, H.L.; Urbano, M.; Vitulli, F.; Ricciardi-Castagnoli, P.; Mortellaro, A. Synergism of NOD2 and NLRP3 activators promotes a unique transcriptional profile in murine dendritic cells. J. Leukoc. Biol. 2010, 88, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.M.; Lee, S.-G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 Patients Upregulate Toll-like Receptor 4-mediated Inflammatory Signaling That Mimics Bacterial Sepsis. J. Korean Med. Sci. 2020, 35. [Google Scholar] [CrossRef]
- Shaath, H.; Vishnubalaji, R.; Elkord, E.; Alajez, N.M. Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe COVID-19. Cells 2020, 9, 2374. [Google Scholar] [CrossRef]
- Van Der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; Heuvel, G.V.D.; Mantere, T.; Kersten, S.; Van Deuren, R.C.; Steehouwer, M.; Van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants Among Young Men with Severe COVID-19. JAMA 2020, 324, 663. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.F.V.D.; Velde, A.A.T. Severe COVID-19: NLRP3 Inflammasome Dysregulated. Front. Immunol. 2020, 11, 1580. [Google Scholar] [CrossRef]
- Lara, P.C.; Macías-Verde, D.; Burgos-Burgos, J. Age-induced NLRP3 Inflammasome Over-activation Increases Lethality of SARS-CoV-2 Pneumonia in Elderly Patients. Aging Dis. 2020, 11, 756–762. [Google Scholar] [CrossRef]
- Toldo, S.; Bussani, R.; Nuzzi, V.; Bonaventura, A.; Mauro, A.G.; Cannatà, A.; Pillappa, R.; Sinagra, G.; Nana-Sinkam, P.; Sime, P.; et al. Inflammasome formation in the lungs of patients with fatal COVID-19. Inflamm. Res. 2021, 70, 7–10. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, H.K. Re-analysis of Single Cell Transcriptome Reveals That the NR3C1-CXCL8-Neutrophil Axis Determines the Severity of COVID-19. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.R.; Mehta, A.; Bhagwati, G.; Lakhchaura, R.; Aiyer, H.; Khamar, B.; Chakrabarti, S. Innate Immune Response Modulation and Resistance to SARS-CoV-2 infection: A Prospective Comparative Cohort Study in High Risk Healthcare Workers. MedRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Arankalle, V.A.; Lole, K.S.; Arya, R.P.; Tripathy, A.S.; Ramdasi, A.Y.; Chadha, M.S.; Sangle, S.A.; Kadam, D.B. Role of Host Immune Response and Viral Load in the Differential Outcome of Pandemic H1N1 (2009) Influenza Virus Infection in Indian Patients. PLoS ONE 2010, 5, e13099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinay, T.; Kim, Y.S.; Howrylak, J.; Hunninghake, G.M.; An, C.H.; Fredenburgh, L.; Massaro, A.F.; Rogers, A.; Gazourian, L.; Nakahira, K.; et al. Inflammasome-regulated Cytokines Are Critical Mediators of Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2012, 185, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.D.; Crother, T.R.; Gonzalez-Villalobos, R.A.; Jupelli, M.; Chen, S.; Dagvadorj, J.; Arditi, M.; Shimada, K. The NLRP3 Inflammasome is Required for the Development of Hypoxemia in LPS/Mechanical Ventilation Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2013, 50, 270–280. [Google Scholar] [CrossRef]
- Nita-Lazar, M.; Banerjee, A.; Feng, C.; Vasta, G.R. Galectins regulate the inflammatory response in airway epithelial cells exposed to microbial neuraminidase by modulating the expression of SOCS1 and RIG1. Mol. Immunol. 2015, 68, 194–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, M.; Fan, J. Pattern Recognition Receptor-Dependent Mechanisms of Acute Lung Injury. Mol. Med. 2010, 16, 69–82. [Google Scholar] [CrossRef]
- Wu, G.; Zhu, Q.; Zeng, J.; Gu, X.; Miao, Y.; Xu, W.; Lv, T.; Song, Y. Extracellular mitochondrial DNA promote NLRP3 inflammasome activation and induce acute lung injury through TLR9 and NF-κB. J. Thorac. Dis. 2019, 11, 4816–4828. [Google Scholar] [CrossRef] [PubMed]
- Bakopoulos, A.; Kapelouzou, A.; Tsilimigras, D.I.; Katsimpoulas, M.; Schizas, D.; Aravanis, C.; Balafas, E.; Mavroidis, M.; Pavlakis, K.; Machairas, A.; et al. Expression of Toll-like receptors (TLRs) in the lungs of an experimental sepsis mouse model. PLoS ONE 2017, 12, e0188050. [Google Scholar] [CrossRef]
- Chen, X.; Wang, T.; Song, L.; Liu, X. Activation of multiple Toll-like receptors serves different roles in sepsis-induced acute lung injury. Exp. Ther. Med. 2019, 18, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, M.; Cao, H.; Zhu, Y.; Zheng, J.; Zhou, H. Extraordinary GU-rich single-strand RNA identified from SARS coronavirus contributes an excessive innate immune response. Microbes Infect. 2013, 15, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Pène, F.; Courtine, E.; Ouaaz, F.; Zuber, B.; Sauneuf, B.; Sirgo, G.; Rousseau, C.; Toubiana, J.; Balloy, V.; Chignard, M.; et al. Toll-like receptors 2 and 4 contribute to sepsis-induced depletion of spleen dendritic cells. Infect. Immun. 2009, 77, 5651–5658. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.; Baggio-Zappia, G.; Brunialti, M.; Assunçao, M.; Azevedo, L.; Machado, F.; Salomao, R. Evaluation of Toll-like, chemokine, and integrin receptors on monocytes and neutrophils from peripheral blood of septic patients and their correlation with clinical outcomes. Braz. J. Med. Biol. Res. 2014, 47, 384–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Härter, L.; Mica, L.; Stocker, R.; Trentz, O.; Keel, M. Increased expression of Toll-like receptor-2 and -4 on leukocytes from patients with sepsis. Shock 2004, 22, 403–409. [Google Scholar] [CrossRef]
- Gonçalves, G.M.; Zamboni, D.S.; Câmara, N.O. The role of innate immunity in septic acute kidney injuries. Shock. 2010, 34 (Suppl. 1), 22–26. [Google Scholar] [CrossRef]
- Weighardt, H.; Holzmann, B. Role of Toll-like receptor responses for sepsis pathogenesis. Immunobiology 2008, 212, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.; Medford, A.R.L.; Hunter, K.J.; Uppington, K.M.; Millar, A.B. Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin. Exp. Immunol. 2004, 136, 312–319. [Google Scholar] [CrossRef]
- Brandl, K.; Glück, T.; Huber, C.; Salzberger, B.; Falk, W.; Hartmann, P. TLR-4 surface display on human monocytes is increased in septic patients. Eur. J. Med. Res. 2005, 10, 319–324. [Google Scholar] [PubMed]
- Asadpour-Behzadi, A.; Kariminik, A. RIG-1 and MDA5 are the important intracellular sensors against bacteria in septicemia suffering patients. J. Appl. Biomed. 2018, 16, 358–361. [Google Scholar] [CrossRef]
- Danielski, L.G.; Della Giustina, A.; Bonfante, S.; Barichello, T.; Petronilho, F. The NLRP3 Inflammasome and Its Role in Sepsis Development. Inflammation 2020, 43, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Atalan, N.; Acar, L.; Yapici, N.; Kudsioglu, T.; Ergen, A.; Yilmaz, S.G.; Isbir, T. The Relationship Between Sepsis-induced Immunosuppression and Serum Toll-like Receptor 9 Level. In Vivo 2018, 32, 1653–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, N.M.; Wang, J.; Redmond, H.P.; Wang, J.H. Current knowledge and future directions of TLR and NOD signaling in sepsis. Mil. Med. Res. 2015, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Rajaee, A.; Barnett, R.; Cheadle, W.G. Pathogen- and Danger-Associated Molecular Patterns and the Cytokine Response in Sepsis. Surg. Infect. 2018, 19, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, D.C.; Baik, C.H.; Travis, O.K.; White, D.L.; Young, C.M.; Pierce, W.A.; Shields, C.A.; Poudel, B.; Williams, J.M. NLRP3 inflammasome activation in platelets in response to sepsis. Physiol. Rep. 2019, 7, e14073. [Google Scholar] [CrossRef] [PubMed]
