
Metal-Bound Methisazone; Novel Drugs Targeting Prophylaxis and Treatment of SARS-CoV-2, a Molecular Docking Study

 ,
,  ,
,  , ,
, ,  ,
,  ,
,  ,
,  , ,
, ,  ,
,  ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Molecular Docking Interaction Data
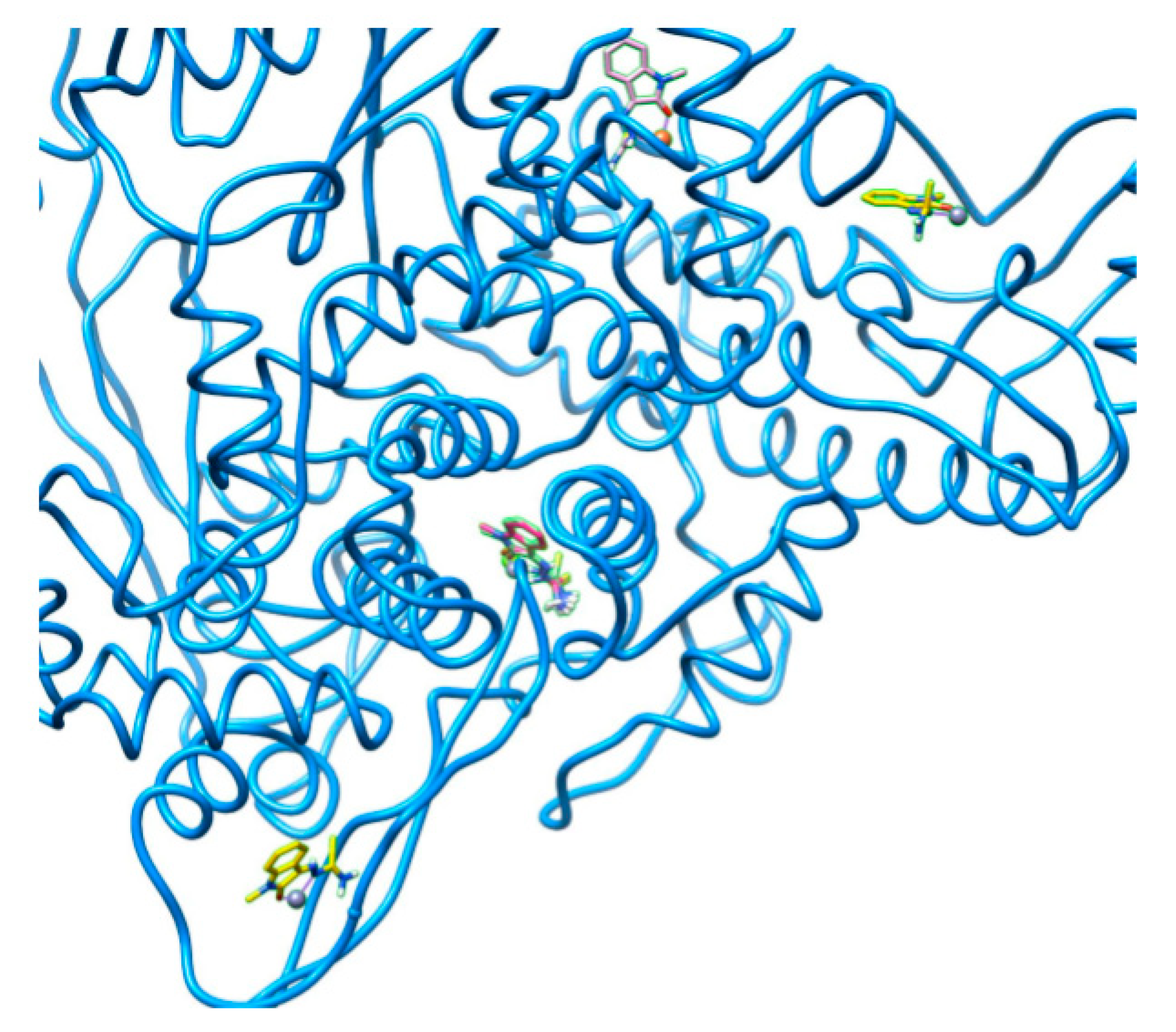
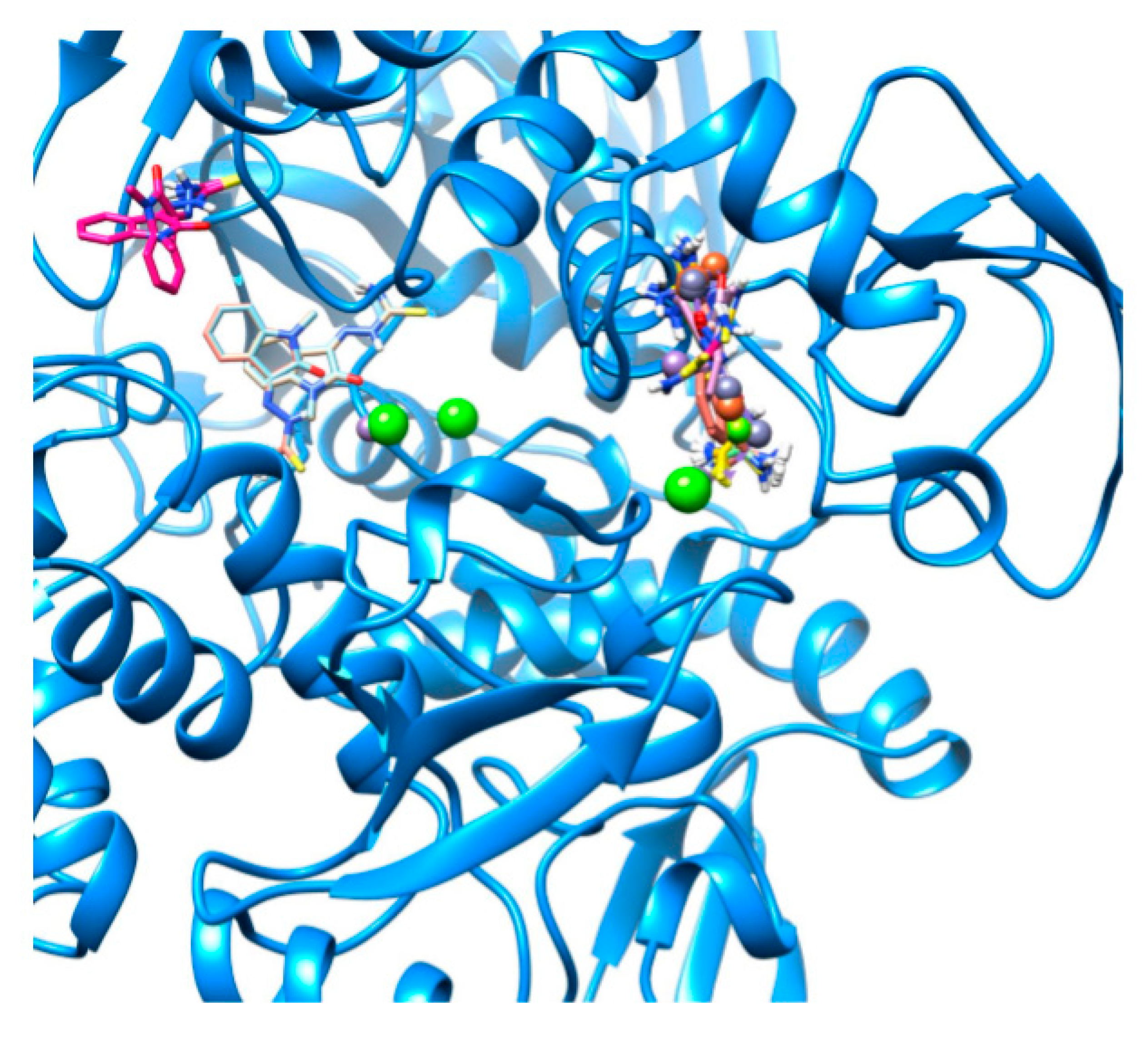
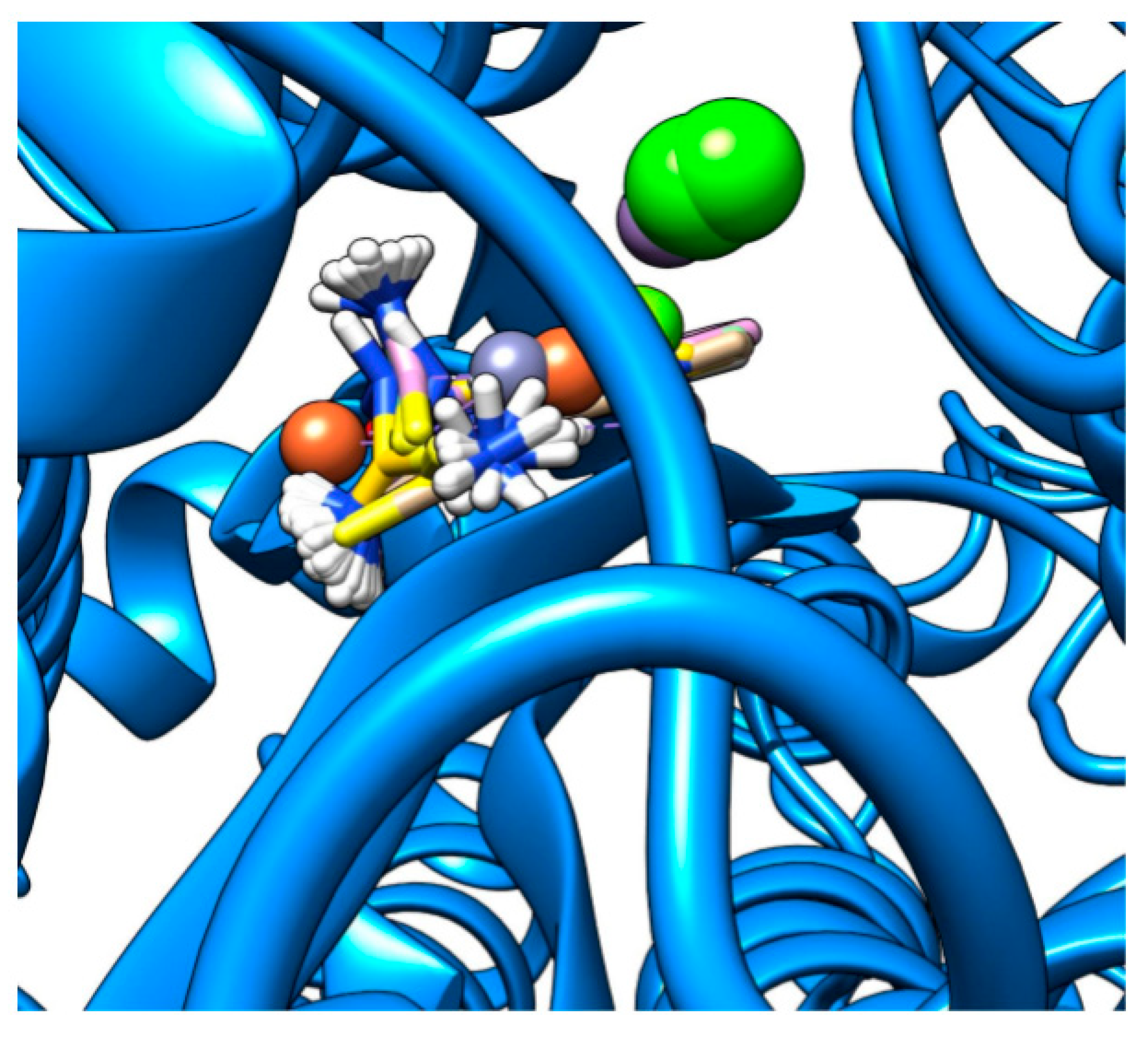
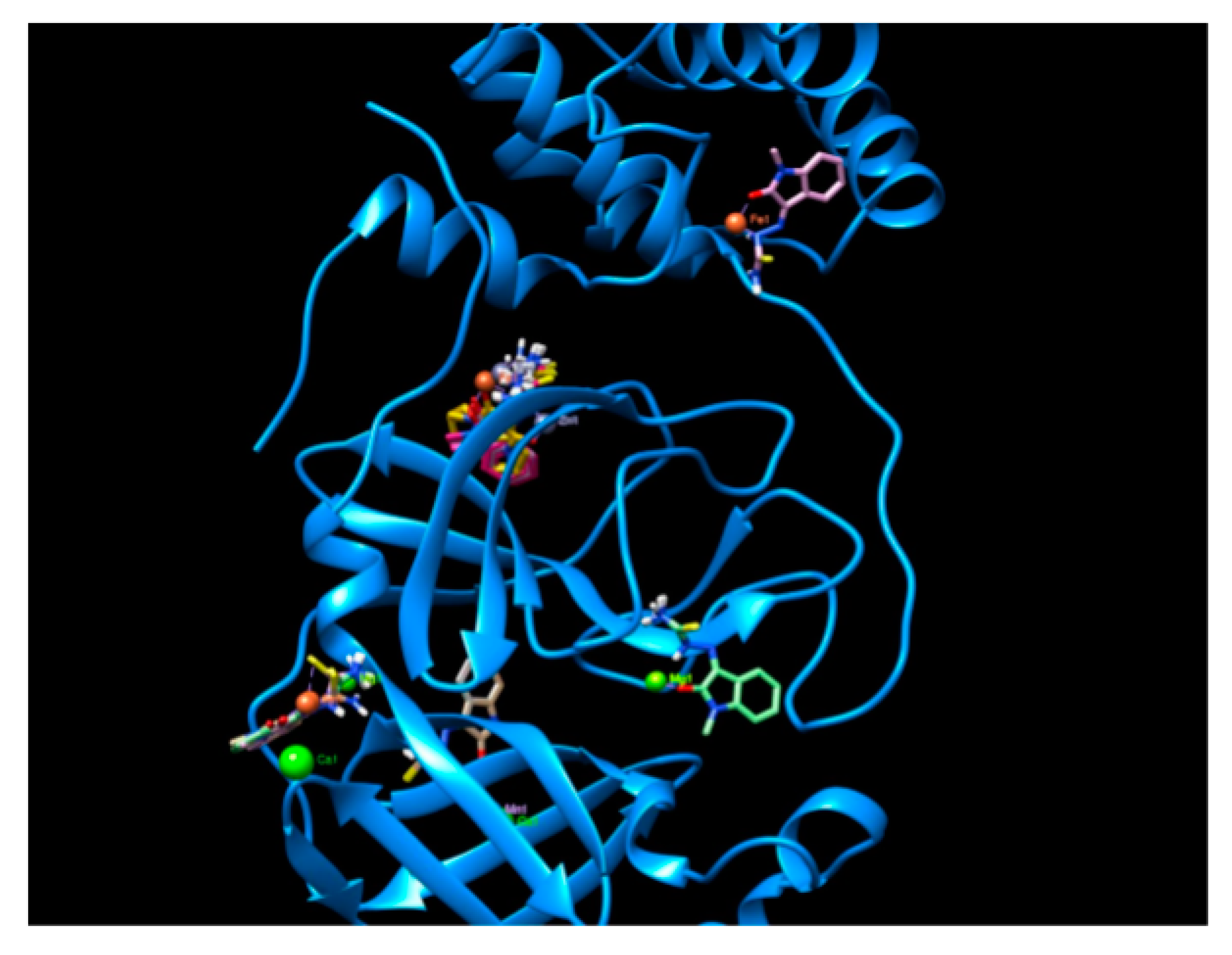
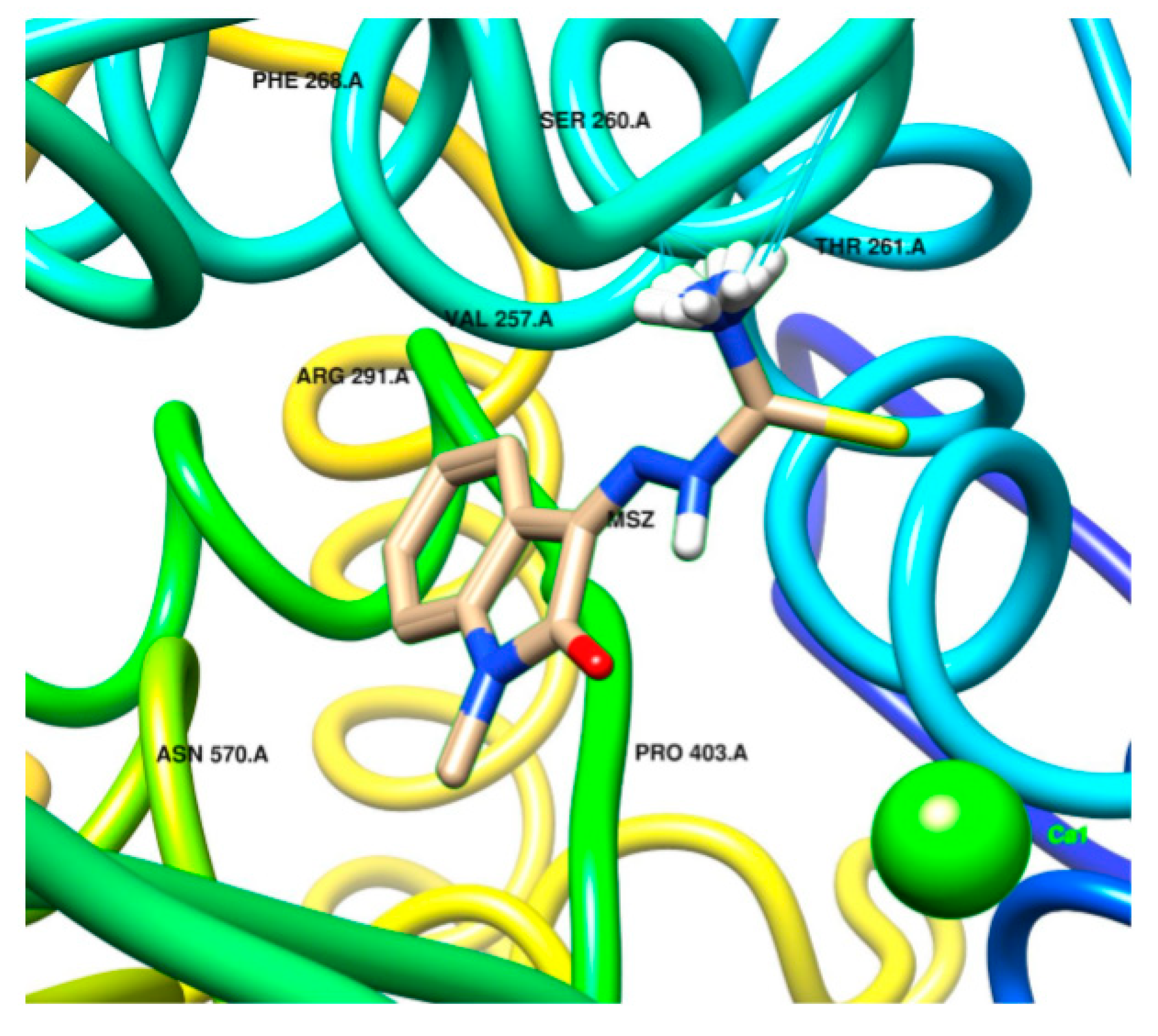
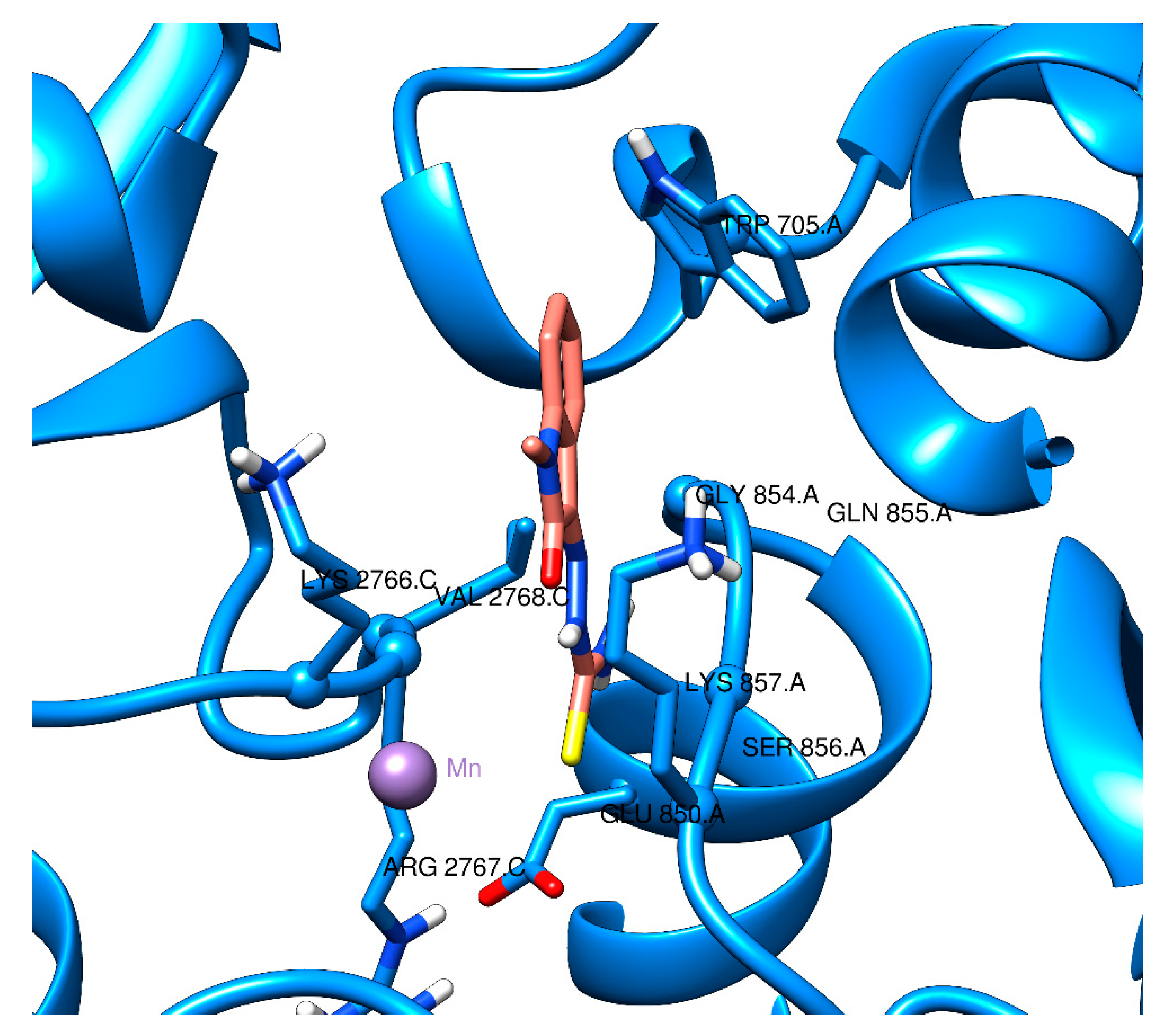
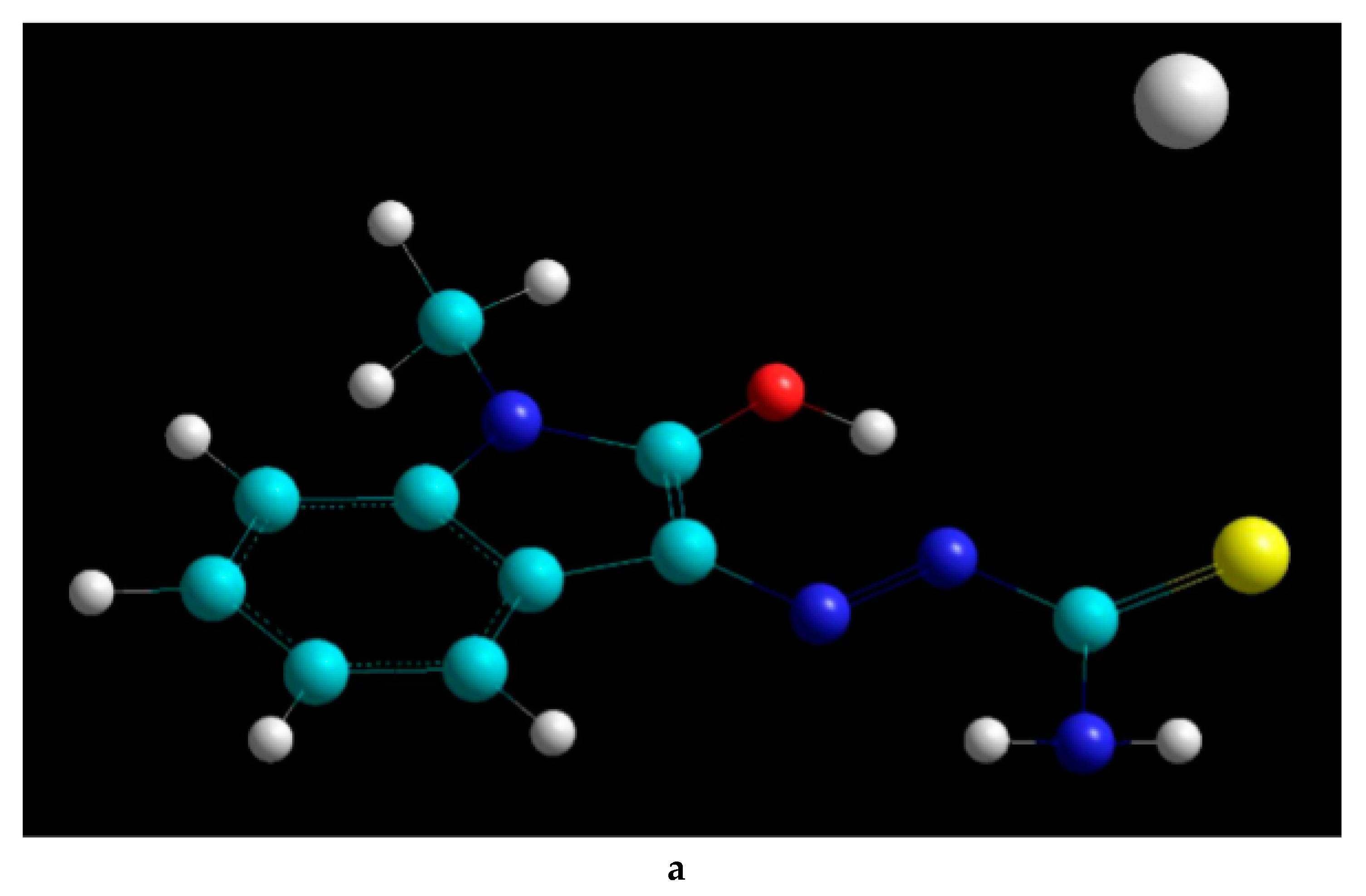
