
Immune Regulatory Processes of the Tumor Microenvironment under Malignant Conditions

 , , ,
, , ,
Abstract
:1. Introduction
2. Composition and Functions of the TME
3. T Cell Function in Malignant Conditions
3.1. T Cell Activation
3.1.1. T Cell Dysfunction in the TME
T Cell Anergy
T Cell Exhaustion
T Cell Senescence
3.1.2. Immune Checkpoints/Inhibitory Co-Stimulatory Signals Causing Immunosuppressive Conditions in the TME
Co-Inhibitory B7-CD28 Family Members
Co-Inhibitory TIM Family Members
Co-Inhibitory Molecules of the Immunoglobulin (Ig) Superfamily
Co-Inhibitory Nectin and Nectin-Like Binding Receptors
Butyrophilins Function as Co-Inhibitory Surface Molecules
Intracellular Factors Acting as Negative Regulators of Anti-Tumor Immunity
3.1.3. Pathways Regulating the Immune Checkpoints and Inhibitory Ligands
3.1.4. Soluble Factors Causing Immunosuppressive Condition in the TME
3.1.4.1. The Role of Cytokines in Immune Evasion
3.1.4.2. Pattern Recognition Receptors Signaling
3.1.4.3. The Role of Prostaglandin E2 (PGE-2) in Immune Evasion
3.1.4.4. Tumor Metabolism and Its Implication in Immune Evasion
3.2. Altered MHC Class I Expression and Immune Evasion
3.2.1. Antigen Presentation
3.2.2. Altered MHC Class I Expression under Malignant Conditions
3.2.3. Non-Classical HLA Class I Molecules in Tumors
3.2.4. The Impact of Epigenetic Mechanisms on Tumor Antigen Presentation
3.2.5. Negative Regulation at the Level of APCs in the TME
3.3. Molecules Suppressing the Anti-Tumor Immune Responses beyond Immune Checkpoints
4. TME-Targeting Therapeutic Approaches
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ab | antibody |
| ADCC | antibody-dependent cellular cytotoxicity |
| AIM2 | absent in melanoma 2 |
| AKT | protein kinase |
| AML | acute myeloid leukemia |
| AP-1 | activator protein 1 |
| APC | antigen-presenting cell |
| ATF | activating transcription factor |
| ATP | adenosine triphosphate |
| B2M | β-2 microglobulin |
| BATF | basic leucine zipper transcription factor |
| BCR | B cell receptor |
| Blimp-1 | B lymphocyte-induced maturation protein-1 |
| BTLA | B and T lymphocyte attenuator |
| BTN | butyrophilin |
| BTNL | butyrophilin-like molecules |
| CAF | cancer-associated fibroblast |
| CAR | chimeric antigen receptor |
| CBL | casitas B-lineage lymphoma |
| CCL | CC-chemokine ligand |
| cDC | conventional dendritic cell |
| CDS | cytosolic DNA sensor |
| CEACAM1/5/6 | carcinoembryonic AG-related cell adhesion molecule 1/5/6 |
| cGas | cyclic GMP-AMP synthase |
| CLEVER-1 | common lymphatic endothelial and vascular endothelial receptor-1 |
| CLR | C-type lectin receptors |
| cNK | conventional natural killer cell |
| COX | cyclooxygenase |
| CSF-1 | Colony Stimulating Factor 1 |
| CTA | cancer/testis antigen |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| CXCL | C-X-C motif chemokine |
| DAI | DNA-dependent activator of IRFs |
| DAMP | damage-associated molecular pattern |
| DC | dendritic cells |
| DCIR | C-type lectin dendritic cell (DC) immunoreceptor |
| DC-SCRIPT | dendritic cell-specific transcript |
| DNA | deoxyribonucleic Acid |
| DNAM-1 | DNAX accessory molecule-1 |
| EAT2 | ewing sarcoma-activated transcript 2 |
| ECM | extracellular matrix |
| Eomes | T-box brain protein 2 |
| ER | endoplasmatic reticulum |
| ERK | extracellular signal-regulated kinase |
| ER-α | nuclear estrogen receptor α |
| FGF-2 | basic fibroblast growth factor |
| FGL1 | fibrinogen-like protein 1 |
| Fos | Fos proto-oncogene, AP-1 transcription factor subunit |