- Sônego, F.; Castanheira, F.V.S.; Czaikoski, P.G.; Kanashiro, A.; Souto, F.O.; França, R.O.; Nascimento, D.C.; Freitas, A.; Spiller, F.; Cunha, L.D.; et al. MyD88-, but Not Nod1- and/or Nod2-Deficient Mice, Show Increased Susceptibility to Polymicrobial Sepsis due to Impaired Local Inflammatory Response. PLoS ONE 2014, 9, e103734. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.; Neder, J.; Cui, P.; Suen, A.; Tanaka, K.; Zou, L.; Chao, W. Toll-like receptors 2 and 7 mediate coagulation activation and coagulopathy in murine sepsis. J. Thromb. Haemost. 2019, 17, 1683–1693. [Google Scholar] [CrossRef]
- Cruz, N.E.R.; Bernal, C.M.; Urióstegui, M.L.C.; Castañoń, J.; Macías, C.L.; Isibasi, A. Toll-like receptors: Dysregulation in vivo in patients with acute respiratory distress syndrome. Rev. Alerg. México 2005, 51, 210–217. [Google Scholar]
- Ichinohe, T. Respective roles of TLR, RIG-I and NLRP3 in influenza virus infection and immunity: Impact on vaccine design. Expert Rev. Vaccines 2010, 9, 1315–1324. [Google Scholar] [CrossRef]
- Lee, N.; Wong, C.K.; Hui, D.S.C.; Lee, S.K.W.; Wong, R.Y.K.; Ngai, K.L.K.; Chan, M.C.W.; Chu, Y.J.; Ho, A.W.Y.; Lui, G.C.Y.; et al. Role of human Toll-like receptors in naturally occurring influenza A infections. Influ. Other Respir. Viruses 2013, 7, 666–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastalerz-Migas, A.; Pokorski, M.; Kilis-Pstrusinska, K.; Doskocz, K.; Sapilak, B.J.; Brydak, L.B. Cytokines and Toll-Like Receptors in the Immune Response to Influenza Vaccination. Adv. Exp. Med. Biol. 2014, 836, 35–40. [Google Scholar] [CrossRef]
- Miettinen, M.; Sareneva, T.; Julkunen, I.; Matikainen, S. IFNs activate toll-like receptor gene expression in viral infections. Genes Immun. 2001, 2, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.P.; Kurt-Jones, E.A.; Finberg, R.W. Innate immunity to respiratory viruses. Cell. Microbiol. 2007, 9, 1641–1646. [Google Scholar] [CrossRef]
- Qi, F.; Wang, D.; Liu, J.; Zeng, S.; Xu, L.; Hu, H.; Liu, B. Respiratory macrophages and dendritic cells mediate respiratory syncytial virus-induced IL-33 production in TLR3- or TLR7-dependent manner. Int. Immunopharmacol. 2015, 29, 408–415. [Google Scholar] [CrossRef]
- Huang, S.; Wei, W.; Yun, Y. Upregulation of TLR7 and TLR3 gene expression in the lung of respiratory syncytial virus infected mice. Acta Microbiol. Sin. 2009, 49, 239–245. [Google Scholar]
- Scagnolari, C.; Midulla, F.; Pierangeli, A.; Moretti, C.; Bonci, E.; Berardi, R.; De Angelis, D.; Selvaggi, C.; Di Marco, P.; Girardi, E.; et al. Gene Expression of Nucleic Acid-Sensing Pattern Recognition Receptors in Children Hospitalized for Respiratory Syncytial Virus-Associated Acute Bronchiolitis. Clin. Vaccine Immunol. 2009, 16, 816–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klouwenberg, P.K.; Tan, L.; Werkman, W.; Van Bleek, G.M.; Coenjaerts, F. The role of Toll-like receptors in regulating the immune response against respiratory syncytial virus. Crit. Rev. Immunol. 2009, 29, 531–550. [Google Scholar] [CrossRef] [PubMed]
- Rallabhandi, P.; Phillips, R.L.; Boukhvalova, M.S.; Pletneva, L.M.; Shirey, K.A.; Gioannini, T.L.; Weiss, J.P.; Chow, J.C.; Hawkins, L.D.; Vogel, S.N.; et al. Respiratory Syncytial Virus Fusion Protein-Induced Toll-Like Receptor 4 (TLR4) Signaling Is Inhibited by the TLR4 Antagonists Rhodobacter sphaeroides Lipopolysaccharide and Eritoran (E5564) and Requires Direct Interaction with MD-2. mBio 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, M.T.; Serra, M.E.; Acosta, P.L.; Marzec, J.; Gibbons, L.; Salim, M.; Rodríguez, A.; Reynaldi, A.; Garcia, A.; Bado, D.; et al. TLR4 genotype and environmental LPS mediate RSV bronchiolitis through Th2 polarization. J. Clin. Investig. 2015, 125, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; Tsai, S.-Y.; Chang, T.-H.; Berton, M.T.; Morris, I.R.; Allen, I.C.; Ting, J.P.-Y.; Bose, S. TLR2/MyD88/NF-κB Pathway, Reactive Oxygen Species, Potassium Efflux Activates NLRP3/ASC Inflammasome during Respiratory Syncytial Virus Infection. PLoS ONE 2012, 7, e29695. [Google Scholar] [CrossRef] [PubMed]
- Marr, N.; Wang, T.-I.; Kam, S.H.Y.; Hu, Y.S.; Sharma, A.A.; Lam, A.; Markowski, J.; Solimano, A.; Lavoie, P.M.; Turvey, S.E. Attenuation of Respiratory Syncytial Virus–Induced and RIG-I–Dependent Type I IFN Responses in Human Neonates and Very Young Children. J. Immunol. 2014, 192, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Fejer, G.; Freudenberg, M.; Greber, U.F.; Gyory, I. Adenovirus-triggered innate signalling pathways. Eur. J. Microbiol. Immunol. 2011, 1, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riad, A.; Westermann, D.; Escher, F.; Becher, P.M.; Savvatis, K.; Lettau, O.; Heimesaat, M.M.; Bereswill, S.; Volk, H.D.; Schultheiss, H.P.; et al. Myeloid differentiation factor-88 contributes to TLR9-mediated modulation of acute coxsackievirus B3-induced myocarditis in vivo. Am. J. Physiol. Circ. Physiol. 2010, 298, H2024–H2031. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Qi, J.; Lei, X.; Wang, J. Interactions Between Enteroviruses and the Inflammasome: New Insights into Viral Pathogenesis. Front. Microbiol. 2019, 10, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, J.; Sun, T.; Yue, Y.; Xiong, S. Macrophage NLRP3 inflammasome activated by CVB3 capsid proteins contributes to the development of viral myocarditis. Mol. Immunol. 2019, 114, 41–48. [Google Scholar] [CrossRef]
- Sajjan, U.S.; Jia, Y.; Newcomb, D.C.; Bentley, J.K.; Lukacs, N.W.; Lipuma, J.J.; Hershenson, M.B. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J. 2006, 20, 2121–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, S.; Zhu, L.; Lee, P.; Lee, W.-M.; Knox, K.; Chen, J.; Di, Y.P.; Chen, Y. Negative Control of TLR3 Signaling by TICAM1 Down-Regulation. Am. J. Respir. Cell Mol. Biol. 2012, 46, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Sykes, A.; Edwards, M.R.; MacIntyre, J.; Del Rosario, A.; Bakhsoliani, E.; Trujillo-Torralbo, M.-B.; Kon, O.M.; Mallia, P.; McHale, M.; Johnston, S.L. Rhinovirus 16–induced IFN-α and IFN-β are deficient in bronchoalveolar lavage cells in asthmatic patients. J. Allergy Clin. Immunol. 2012, 129, 1506–1514.e6. [Google Scholar] [CrossRef]
- Sallenave, J.-M.; Guillot, L. Innate Immune Signaling and Proteolytic Pathways in the Resolution or Exacerbation of SARS-CoV-2 in Covid-19: Key Therapeutic Targets? Front. Immunol. 2020, 11, 1229. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-S.; Ko, B.-H.; Ju, J.-C.; Chang, H.-H.; Huang, S.-H.; Lin, C.-W. SARS Unique Domain (SUD) of Severe Acute Respiratory Syndrome Coronavirus Induces NLRP3 Inflammasome-Dependent CXCL10-Mediated Pulmonary Inflammation. Int. J. Mol. Sci. 2020, 21, 3179. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Bujko, K.; Ciechanowicz, A.; Sielatycka, K.; Cymer, M.; Marlicz, W.; Kucia, M. SARS-CoV-2 Entry Receptor ACE2 Is Expressed on Very Small CD45−Precursors of Hematopoietic and Endothelial Cells and in Response to Virus Spike Protein Activates the Nlrp3 Inflammasome. Stem Cell Rev. Rep. 2020, 17, 266–277. [Google Scholar] [CrossRef]