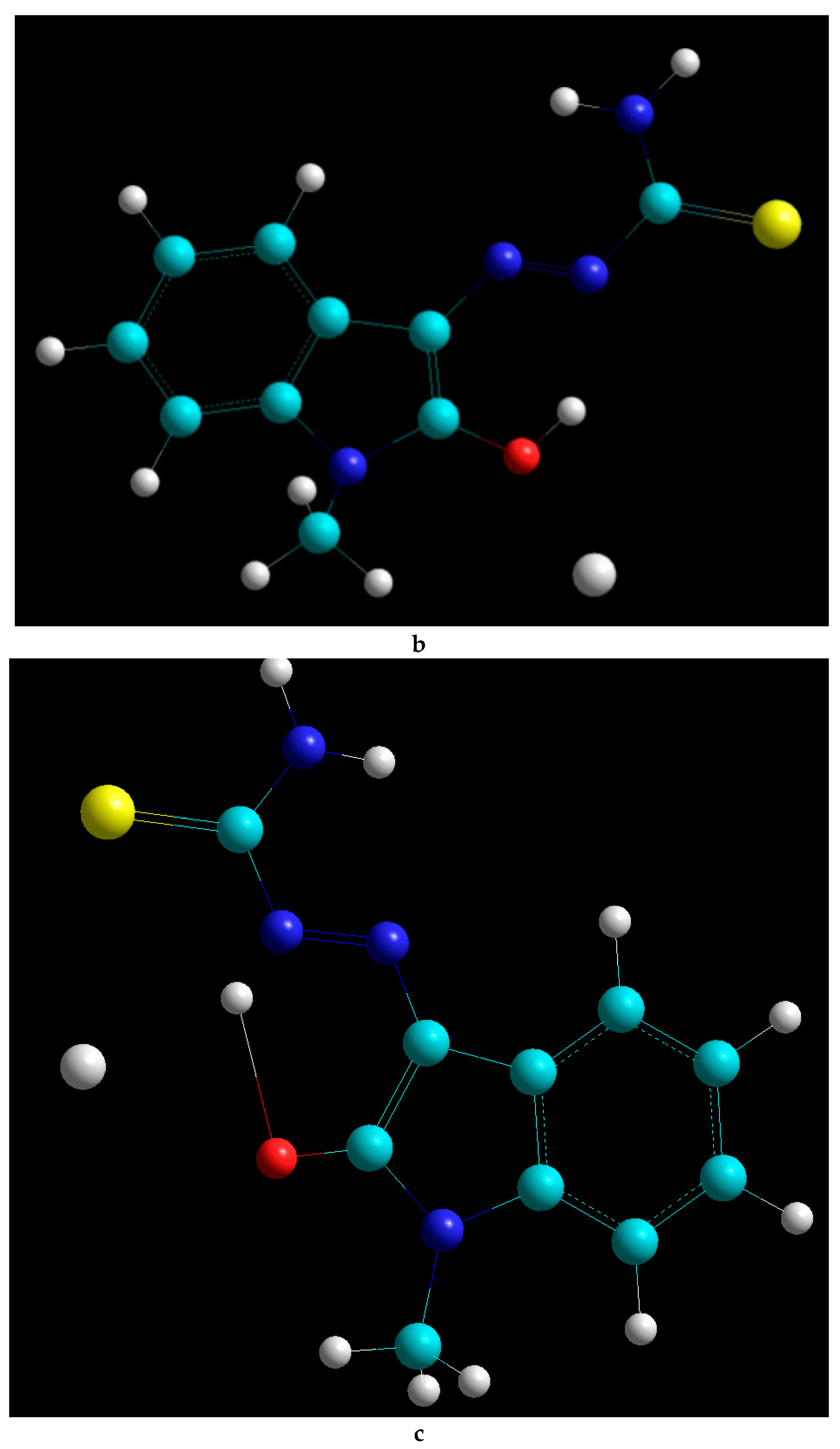
2.2. Docking Visualization
3. Discussion
- -
- Methisazone and the metal were placed in random positions (at least 20 different ones).
- -
- A geometry optimization reached a local energetic minimum.
- -
- A molecular dynamics simulation was employed to move the Methisazone–metal system from equilibrium.
- -
- If the attempt was not successful, the local minimum was the global minimum, and the complex represented the real structure. All properties of the complex were fed to Chimera for the molecular docking investigation (end of optimization).
- -
- If the attempt was successful, a new geometry optimization was run to find a new energetic minimum.
- -
- The process was continued until the global minimum was reached, and all properties were fed to Chimera for the molecular docking investigation (end of optimization).
4. Materials and Methods
4.1. Structure Preparation
4.2. Docking Analysis
4.3. Drug Candidate Analysis
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- World Health Organization. Coronavirus Disease 2019 (COVID-19): Situation Report. Available online: https://covid19.who.int/ (accessed on 10 January 2021).
- CDC. 2009 H1N1 Pandemic (H1N1pdm09 Virus) | Pandemic Influenza (Flu). Available online: https://www.cdc.gov/flu/pandemic-resources/2009-h1n1-pandemic.html (accessed on 29 April 2020).