| FOXP3 | forkhead box P3 |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GARP | glycoprotein-A repetitions predominant |
| Gata3 | GATA binding protein 3 |
| GITR9 | glucocorticoid-induced TNFR-related protein 9 |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| HCC | hepatocellular carcinoma |
| HERV | human genome consists of endogenous retroviruses |
| HIF-1a | hypoxia-inducible factor 1-alpha |
| HLA-DR | human leukocyte antigen—DR isotype |
| HLA | human leukocyte antigen |
| HMGB1 | high mobility group box 1 |
| HVEM | herpes virus entry mediator |
| ICAM | intercellular adhesion molecule |
| ICB | immune checkpoint blockade |
| ICOS | inducible T cell co-stimulator |
| IDO | indoleamine-pyrrole 2,3-dioxygenase |
| IFN | interferon |
| Ig | immunoglobulin |
| IKAROS | ikaros family zinc finger protein 1 |
| IL | interleukin |
| ILT2/4 | immunoglobulin-like transcript 2/4 |
| iNOS | inducible nitric oxide synthases |
| IRF | interferon regulatory factor |
| JAK | janus kinase |
| Jun | Jun proto-oncogene, AP-1 transcription factor subunit |
| KIR2DL4 | killer immunoglobulin-like receptor (KIR) 2DL4 |
| KIR3DL2 | killer cell immunoglobulin-like receptor (KIR) 3DL2 |
| KLRG-1 | killer cell lectin like receptor G1 |
| LAG-3 | lymphocyte activation gene 3 |
| LGP2 | laboratory of genetics and physiology 2 |
| LIGHT/TNFSF14 | tumor necrosis factor superfamily member 14 |
| LIF | leukemia inhibitory factor |
| lncRNA | long non-coding RNA |
| LPS | lipopolysaccharides |
| LTα | lymphotoxin α |
| mAb | monoclonale antibody |
| Maf | musculoaponeurotic fibrosarcoma |
| MAPK | mitogen activated protein kinases |
| MDA5 | melanoma differentiation-associated protein 5 |
| MDSC | myeloid-derived suppressor cells |
| MHC | major histocompatibility complex |
| MICL | myeloid inhibitory C-type lectin-like receptor |
| Mincle | macrophage inducible Ca2+-dependent lectin receptor |
| MMP9 | matrix metallopeptidase 9 |
| mTOR | mammalian target of rapamycin complex 1 |
| MYC | myelocytomatosis oncogene |
| MyD88 | myeloid differentiation primary response 88 |
| Necls | nectin and nectin-like molecules |
| NFAT | nuclear factor of activated T cells |
| NF-КB | nuclear factor kappa light chain enhancer of activated B cells |
| NK cell | natural killer cell |
| NKG2A | natural killer group 2A |
| NKX2-1-AS1 | NKX2-1 antisense RNA 1 |
| NLR | nucleotide binding oligomerization domain-like receptor |
| NLRC5 | NOD-like receptor family CARD domain containing 5 |
| NO | nitrogen monoxide |
| NR2F2/6 | nuclear receptor subfamily 2 group F member 2/6 |
| NR4A | nuclear receptor 4A |
| NSCLC | non-small cell lung cancer |
| PAMP | pathogen-associated molecular pattern |
| PC | plasma cell |
| PCPB1 | poly(rC)-binding protein 1 |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed cell death 1 ligand 1 |
| PD-L2 | programmed cell death 1 ligand 2 |
| PGE | prostaglandin E synthase |
| PI3K | phosphatidylinositol-3-kinase |
| PKC | protein kinase C |
| PLXNB1/2 | plexin B1/2 |
| Prdm1 | PR domain zinc finger protein 1 |
| PRR | pattern recognition receptors |
| PS | phosphatidylserine |
| PtdSer | phosphatidylserine |
| PVRIG | poliovirus receptor related immunoglobulin |
| RIG-I | retinoic acid-inducible gene I |
| RGS-5 | regulator of G-protein signaling-5 |
| RLR | retinoic acid-inducible gene I (RIG-I)-like receptors |
| RORγt | RAR-related orphan receptor gamma |
| ROS | reactive oxygen species |
| SAP | SLAM-associated protein |
| SEMA3A/3B/4D | semaphorin 3A/3B/4D |
| SIRPα | signal regulatory protein α |