- Totura, A.L.; Whitmore, A.C.; Agnihothram, S.; Schäfer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2015, 6, e00638-15. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Yen, Y.-T.; Singh, S.; Kao, C.-L.; Wu-Hsieh, B.A. SARS-CoV Regulates Immune Function-Related Gene Expression in Human Monocytic Cells. Viral Immunol. 2012, 25, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.-Y.; Moriyama, M.; Chang, M.-F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, K.; Yuen, K.; Castano-Rodriguez, C.; Ye, Z.; Yeung, M.; Fung, S.; Yuan, S.; Chan, C.; Yuen, K.; Enjuanes, L.; et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Anderson, D.E.; Zhang, Q.; Tan, C.W.; Lim, B.L.; Luko, K.; Wen, M.; Ni Chia, W.; Mani, S.; Wang, L.C.; et al. Dampened NLRP3-mediated inflammation in bats and implications for a special viral reservoir host. Nat. Microbiol. 2019, 4, 789–799. [Google Scholar] [CrossRef]
- Kim, J.; Yang, Y.L.; Jang, Y.-S. Human β-defensin 2 is involved in CCR2-mediated Nod2 signal transduction, leading to activation of the innate immune response in macrophages. Immunobiology 2019, 224, 502–510. [Google Scholar] [CrossRef]
- Srivastava, S.; Kamthania, M.; Singh, S.; Saxena, A.K.; Sharma, N. Structural basis of development of multi-epitope vaccine against Middle East respiratory syndrome using in silico approach. Infect. Drug Resist. 2018, 11, 2377–2391. [Google Scholar] [CrossRef] [Green Version]
- Scheuplein, V.A.; Seifried, J.; Malczyk, A.H.; Miller, L.; Höcker, L.; Vergara-Alert, J.; Dolnik, O.; Zielecki, F.; Becker, B.; Spreitzer, I.; et al. High Secretion of Interferons by Human Plasmacytoid Dendritic Cells upon Recognition of Middle East Respiratory Syndrome Coronavirus. J. Virol. 2015, 89, 3859–3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Tian, J.; Kang, H.; Guo, D.; Liu, J.; Liu, D.; Jiang, Q.; Xiaoliang, H.; Qu, J.; Qu, L. Transmissible Gastroenteritis Virus Papain-Like Protease 1 Antagonizes Production of Interferon-βthrough Its Deubiquitinase Activity. BioMed Res. Int. 2017, 2017, 7089091. [Google Scholar] [CrossRef] [Green Version]
- Al-Qahtani, A.A.; Lyroni, K.; Aznaourova, M.; Tseliou, M.; Al-Anazi, M.R.; Al-Ahdal, M.N.; Alkahtani, S.; Sourvinos, G.; Tsatsanis, C. Middle east respiratory syndrome corona virus spike glycoprotein suppresses macrophage responses via DPP4-mediated induction of IRAK-M and PPARγ. Oncotarget 2017, 8, 9053–9066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosch, S.F.; Mahajan, S.D.; Collins, A.R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-κB pathway in human monocyte macrophages in vitro. Virus Res. 2009, 142, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Law, H.K.W.; Cheung, C.Y.; Sia, S.F.; Chan, Y.O.; Peiris, J.S.M.; Lau, Y.L. Toll-like receptors, chemokine receptors and death receptor ligands responses in SARS coronavirus infected human monocyte derived dendritic cells. BMC Immunol. 2009, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Yang, Y.L.; Jang, S.-H.; Jang, Y.-S. Human β-defensin 2 plays a regulatory role in innate antiviral immunity and is capable of potentiating the induction of antigen-specific immunity. Virol. J. 2018, 15, 124. [Google Scholar] [CrossRef] [Green Version]
- Baral, P.; Batra, S.; Zemans, R.L.; Downey, G.P.; Jeyaseelan, S. Divergent functions of toll-like receptors during bacterial lung infections. Am. J. Respir. Crit. Care Med. 2014, 190, 722–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, M.; Veckman, V.; Latvala, S.; Sareneva, T.; Matikainen, S.; Julkunen, I. Live Lactobacillus rhamnosus and Streptococcus pyogenes differentially regulate Toll-like receptor (TLR) gene expression in human primary macro-phages. J. Leukoc. Biol. 2008, 84, 1092–1100. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H.; Paludan, S.R.; Kilian, M.; Østergaard, L. Live Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis activate the inflammatory response through Toll-like receptors 2, 4, and 9 in species-specific patterns. J. Leukoc. Biol. 2006, 80, 267–277. [Google Scholar] [CrossRef]
- Liadaki, K.; Petinaki, E.; Skoulakis, C.; Tsirevelou, P.; Klapsa, D.; Germenis, A.E.; Speletas, M. Toll-Like Receptor 4 Gene (TLR4), but NotTLR2, Polymorphisms Modify the Risk of Tonsillar Disease Due toStreptococcus pyogenesandHaemophilus influenzae. Clin. Vaccine Immunol. 2010, 18, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Schröder, N.W.; Morath, S.; Alexander, C.; Hamann, L.; Hartung, T.; Zähringer, U.; Göbel, U.B.; Weber, J.R.; Schumann, R.R. Lipoteichoic Acid (LTA) of Streptococcus pneumoniaeand Staphylococcus aureus Activates Immune Cells via Toll-like Receptor (TLR)-2, Lipopolysaccharide-binding Protein (LBP), and CD14, whereas TLR-4 and MD-2 Are Not Involved. J. Biol. Chem. 2003, 278, 15587–15594. [Google Scholar] [CrossRef] [Green Version]
- Schmeck, B.; Huber, S.; Moog, K.; Zahlten, J.; Hocke, A.C.; Opitz, B.; Hammerschmidt, S.; Mitchell, T.J.; Kracht, M.; Rosseau, S.; et al. Pneumococci induced TLR- and Rac1-dependent NF-κB-recruitment to the IL-8 promoter in lung epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L730–L737. [Google Scholar] [CrossRef]
- Harder, J.; Franchi, L.; Muñoz-Planillo, R.; Park, J.H.; Reimer, T.; Núñez, G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J. Immunol. 2009, 183, 5823–5829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinhuis, B.; Koenders, M.I.; Van De Loo, F.A.; Van Lent, P.L.E.M.; Kim, S.-H.; Dinarello, C.A.; Joosten, L.A.B.; Berg, W.B.V.D. IL-32 and Streptococcus pyogenes cell wall fragments synergise for IL-1-dependent destructive arthritis via upregulation of TLR-2 and NOD2. Ann. Rheum. Dis. 2010, 69, 1866–1872. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Oshikawa, T.; Ohe, G.; Nishikawa, H.; Furuichi, S.; Tano, T.; Moriya, Y.; Saito, M.; Sato, M. Severe impairment of anti-cancer effect of lipoteichoic acid-related molecule isolated from a penicillin-killed Streptococcus pyogenes in toll-like receptor 4-deficient mice. Int. Immunopharmacol. 2001, 1, 1789–1795. [Google Scholar] [CrossRef]
- Thorburn, A.N.; Tseng, H.-Y.; Donovan, C.; Hansbro, N.G.; Jarnicki, A.G.; Foster, P.S.; Gibson, P.G.; Hansbro, P.M. TLR2, TLR4 AND MyD88 Mediate Allergic Airway Disease (AAD) and Streptococcus pneumoniae-Induced Suppression of AAD. PLoS ONE 2016, 11, e0156402. [Google Scholar] [CrossRef]
- Lemire, P.; Roy, D.; Fittipaldi, N.; Okura, M.; Takamatsu, D.; Bergman, E.; Segura, M. Implication of TLR- but Not of NOD2-Signaling Pathways in Dendritic Cell Activation by Group B Streptococcus Serotypes III and V. PLoS ONE 2014, 9, e113940. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Gupta, R.; Signorino, G.; Malara, A.; Cardile, F.; Biondo, C.; Midiri, A.; Galbo, R.; Trieu-Cuot, P.; Papasergi, S.; et al. Activation of the NLRP3 Inflammasome by Group B Streptococci. J. Immunol. 2012, 188, 1953–1960. [Google Scholar] [CrossRef]