- Katul, G.G.; Mrad, A.; Bonetti, S.; Manoli, G.; Parolari, A.J. Global convergence of COVID-19 basic reproduction number and estimation from early-time SIR dynamics. PLoS ONE 2020, 15, e0239800. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gayle, A.A.; Wilder-Smith, A.; Rocklöv, J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J. Travel Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in Hospitalized Patients with Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Jorgensen, S.C.J.; Kebriaei, R.; Dresser, L.D. Remdesivir: Review of Pharmacology, Pre-clinical Data, and Emerging Clinical Experience for COVID-19. Pharmacotherapy 2020, 40, 659–671. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Toor, H.G.; Banerjee, D.I.; Lipsa Rath, S.; Darji, S.A. Computational drug re-purposing targeting the spike glycoprotein of SARS-CoV-2 as an effective strategy to neutralize COVID-19. Eur. J. Pharmacol. 2021, 890, 173720. [Google Scholar] [CrossRef] [PubMed]
- Wise, J. Covid-19: New coronavirus variant is identified in UK. BMJ 2020, 371, m4857. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, T.; Rarey, M. Computational methods for biomolecular docking. Curr. Opin. Struct. Biol. 1996, 6, 402–406. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef] [Green Version]
- Shah, B.; Modi, P.; Sagar, S.R. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020, 252, 117652. [Google Scholar] [CrossRef]
- Oxford, J.S.; Öberg, B. Conquest of Viral Diseases: A Topical Review of Drugs and Vaccines; Elsevier: Amsterdam, The Netherlands, 1985. [Google Scholar]
- Bauer, D.; St Vincent, L. Prophylactic Treatment of Smallpox Contacts with N-Methylisatin (β-Thiosemicarbazone (Compound 33T57, Marboran). Lancet 1963, 282, 494–496. [Google Scholar] [CrossRef]
- Bauer, D.; Vincent, L.; Kempe, C.; Young, P.; Downie, A. Prophylaxis of smallpox with methisazone. Am. J. Epidemiol. 1969, 90, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.L. Some observations on the role of methisazone (‘Marboran’) in the prophylaxis of smallpox in a rural area. South Afr. Med J. 1964, 38, 868–869. [Google Scholar]
- Khan, S.A.; Asiri, A.M.; Al-Amry, K.; Malik, M.A. Synthesis, characterization, electrochemical studies, and in vitro antibacterial activity of novel thiosemicarbazone and its Cu(II), Ni(II), and Co(II) complexes. Sci. World J. 2014, 2014, 592375. [Google Scholar] [CrossRef] [Green Version]
- Shityakov, S.; Förster, C. In silico predictive model to determine vector-mediated transport properties for the blood-brain barrier choline transporter. Adv. Appl. Bioinform. Chem. 2014, 7, 23–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, L. In Silico Molecular Dynamics Docking of Drugs to the Inhibitory Active Site of SARS-CoV-2 Protease and Their Predicted Toxicology and ADME. SSRN Electron. J. 2020. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | 6M71_A PDB: 6M71 | 6VYB PDB: 6VYB | 6W9C PDB: 6W9C | 6Y2E_A PDB: 6Y2E |
|---|---|---|---|---|
| MSZ | −7.1 | −7.7 | −6.8 | −6.2 |
| Ca–MSZ | −7.1 | −8 | −6.9 | −6.4 |
| Fe–MSZ | −6.9 | −7.9 | −7.4 | −6.6 |
| Mg–MSZ | −6.8 | −7.9 | −7.3 | −6.4 |
| Mn–MSZ | −7.1 | −8.3 | −7.1 | −6.3 |
| Zn–MSZ | −7 | −8 | −7.1 | −6.7 |
| Complex | 6M71_A | 6VYB | 6W9C | 6Y2E_A |
|---|---|---|---|---|
| Ca–MSZ | 0 | −0.3 | −0.1 | −0.2 |
| Fe–MSZ | 0.2 | −0.2 | −0.6 | −0.4 |
| Mg–MSZ | 0.3 | −0.2 | −0.5 | −0.2 |
| Mn–MSZ | 0 | −0.6 | −0.3 | −0.1 |
| Zn–MSZ | 0.1 | −0.3 | −0.3 | −0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelaal Ahmed Mahmoud M. Alkhatip, A.; Georgakis, M.; Montero Valenzuela, L.R.; Hamza, M.; Farag, E.; Hodgkinson, J.; Hosny, H.; Kamal, A.M.; Wagih, M.; Naguib, A.; et al. Metal-Bound Methisazone; Novel Drugs Targeting Prophylaxis and Treatment of SARS-CoV-2, a Molecular Docking Study. Int. J. Mol. Sci. 2021, 22, 2977. https://doi.org/10.3390/ijms22062977
Abdelaal Ahmed Mahmoud M. Alkhatip A, Georgakis M, Montero Valenzuela LR, Hamza M, Farag E, Hodgkinson J, Hosny H, Kamal AM, Wagih M, Naguib A, et al. Metal-Bound Methisazone; Novel Drugs Targeting Prophylaxis and Treatment of SARS-CoV-2, a Molecular Docking Study. International Journal of Molecular Sciences. 2021; 22(6):2977. https://doi.org/10.3390/ijms22062977
Chicago/Turabian StyleAbdelaal Ahmed Mahmoud M. Alkhatip, Ahmed, Michail Georgakis, Lucio R. Montero Valenzuela, Mohamed Hamza, Ehab Farag, Jaqui Hodgkinson, Hisham Hosny, Ahmed M. Kamal, Mohamed Wagih, Amr Naguib, and et al. 2021. "Metal-Bound Methisazone; Novel Drugs Targeting Prophylaxis and Treatment of SARS-CoV-2, a Molecular Docking Study" International Journal of Molecular Sciences 22, no. 6: 2977. https://doi.org/10.3390/ijms22062977