- Henneke, P.; Takeuchi, O.; Malley, R.; Lien, E.; Ingalls, R.R.; Freeman, M.W.; Mayadas, T.; Nizet, V.; Akira, S.; Kasper, D.L.; et al. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J. Immunol. 2002, 169, 3970–3977. [Google Scholar] [CrossRef] [Green Version]
- Askarian, F.; Wagner, T.; Johannessen, M.; Nizet, V. Staphylococcus aureus modulation of innate immune responses through Toll-like (TLR), (NOD)-like (NLR) and C-type lectin (CLR) receptors. FEMS Microbiol. Rev. 2018, 42, 656–671. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, B.; Mao, W.; Feng, S.; Yao, Y.; Bai, F.; Shen, Y.; Guleng, A.; Jirigala, B.; Cao, J. Prostaglandin E2 Regulates Activation of Mouse Peritoneal Macrophages by Staphylococcus aureus through Toll-Like Receptor 2, Toll-Like Receptor 4, and NLRP3 Inflammasome Signaling. J. Innate Immun. 2020, 12, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, K.S.; Beckwith, M.S.; Ullmann, S.; Sætra, R.S.; Kim, H.; Marstad, A.; Åsberg, S.E.; Strand, T.A.; Haug, M.; Niederweis, M.; et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat. Commun. 2020, 11, 2270. [Google Scholar] [CrossRef] [PubMed]
- Wiese, K.M.; Coates, B.M.; Ridge, K.M. The Role of Nucleotide-Binding Oligomerization Domain–Like Receptors in Pulmonary Infection. Am. J. Respir. Cell Mol. Biol. 2017, 57, 151–161. [Google Scholar] [CrossRef]
- Regueiro, V.; Moranta, D.; Campos, M.A.; Margareto, J.; Garmendia, J.; Bengoechea, J.A. Klebsiella pneumoniae Increases the Levels of Toll-Like Receptors 2 and 4 in Human Airway Epithelial Cells. Infect. Immun. 2008, 77, 714–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieland, C.W.; Van Lieshout, M.H.P.; Hoogendijk, A.J.; Van Der Poll, T. Host defence during Klebsiella pneumonia relies on haematopoietic-expressed Toll-like receptors 4 and 2. Eur. Respir. J. 2010, 37, 848–857. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.-Y.; Park, J.-H.; Park, J.-I.; Kim, J.-Y.; Seo, S.-M.; Ham, S.-H.; Jeong, E.-S.; Choi, Y.-K. Cooperative Interactions between Toll-Like Receptor 2 and Toll-Like Receptor 4 in Murine Klebsiella pneumoniae Infections. J. Microbiol. Biotechnol. 2017, 27, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Bhan, U.; Lukacs, N.W.; Osterholzer, J.J.; Newstead, M.W.; Zeng, X.; Moore, T.A.; McMillan, T.R.; Krieg, A.M.; Akira, S.; Standiford, T.J. TLR9 Is Required for Protective Innate Immunity in Gram-Negative Bacterial Pneumonia: Role of Dendritic Cells. J. Immunol. 2007, 179, 3937–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, K.-F.; Yang, F.-L.; Chiu, H.-W.; Chou, J.-C.; Dong, W.-C.; Lin, C.-N.; Lin, C.-Y.; Wang, J.-T.; Li, L.-H.; Chiu, H.-W.; et al. Capsular Polysaccharide Is Involved in NLRP3 Inflammasome Activation by Klebsiella pneumoniae Serotype K1. Infect. Immun. 2015, 83, 3396–3409. [Google Scholar] [CrossRef] [Green Version]
- March, C.; Moranta, D.; Regueiro, V.; Llobet, E.; Tomás, A.; Garmendia, J.; Bengoechea, J.A. Klebsiella pneumoniae Outer Membrane Protein A Is Required to Prevent the Activation of Airway Epithelial Cells. J. Biol. Chem. 2011, 286, 9956–9967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galdiero, M.; Galdiero, M.; Finamore, E.; Rossano, F.; Gambuzza, M.; Catania, M.R.; Teti, G.; Midiri, A.; Mancuso, G. Haemophilus influenzae Porin Induces Toll-Like Receptor 2-Mediated Cytokine Production in Human Monocytes and Mouse Macrophages. Infect. Immun. 2004, 72, 1204–1209. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Leichtle, A.; Zuckerman, E.; Pak, K.; Spriggs, M.; Wasserman, S.I.; Kurabi, A. NOD1/NOD2-mediated recognition of non-typeable Haemophilus influenzae activates innate immunity during otitis media. Innate Immun. 2019, 25, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loria, J.R.D.; Rohmann, K.; Droemann, D.; Kujath, P.; Rupp, J.; Goldmann, T.; Dalhoff, K. Nontypeable Haemophilus Influenzae Infection Upregulates the NLRP3 Inflammasome and Leads to Caspase-1-Dependent Secretion of Interleukin-1β—A Possible Pathway of Exacerbations in COPD. PLoS ONE 2013, 8, e66818. [Google Scholar] [CrossRef] [Green Version]
- Wieland, C.W.; Florquin, S.; Van Der Poll, T. Toll-like receptor 9 is not important for host defense against Haemophilus influenzae. Immunobiology 2010, 215, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Xu, Z.; Zhang, R.; Wu, Z.; Lim, J.-H.; Koga, T.; Li, J.-D.; Shen, H. Nontypeable Haemophilus influenzae induces COX-2 and PGE2 expression in lung epithelial cells via activation of p38 MAPK and NF-kappa B. Respir. Res. 2008, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akamine, M.; Higa, F.; Arakaki, N.; Kawakami, K.; Takeda, K.; Akira, S.; Saito, A. Differential Roles of Toll-Like Receptors 2 and 4 in In Vitro Responses of Macrophages to Legionella pneumophila. Infect. Immun. 2005, 73, 352–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Cao, X.; Yang, Z. [Recombinant Legionella pneumophila flagella protein A (rflaA) induces the secretion of IL-6 and IL-1β in RAW264.7 cells in vitro]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2017, 33, 601–605. (In Chinese) [Google Scholar]
- He, X.; Mekasha, S.; Mavrogiorgos, N.; Fitzgerald, K.A.; Lien, E.; Ingalls, R.R. Inflammation and Fibrosis duringChlamydia pneumoniaeInfection Is Regulated by IL-1 and the NLRP3/ASC Inflammasome. J. Immunol. 2010, 184, 5743–5754. [Google Scholar] [CrossRef] [Green Version]
- Massari, P.; Toussi, D.N.; Tifrea, D.F.; De La Maza, L.M. Toll-Like Receptor 2-Dependent Activity of Native Major Outer Membrane Protein Proteosomes of Chlamydia trachomatis. Infect. Immun. 2012, 81, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Joyee, A.G. Role of toll-like receptors in immune responses to chlamydial infections. Curr. Pharm. Des. 2008, 14, 593–600. [Google Scholar] [CrossRef]
- Itoh, R.; Murakami, I.; Chou, B.; Ishii, K.; Soejima, T.; Suzuki, T.; Hiromatsu, K. Chlamydia pneumoniae harness host NLRP3 inflammasome-mediated caspase-1 activation for optimal intracellular growth in murine macrophages. Biochem. Biophys. Res. Commun. 2014, 452, 689–694. [Google Scholar] [CrossRef]
- Idosa, B.A.; Kelly, A.; Jacobsson, S.; Demirel, I.; Fredlund, H.; Särndahl, E.; Persson, A. Neisseria meningitidis-Induced Caspase-1 Activation in Human Innate Immune Cells Is LOS-Dependent. J. Immunol. Res. 2019, 2019, 6193186. [Google Scholar] [CrossRef] [Green Version]
- Woodhams, K.L.; Chan, J.M.; Lenz, J.D.; Hackett, K.T.; Dillard, J.P. Peptidoglycan Fragment Release from Neisseria meningitidis. Infect. Immun. 2013, 81, 3490–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhazmi, A. NOD-like receptor(s) and host immune responses with Pseudomonas aeruginosa infection. Inflamm. Res. 2018, 67, 479–493. [Google Scholar] [CrossRef]
- Pène, F.; Grimaldi, D.; Zuber, B.; Sauneuf, B.; Rousseau, C.; El Hachem, C.; Martin, C.; Belaïdouni, N.; Balloy, V.; Mira, J.-P.; et al. Toll-Like Receptor 2 Deficiency Increases Resistance to Pseudomonas aeruginosa Pneumonia in the Setting of Sepsis-Induced Immune Dysfunction. J. Infect. Dis. 2012, 206, 932–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugliandolo, E.; Fusco, R.; Ginestra, G.; D’Amico, R.; Bisignano, C.; Mandalari, G.; Cuzzocrea, S.; Di Paola, R. Involvement of TLR4 and PPAR-α Receptors in Host Response and NLRP3 Inflammasome Activation, Against Pulmonary Infection with Pseudomonas Aeruginosa. Shock 2019, 51, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Segovia, J.A.; Chang, T.-H.; Winter, V.T.; Coalson, J.J.; Cagle, M.P.; Pandranki, L.; Bose, S.; Baseman, J.B.; Kannan, T.R. NLRP3 Is a Critical Regulator of Inflammation and Innate Immune Cell Response during Mycoplasma pneumoniae Infection. Infect. Immun. 2017, 86, e00548-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T. Inflammation-inducing Factors of Mycoplasma pneumoniae. Front. Microbiol. 2016, 7, 414. [Google Scholar] [CrossRef] [Green Version]
- You, X.-X.; Zeng, Y.-H.; Wu, Y.-M. Interactions between mycoplasma lipid-associated membrane proteins and the host cells. J. Zhejiang Univ. Sci. B 2006, 7, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, C.; Kuchler, K. Fungal pathogens—a sweet and sour treat for toll-like receptors. Front. Cell. Infect. Microbiol. 2012, 2, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patin, E.C.; Thompson, A.; Orr, S.J. Pattern recognition receptors in fungal immunity. Semin. Cell Dev. Biol. 2019, 89, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Jannuzzi, G.P.; De Almeida, J.R.F.; Paulo, L.N.M.; De Almeida, S.R.; Ferreira, K.S. Intracellular PRRs Activation in Targeting the Immune Response Against Fungal Infections. Front. Cell. Infect. Microbiol. 2020, 10, 591970. [Google Scholar] [CrossRef] [PubMed]
- Van Der Graaf, C.A.A.; Netea, M.G.; Franke, B.; Girardin, S.E.; Van Der Meer, J.W.M.; Kullberg, B.J. Nucleotide Oligomerization Domain 2 (Nod2) Is Not Involved in the Pattern Recognition of Candida albicans. Clin. Vaccine Immunol. 2006, 13, 423–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehata, M.M.; Mostafa, A.; Teubner, L.; Mahmoud, S.H.; Kandeil, A.; Elshesheny, R.; Boubak, T.A.; Frantz, R.; La Pietra, L.; Pleschka, S.; et al. Bacterial Outer Membrane Vesicles (OMVs)-Based Dual Vaccine for Influenza a H1N1 Virus and MERS-CoV. Vaccines 2019, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Shim, S.-M.; Song, E.-J.; Song, D.; Lee, T.-Y.; Kim, O.-J.; Nam, J.-H.; Jeong, D.G.; Lee, C.-K.; Kim, S.-H.; Kim, J.-K. Nontoxic outer membrane vesicles efficiently increase the efficacy of an influenza vaccine in mice and ferrets. Vaccine 2017, 35, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.-H.; Seo, S.H.; Kim, C.-U.; Jang, M.S.; Song, M.-S.; Lee, T.-Y.; Jeong, Y.-J.; Lee, M.-S.; Park, J.-H.; Lee, P.; et al. Bacterial Outer Membrane Vesicles Provide Broad-Spectrum Protection against Influenza Virus Infection via Recruitment and Activation of Macrophages. J. Innate Immun. 2019, 11, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Cocon, L.; Diaz, O.; Jacquemin, C.; Barthel, V.; Ogire, E.; Ramière, C.; André, P.; Lotteau, V.; Vidalain, P.-O. The current landscape of corona-virus-host protein-protein interactions. J. Transl. Med. 2020, 18, 319. [Google Scholar] [CrossRef]
- Lopez, L.; Sang, P.C.; Tian, Y.; Sang, Y. Dysregulated Interferon Response Underlying Severe COVID-19. Viruses 2020, 12, 1433. [Google Scholar] [CrossRef] [PubMed]
- Remy, K.E.; Mazer, M.; Striker, D.A.; Ellebedy, A.H.; Walton, A.H.; Unsinger, J.; Blood, T.M.; Mudd, P.A.; Yi, D.J.; Mannion, D.A.; et al. Severe immunosuppression and not a cytokine storm characterizes COVID-19 infections. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Acharya, D.; Liu, G.; Gack, M.U. Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol. 2020, 20, 397–398. [Google Scholar] [CrossRef] [PubMed]
- Jamilloux, Y.; Henry, T.; Belot, A.; Viel, S.; Fauter, M.; El Jammal, T.; Walzer, T.; François, B.; Sève, P. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun. Rev. 2020, 19, 102567. [Google Scholar] [CrossRef]
- Chen, X.; Liao, B.; Cheng, L.; Peng, X.; Xu, X.; Li, Y.; Hu, T.; Li, J.; Zhou, X.; Ren, B. The microbial coinfection in COVID-19. Appl. Microbiol. Biotechnol. 2020, 104, 7777–7785. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Troise, O.; Donaldson, H.; Mughal, N.; Moore, L. Bacterial and fungal coinfection among hospitalized patients with COVID-19: A retrospective cohort study in a UK secondary-care setting. Clin. Microbiol. Infect. 2020, 26, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Fattorini, L.; Creti, R.; Palma, C.; Pantosti, A. Unit of Antibiotic Resistance and Special Pathogens; Unit of Antibiotic Resistance and Special Pathogens of the Department of Infectious Diseases; Istituto Superiore di Sanità; Rome. Bac-terial coinfections in COVID-19: An underestimated adversary. Ann. Ist. Super. Sanita 2020, 56, 359–364. [Google Scholar] [CrossRef]
- Monnet, D.L.; Harbarth, S. Will coronavirus disease (COVID-19) have an impact on antimicrobial resistance? Eurosurveillance 2020, 25, 2001886. [Google Scholar] [CrossRef] [PubMed]
- Rawson, T.M.; Moore, L.S.P.; Castro-Sanchez, E.; Charani, E.; Davies, F.; Satta, G.; Ellington, M.J.; Holmes, A.H. COVID-19 and the potential long-term impact on antimicrobial resistance. J. Antimicrob. Chemother. 2020, 75, 1681–1684. [Google Scholar] [CrossRef]
- Koehler, P.; Cornely, O.A.; Böttiger, B.W.; Dusse, F.; Eichenauer, D.A.; Fuchs, F.; Hallek, M.; Jung, N.; Klein, F.; Persigehl, T.; et al. COVID-19 associated pulmonary aspergillosis. Mycoses 2020, 63, 528–534. [Google Scholar] [CrossRef]
- Nasir, N.; Farooqi, J.; Mahmood, S.F.; Jabeen, K. COVID-19-associated pulmonary aspergillosis (CAPA) in patients admitted with severe COVID-19 pneumonia: An observational study from Pakistan. Mycoses 2020, 63, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Verweij, P.E.; Rijnders, B.J.A.; Brüggemann, R.J.M.; Azoulay, E.; Bassetti, M.; Blot, S.; Calandra, T.; Clancy, C.J.; Cornely, O.A.; Chiller, T.; et al. Review of influenza-associated pulmonary aspergillosis in ICU patients and proposal for a case definition: An expert opinion. Intensive Care Med. 2020, 46, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.; Valerio, M.; Álvarez-Uría, A.; Olmedo, M.; Veintimilla, C.; Padilla, B.; De La Villa, S.; Guinea, J.; Escribano, P.; Ruiz-Serrano, M.J.; et al. Invasive pulmonary aspergillosis in the COVID-19 era: An expected new entity. Mycoses 2021, 64, 132–143. [Google Scholar] [CrossRef]
- Langford, B.J.; So, M.; Raybardhan, S.; Leung, V.; Soucy, J.-P.R.; Westwood, D.; Daneman, N.; MacFadden, D.R. Antibiotic prescribing in patients with COVID-19: Rapid review and meta-analysis. Clin. Microbiol. Infect. 2021. [Google Scholar] [CrossRef] [PubMed]
- Seaton, R.A.; Gibbons, C.L.; Cooper, L.; Malcolm, W.; McKinney, R.; Dundas, S.; Griffith, D.; Jeffreys, D.; Hamilton, K.; Choo-Kang, B.; et al. Survey of antibiotic and antifungal prescribing in patients with suspected and confirmed COVID-19 in Scottish hospitals. J. Infect. 2020, 81, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.S.-K.; Chau, S.K.-Y.; Tso, E.Y.-K.; Tsang, S.W.-C.; Li, I.Y.-F.; Wong, B.K.-C.; Fung, K.S.-C. Bacterial co-infections and antibiotic prescribing practice in adults with COVID-19: Experience from a single hospital cluster. Ther. Adv. Infect. Dis. 2020, 7. [Google Scholar] [CrossRef]
- Shope, R.E. Swine influenza: III. Filtration experiments and etiology. J. Exp. Med. 1931, 54, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Shope, R.E. The etiology of swine influenza. Science 1931, 73, 214–215. [Google Scholar] [CrossRef]
- Brundage, J.F. Interactions between influenza and bacterial respiratory pathogens: Implications for pandemic pre-paredness. Lancet 2006, 6, 303–312. [Google Scholar] [CrossRef]
- Brundage, J.F.; Shanks, D.G. Deaths from bacterial pneumonia in the 1918-19 influenza pandemic. Emerg. Infect. Dis. 2008, 14, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Bime, C.; Casanova, N.G.; Nikolich-Zugich, J.; Knox, K.S.; Camp, S.M.; Garcia, J.G. Strategies to DAMPen COVID-19-mediated lung and systemic inflammation and vascular injury. Transl. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cicco, S.; Cicco, G.; Racanelli, V.; Vacca, A. Neutrophil Extracellular Traps (NETs) and Damage-Associated Molecular Patterns (DAMPs): Two Potential Targets for COVID-19 Treatment. Mediat. Inflamm. 2020, 2020, 7527953. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Birukov, K.G.; Birukova, A.A. EXPRESS: Extracellular histones in lung dysfunction: A new biomarker and therapeutic target? Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef]
- Tsujimoto, H.; Ono, S.; Majima, T.; Kawarabayashi, N.; Takayama, E.; Kinoshita, M.; Seki, S.; Hiraide, H.; Moldawer, L.L.; Mochizuki, H. NEUTROPHIL ELASTASE, MIP-2, AND TLR-4 EXPRESSION DURING HUMAN AND EXPERIMENTAL SEPSIS. Shock 2005, 23, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Paunel-Görgülü, A.; Wacker, M.; El Aita, M.; Hassan, S.; Schlachtenberger, G.; Deppe, A.; Choi, Y.-H.; Kuhn, E.; Mehler, T.O.; Wahlers, T. cfDNA correlates with endothelial damage after cardiac surgery with prolonged cardiopulmonary bypass and amplifies NETosis in an intracellular TLR9-independent manner. Sci. Rep. 2017, 7, 17421. [Google Scholar] [CrossRef] [PubMed]
- Huckriede, J.; Anderberg, S.B.; Morales, A. Markers of NETosis and DAMPs are altered in critically ill COVID-19 patients. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Andersson, U.; Ottestad, W.; Tracey, K.J. Extracellular HMGB1: A therapeutic target in severe pulmonary inflammation including COVID-19? Mol. Med. 2020, 26, 42. [Google Scholar] [CrossRef] [PubMed]
- Köseler, A.; Sabirli, R.; Gören, T.; Türkçüer, I.; Kurt, Ö. Endoplasmic Reticulum Stress Markers in SARS-COV-2 Infection and Pneumonia: Case-Control Study. In Vivo 2020, 34, 1645–1650. [Google Scholar] [CrossRef]
- Sabirli, R.; Koseler, A.; Goren, T.; Turkcuer, I.; Kurt, O. High GRP78 levels in Covid-19 infection: A case-control study. Life Sci. 2021, 265, 118781. [Google Scholar] [CrossRef] [PubMed]
- Preissner, K.T.; Fischer, S.; Deindl, E. Extracellular RNA as a Versatile DAMP and Alarm Signal That Influences Leukocyte Recruitment in Inflammation and Infection. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Shaver, C.M.; Landstreet, S.R.; Pugazenthi, S.; Scott, F.; Putz, N.; Ware, L.B.; Bastarache, J.A. The NLRP3 inflammasome in macrophages is stimulated by cell-free hemoglobin. Physiol. Rep. 2020, 8, e14589. [Google Scholar] [CrossRef]
- Meegan, J.E.; Shaver, C.M.; Putz, N.D.; Jesse, J.J.; Landstreet, S.R.; Lee, H.N.R.; Sidorova, T.N.; McNeil, J.B.; Wynn, J.L.; Cheung-Flynn, J.; et al. Cell-free hemoglobin increases inflammation, lung apoptosis, and microvascular permeability in murine polymicrobial sepsis. PLoS ONE 2020, 15, e0228727. [Google Scholar] [CrossRef]
- Zhou, H.; Li, C.; Hu, T.; Liu, T.; Ni, N.; Chen, W.; Zhao, H.; Ruan, S.; Li, J.; Wu, H.; et al. Total infectomes of 162 SARS-CoV-2 cases using meta-transcriptomic sequencing. J. Infect. 2021, 82, e44–e48. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, T.; Weißbrich, B.; Wedekink, F.; Notz, Q.; Herrmann, J.; Krone, M.; Sitter, M.; Schmid, B.; Kredel, M.; Stumpner, J.; et al. Biodistribution and serologic response in SARS-CoV-2 induced ARDS: A cohort study. PLoS ONE 2020, 15, e0242917. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Cocon, L.; Aublin-Gex, A.; Sestito, S.E.; Shirey, K.A.; Patel, M.C.; André, P.; Blanco, J.C.; Vogel, S.N.; Peri, F.; Lotteau, V. TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Sci. Rep. 2017, 7, srep40791. [Google Scholar] [CrossRef] [PubMed]
- Shinya, K.; Ito, M.; Makino, A.; Tanaka, M.; Miyake, K.; Eisfeld, A.J.; Kawaoka, Y. The TLR4-TRIF Pathway Protects against H5N1 Influenza Virus Infection. J. Virol. 2011, 86, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; Van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.C.; Wang, H.; et al. Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Lester, S.N.; Li, K. Toll-Like Receptors in Antiviral Innate Immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Thindwa, D.; Quesada, M.G.; Liu, Y.; Bennett, J.; Cohen, C.; Knoll, M.D.; von Gottberg, A.; Hayford, K.; Flasche, S. Use of seasonal influenza and pneumococcal polysaccharide vaccines in older adults to reduce COVID-19 mortality. Vaccine 2020, 38, 5398–5401. [Google Scholar] [CrossRef] [PubMed]
- Jehi, L.; Ji, X.; Milinovich, A.; Erzurum, S.; Rubin, B.P.; Gordon, S.; Young, J.B.; Kattan, M.W. Individualizing Risk Prediction for Positive Coronavirus Disease 2019 Testing. Chest 2020, 158, 1364–1375. [Google Scholar] [CrossRef]
- Noale, M.; Trevisan, C.; Maggi, S.; Incalzi, R.A.; Pedone, C.; Di Bari, M.; Adorni, F.; Jesuthasan, N.; Sojic, A.; Galli, M.; et al. The Association between Influenza and Pneumococcal Vaccinations and SARS-Cov-2 Infection: Data from the EPICOVID19 Web-Based Survey. Vaccines 2020, 8, 471. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.C.; Cutland, C.L.; Klugman, K.P.; Madhi, S.A. Pneumococcal Conjugate Vaccine Protection against Coronavirus-Associated Pneumonia Hospitalization in Children Living with and without HIV. mBio 2021, 12. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Age and Location in Severity of COVID-19 Pathology: Do Lactoferrin and Pneumococcal Vaccination Explain Low Infant Mortality and Regional Differences? BioEssays 2020, 42. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Possible Cross-Reactivity between SARS-CoV-2 Proteins, CRM197 and Proteins in Pneumococcal Vaccines May Protect Against Symptomatic SARS-CoV-2 Disease and Death. Vaccines 2020, 8, 559. [Google Scholar] [CrossRef]
- Pawlowski, C.; Puranik, A.; Bandi, H.; Venkatakrishnan, A.J.; Agarwal, V.; Kennedy, R.; O’Horo, J.C.; Gores, P.G.J.; Williams, A.W.; Halamka, P.J.; et al. Exploratory analysis of immunization records highlights decreased SARS-CoV-2 rates in individuals with recent non-COVID-19 vaccina-tions. medRxiv 2020. [Google Scholar] [CrossRef]
- Sumbul, B.; Sumbul, H.E.; Okyay, R.A.; Gülümsek, E.; Şahin, A.R.; Boral, B.; Koçyiğit, B.F.; Alfishawy, M.; Gold, J.; Tasdogan, A.M. Is there a link between pre-existing antibodies acquired due to childhood vaccinations or past infections and COVID-19? A case control study. PeerJ 2021, 9, e10910. [Google Scholar] [CrossRef]
- Blot, M.; Jacquier, M.; Manoha, C.; Piroth, L.; Charles, P.-E.; Glele, L.-S.A.; Beltramo, G.; Nguyen, M.; Bonniaud, P.; Prin, S.; et al. Alveolar SARS-CoV-2 Viral Load Is Tightly Correlated with Severity in COVID-19 ARDS. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Bitker, L.; Dhelft, F.; Chauvelot, L.; Frobert, E.; Folliet, L.; Mezidi, M.; Trouillet-Assant, S.; Belot, A.; Lina, B.; Wallet, F.; et al. Protracted viral shedding and viral load are associated with ICU mortality in Covid-19 patients with acute respiratory failure. Ann. Intensiv. Care 2020, 10, 167. [Google Scholar] [CrossRef] [PubMed]
- Terán-Cabanillas, E.; Montalvo-Corral, M.; Silva-Campa, E.; Caire-Juvera, G.; Moya-Camarena, S.Y.; Hernández, J. Production of interferon α and β, pro-inflammatory cytokines and the expression of suppressor of cytokine signaling (SOCS) in obese subjects infected with influenza A/H1N1. Clin. Nutr. 2014, 33, 922–926. [Google Scholar] [CrossRef]
- Kleszczyński, K.; Slominski, A.T.; Steinbrink, K.; Reiter, R.J. Clinical Trials for Use of Melatonin to Fight against COVID-19 Are Urgently Needed. Nutrients 2020, 12, 2561. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P.; Brown, G.M.; Reiter, R.J.; Pandi-Perumal, S.R. Elderly as a High-risk Group during COVID-19 Pandemic: Effect of Circadian Misalignment, Sleep Dysregulation and Melatonin Administration. Sleep Vigil. 2020, 4, 81–87. [Google Scholar] [CrossRef]
- El-Missiry, M.A.; El-Missiry, Z.M.; Othman, A.I. Melatonin is a potential adjuvant to improve clinical outcomes in individuals with obesity and diabetes with coexistence of Covid-19. Eur. J. Pharmacol. 2020, 882, 173329. [Google Scholar] [CrossRef] [PubMed]
- Juybari, K.B.; Pourhanifeh, M.H.; Hosseinzadeh, A.; Hemati, K.; Mehrzadi, S. Melatonin potentials against viral infections including COVID-19: Current evidence and new findings. Virus Res. 2020, 287, 198108. [Google Scholar] [CrossRef] [PubMed]
- Artigas, L.; Coma, M.; Matos-Filipe, P.; Aguirre-Plans, J.; Farrés, J.; Valls, R.; Fernandez-Fuentes, N.; De La Haba-Rodriguez, J.; Olvera, A.; Barbera, J.; et al. In-silico drug repurposing study predicts the combination of pirfenidone and melatonin as a promising candidate therapy to reduce SARS-CoV-2 infection progression and respiratory distress caused by cytokine storm. PLoS ONE 2020, 15, e0240149. [Google Scholar] [CrossRef] [PubMed]
- Ramlall, V.; Zucker, J.; Tatonetti, N. Melatonin is significantly associated with survival of intubated COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Grailer, J.J.; Wang, N.; Wang, M.; Yao, J.; Zhong, R.; Gao, G.F.; Ward, P.A.; Tan, D.-X.; et al. Melatonin alleviates acute lung injury through inhibiting the NLRP3 inflammasome. J. Pineal Res. 2016, 60, 405–414. [Google Scholar] [CrossRef]
- Wu, H.-M.; Zhao, C.-C.; Xie, Q.-M.; Xu, J.; Fei, G.-H. TLR2-Melatonin Feedback Loop Regulates the Activation of NLRP3 Inflammasome in Murine Allergic Airway Inflammation. Front. Immunol. 2020, 11, 172. [Google Scholar] [CrossRef] [Green Version]
- Bonomini, F.; Dos Santos, M.; Veronese, F.V.; Rezzani, R. NLRP3 Inflammasome Modulation by Melatonin Supplementation in Chronic Pristane-Induced Lupus Nephritis. Int. J. Mol. Sci. 2019, 20, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-H.; Cao, X.-J.; Wei, W. Melatonin decreases TLR3-mediated inflammatory factor expression via inhibition of NF-B activation in respiratory syncytial virus-infected RAW264.7 macrophages. J. Pineal Res. 2008, 45, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, A.D.; Yalcin, A.N. Future perspective: Biologic agents in patients with severe COVID-19. Immunopharmacol. Immunotoxicol. 2021, 43, 1–7. [Google Scholar] [CrossRef]
- Della-Torre, E.; Campochiaro, C.; Cavalli, G.; De Luca, G.; Napolitano, A.; La Marca, S.; Boffini, N.; Da Prat, V.; Di Terlizzi, G.; Lanzillotta, M.; et al. Interleukin-6 blockade with sarilumab in severe COVID-19 pneumonia with systemic hyperinflammation: An open-label cohort study. Ann. Rheum. Dis. 2020, 79, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Sarfraz, A.; Sarfraz, Z.; Sarfraz, M.; Aftab, H.; Pervaiz, Z. Tocilizumab and COVID-19: A Meta-Analysis of 2120 Patients with Severe Disease and Implications for Clinical Trial Methodologies. Turk. J. Med. Sci. 2021. [Google Scholar] [CrossRef]
- Salvarani, C.; Dolci, G.; Massari, M.; Merlo, D.F.; Cavuto, S.; Savoldi, L.; Bruzzi, P.; Boni, F.; Braglia, L.; Turrà, C.; et al. Effect of Tocilizumab vs Standard Care on Clinical Worsening in Patients Hospitalized With COVID-19 Pneumonia. JAMA Intern. Med. 2021, 181, 24. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Julián, E.; López-Veloso, M.; De-La-Torre-Ferrera, N.; Barraza-Vengoechea, J.C.; Delgado-López, P.D.; Colazo-Burlato, M.; Ubeira-Iglesias, M.; Montero-Baladía, M.; Lorenzo-Martín, A.; Minguito-De-La-Iglesia, J.; et al. High dose subcutaneous Anakinra to treat acute respiratory distress syndrome secondary to cytokine storm syndrome among severely ill COVID-19 patients. J. Autoimmun. 2020, 115, 102537. [Google Scholar] [CrossRef] [PubMed]
- Aomar-Millán, I.F.; Salvatierra, J.; Torres-Parejo, Ú.; Faro-Miguez, N.; Callejas-Rubio, J.L.; Ceballos-Torres, Á.; Cruces-Moreno, M.T.; Gómez-Jiménez, F.J.; Hernández-Quero, J.; Anguita-Santos, F. Anakinra after treatment with corticosteroids alone or with tocilizumab in patients with severe COVID-19 pneumonia and moderate hyperinflammation. A retrospective cohort study. Intern. Emerg. Med. 2021, 1–10. [Google Scholar] [CrossRef]
- Demidowich, A.P.; Davis, A.I.; Dedhia, N.; Yanovski, J.A. Colchicine to decrease NLRP3-activated inflammation and improve obesity-related metabolic dysregulation. Med. Hypotheses 2016, 92, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Reyes, A.Z.; Hu, K.A.; Teperman, J.; Muskardin, T.L.W.; Tardif, J.-C.; Shah, B.; Pillinger, M.H. Anti-inflammatory therapy for COVID-19 infection: The case for colchicine. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Lauterio, T.J.; Wolfe, H.R. Mechanism of Action of Colchicine in the Treatment of Gout. Clin. Ther. 2014, 36, 1465–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrachatis, D.A.; Giannopoulos, G.V.; Giotaki, S.G.; Raisakis, K.; Kossyvakis, C.; Iliodromitis, K.E.; Reimers, B.; Tousoulis, D.; Cleman, M.; Stefanadis, C.; et al. Impact of colchicine on mortality in patients with COVID-19: A meta-analysis. Hell. J. Cardiol. 2021, 6. [Google Scholar] [CrossRef]
- Patra, M.C.; Choi, S. Recent progress in the development of Toll-like receptor (TLR) antagonists. Expert Opin. Ther. Patents 2016, 26, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Chemokines and chemokine receptors during COVID-19 infection. Comput. Struct. Biotechnol. J. 2021, 19, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Andreakos, E.; Tsiodras, S. COVID-19: Lambda interferon against viral load and hyperinflammation. EMBO Mol. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Nile, S.H.; Nile, A.; Qiu, J.; Li, L.; Jia, X.; Kai, G. COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020, 53, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Vanderheiden, A.; Ralfs, P.; Chirkova, T.; Upadhyay, A.A.; Zimmerman, M.G.; Bedoya, S.; Aoued, H.; Tharp, G.M.; Pellegrini, K.L.; Manfredi, C.; et al. Type I and Type III IFN Restrict SARS-CoV-2 Infection of Human Airway Epithelial Cultures. J. Virol. 2020, 94, e00985-20. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, I.; Fernbach, S.; Pohl, M.O.; Karakus, U.; Huber, M.; Trkola, A.; Stertz, S.; Hale, B.G. Antiviral Activity of Type I, II, and III Interferons Counterbalances ACE2 Inducibility and Restricts SARS-CoV-2. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Kandel, C.; Biondi, M.J.; Kozak, R.A.; Zahoor, M.A.; Lemieux, C.; Borgia, S.M.; Boggild, A.K.; Powis, J.; McCready, J.; et al. Peginterferon lambda for the treatment of outpatients with COVID-19: A phase 2, placebo-controlled randomised trial. Lancet Respir. Med. 2021. Available online: https://www.thelancet.com/journals/lanres/article/PIIS2213-2600(20)30566-X/fulltext (accessed on 17 February 2021). [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptors | TLR1 | TLR2 | TLR3 | TLR4 | TLR5 | TLR6 | TLR7 | TLR8 | TLR9 | TLR10 | NOD1 | NOD2 | NL-RP3 | RIG1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Activated by: | Lipo-peptides | Gram-PGN, LTA & HSP, HMGB1 | ds-RNA, polyI:C, DAMP | Gram+ LPS & HSP, HMGB1 | Flagellin | LTA, lipopeps | ss-RNA | ss- RNA & pyogenic Bacteria | CpG DNA & mtDNA | Retroviral RNA | Meso-DAP | MDP | ? | Viral RNA |
| COVID-19 SEVERE | - | ^ | +/- | ^ | - | - | ^ | - | +/- | - | - | - | ^ | - |
| INFLUENZA-ASSOCIATED ALI/ARDS | v | ^ | ^ | ^ | - | ^ | +/- | ^ | ^ | v | ||||
| SEPSIS/ALI (murine) | ^ | +/- | ^ | - | ^ | ^ | - | - | ^ | |||||
| SEPSIS human patients | - | +/- | - | ^ | +/- | - | +/- | - | -/v | - | - | - | ^ | |
| Polymicrobial SEPSIS murine model | - | ^ | - | ^ | - | - | ^ | - | - | - | - | ^ | ||
| SEPSIS CONCENSUS | - | ^ | - | ^ | - | - | ^ | - | - | - | - | ^ |
| Receptor: | TLR 1 | TLR 2 | TLR 3 | TLR 4 | TLR 5 | TLR 6 | TLR 7 | TLR 8 | TLR 9 | TLR 10 | NOD 1 | NOD 2 | NL-RP 3 | RIG 1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Activated by: | Lipopeptides | Gram- PGN, LTA & HSP, HMGB1 | ds-RNA, polyI:C, DAMP | Gram+ LPS & HSP, HMGB1 | Flagellin | LTA, lipopeps | ss-RNA | ss- RNA & pyogenic Bacteria | CpG DNA & mtDNA | Retroviral RNA | Meso-DAP | MDP | ? | Viral RNA |
| CoV 229E | - | - | - | ^ | - | v | ||||||||
| SARS-CoV-1 | - | +/- | ^ | - | - | - | ^ | ^ | v | - | ^ | v | ||
| MERS | ^ | v | ^ | +/- | ^ | v | ||||||||
| SARS-CoV-2 | +/- | ^ | ^ | ^ | ^ | v | ||||||||
| Coronavirus CONCENSUS | ^ | ^ | ^ | v | ||||||||||
| Influenza A viruses | - | v | ^ | v | - | - | ^ | ^ | ^ | - | ^ | ^ | ^ | |
| Rhinoviruses | ^ | ^ | - | +/- | ||||||||||
| Respiratory syncytial virus | - | - | ^ | +/- | - | ^ | ^ | +/- | ^ | ^ | ^ | |||
| Adenovirus | ^ | ^ | ^ | ^ | ^ | |||||||||
| Coxsackie-viruses | ^ | ^ | ^ | ^ | ^ | ^ | ^ | |||||||
| RESP VIRUS CONCENSUS | - | - | ^ | - | - | - | ^ | ^ | ^ | ^ | ^ | ^ |
| TLR 1 | TLR 2 | TLR 3 | TLR 4 | TLR 5 | TLR 6 | TLR 7 | TLR 8 | TLR 9 | TLR 10 | NOD 1 | NOD 2 | NL-RP3 | RIG 1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipopeptides | Gram- PGN, LTA & HSP, HMGB1 | ds-RNA, polyI:C, DAMP | Gram+ LPS & HSP, HMGB1 | Flagellin | LTA, lipopeps | ss-RNA | ss- RNA & pyogenic Bacteria | CpG DNA & mtDNA | Retroviral RNA | Meso-DAP | MDP | ? | Viral RNA | |
| GRAM POS | ||||||||||||||
| Group A Streptococci | ^ | ^ | - | ^ | - | - | - | ^ | - | - | ^ | ^ | ||
| Group B Streptococci | - | ^ | ^ | ^ | - | - | ^ | |||||||
| Staphylococcus aureus | ^ | ^ | ^ | ^ | - | ^ | ^ | |||||||
| GRAM AMBI | ||||||||||||||
| Mycobacterium tuberculosis | ^ | ^ | +/- | ^ | ^ | ^ | ^ | |||||||
| GRAM NEG | ||||||||||||||
| Klebsiella pneumoniae | ^ | ^ | ^ | ^ | ^ | |||||||||
| Haemophilus influenzae | ^ | +/- | - | ^ | ^ | ^ | ||||||||
| Legionella pneumophila | ^ | ^ | ^ | - | - | ^ | ||||||||
| Chlamydia pneumoniae | ^ | ^ | ^ | ^ | ^ | |||||||||
| Neisseria meningitidis | ^ | ^ | ^ | ^ | ^ | ^ | ||||||||
| Pseudomonas aeruginosa | ^ | ^ | ^ | ^ | ^ | ^ | ||||||||
| NO GRAM | ||||||||||||||
| Mycoplasma pneumoniae | ^ | ^ | ^ | ^ | - | - | ^ | |||||||
| BACTERIA CONCENSUS | ^ | ^ | ^ | ^ | +/- | ^ | ^ |
| TLR 1 | TLR 2 | TLR 3 | TLR 4 | TLR 5 | TLR 6 | TLR 7 | TLR 8 | TLR 9 | TLR 10 | NOD 1 | NOD 2 | NL-RP 3 | RIG 1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipopeptides | Gram- PGN, LTA & HSP, HMGB1 | ds-RNA, polyI:C, DAMP | Gram+ LPS & HSP, HMGB1 | Flagellin | LTA, lipopeps | ss-RNA | ss-RNA & pyogenic Bacteria | CpG DNA & mtDNA | Retroviral RNA | Meso-DAP | MDP | ? | Viral RNA | |
| Aspergillus spp. | ^ | ^ | ^ | ^ | ^ | ^ | ^ | |||||||
| Candida spp. | ^ | ^ | ^ | ^ | ^ | - | ^ | |||||||
| Cryptococcus spp. | ^ | ^ | ^ | ^ | ^ | |||||||||
| FUNGI CONSENSUS | ^ | ^ | ^ | ^ | ^ |
| TLR 1 | TLR 2 | TLR 3 | TLR 4 | TLR 5 | TLR 6 | TLR 7 | TLR 8 | TLR 9 | TLR 10 | NOD 1 | NOD 2 | NLRP 3 | RIG 1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| @@ | Lipo-peptides | Gram- PGN, LTA & HSP, HMGB1 | ds-RNA, polyI:C, DAMP | Gram+ LPS & HSP, HMGB1 | Flagellin | LTA, lipopeps | ss-RNA | ss- RNA & pyogenic Bacteria | CpG DNA & mtDNA | Retroviral RNA | Meso-DAP | MDP | ? | Viral RNA |
| Coronavirus CONSENSUS | ^ | ^ | ^ | v | ||||||||||
| RESP VIRUS CONCENSUS | - | - | ^ | - | - | - | ^ | ^ | ^ | ^ | ^ | ^ | ||
| BACTERIA CONSENSUS | ^ | ^ | ^ | G+ G- | ^ | ^ | ||||||||
| FUNGI CONSENSUS | ^ | ^ | ^ | ^ | ^ | |||||||||
| COVID-19 SEVERE CONSENSUS | - | ^ | +/- | ^ | - | - | ^ | - | +/- | - | - | - | ^ | - |
| INFLUENZA-ASSOCIATED ALI/ARDS | v | ^ | ^ | ^ | - | ^ | +/- | ^ | ^ | v | ||||
| SEPSIS human patients | - | +/- | - | ^ | +/- | - | +/- | - | -/v | - | - | - | ^ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root-Bernstein, R. Innate Receptor Activation Patterns Involving TLR and NLR Synergisms in COVID-19, ALI/ARDS and Sepsis Cytokine Storms: A Review and Model Making Novel Predictions and Therapeutic Suggestions. Int. J. Mol. Sci. 2021, 22, 2108. https://doi.org/10.3390/ijms22042108
Root-Bernstein R. Innate Receptor Activation Patterns Involving TLR and NLR Synergisms in COVID-19, ALI/ARDS and Sepsis Cytokine Storms: A Review and Model Making Novel Predictions and Therapeutic Suggestions. International Journal of Molecular Sciences. 2021; 22(4):2108. https://doi.org/10.3390/ijms22042108
Chicago/Turabian StyleRoot-Bernstein, Robert. 2021. "Innate Receptor Activation Patterns Involving TLR and NLR Synergisms in COVID-19, ALI/ARDS and Sepsis Cytokine Storms: A Review and Model Making Novel Predictions and Therapeutic Suggestions" International Journal of Molecular Sciences 22, no. 4: 2108. https://doi.org/10.3390/ijms22042108