| Sirt1 | regulatory protein SIR2 homolog |
| SLAMF4 | signaling lymphocyte activation molecule F4 |
| STAT | signal transducer and activator of transcription |
| STING | stimulator of interferon genes |
| TAM | tumor-associated macrophages |
| TAMC | tumor-associated mast cell |
| TAN | tumor-associated neutrophils |
| TAP1/2 | antigen processing 1/2 |
| T-bet | T-box expressed in T cells |
| TCR | T cell receptor |
| TDO | tryptophan-2,3-dioxygenase |
| Tfh cells | follicular helper T cells |
| TGF | transforming growth factor |
| Th cells | T helper cells |
| TIGIT | T cell immunoreceptor with Ig and ITIM domains |
| TILs | tumor-infiltrating lymphocytes |
| TIM-3 | T cell immunoglobulin domain and mucin domain containing protein-3 |
| TLR | toll-like receptor |
| TME | tumor microenvironment |
| TNF | tumor necrosis factor |
| TNFR | tumor necrosis factor receptor |
| TOX | thymocyte selection-associated HMG BOX |
| Tregs | regulatory T cells |
| VEGF | vascular endothelial growth factor |
| VISTA | V-domain immunoglobulin suppressor of T cell activation |
| VSIG-3 | V-Set and immunoglobulin domain-containing protein 3 |
| Wnt | wingless-related integration site |
References
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular Aspects and Future Perspectives of Cytokine-Based Anti-Cancer Immunotherapy. Front. Cell Dev. Biol. 2020, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural Killer Cells in Cancer Biology and Therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Mhaidly, R.; Mechta-Grigoriou, F. Fibroblast Heterogeneity in Tumor Micro-Environment: Role in Immunosuppression and New Therapies. Semin. Immunol. 2020, 48, 101417. [Google Scholar] [CrossRef] [PubMed]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Peltanova, B.; Raudenska, M.; Masarik, M. Effect of Tumor Microenvironment on Pathogenesis of the Head and Neck Squamous Cell Carcinoma: A Systematic Review. Mol. Cancer 2019, 18, 63. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Lucarini, V.; Marone, G.; Mattei, F.; Marone, G.; Schiavoni, G. Eosinophils: The Unsung Heroes in Cancer? OncoImmunology 2018, 7, e1393134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, K.L.; Storkus, W.J. Immunotherapeutic Targeting of Tumor-Associated Blood Vessels. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2017; Volume 1036, pp. 191–211. [Google Scholar]
- Bose, A.; Barik, S.; Banerjee, S.; Ghosh, T.; Mallick, A.; Bhattacharyya Majumdar, S.; Goswami, K.K.; Bhuniya, A.; Banerjee, S.; Baral, R.; et al. Tumor-Derived Vascular Pericytes Anergize Th Cells. J. Immunol. 2013, 191, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.F.; Rao, Z.; Bell, J.I. Molecular Coordination of Aβ T-Cell Receptors and Coreceptors CD8 and CD4 in Their Recognition of Peptide-MHC Ligands. Trends Immunol. 2002, 23, 408–413. [Google Scholar] [CrossRef]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. CD28-Mediated Signalling Co-Stimulates Murine T Cells and Prevents Induction of Anergy in T-Cell Clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Lins, D.C.; Mescher, M.F. Signal 3 Determines Tolerance versus Full Activation of Naive CD8 T Cells: Dissociating Proliferation and Development of Effector Function. J. Exp. Med. 2003, 197, 1141–1151. [Google Scholar] [CrossRef]
- Maxwell, J.R.; Weinberg, A.; Prell, R.A.; Vella, A.T. Danger and OX40 Receptor Signaling Synergize to Enhance Memory T Cell Survival by Inhibiting Peripheral Deletion. J. Immunol. 2000, 164, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubin, M.; Kamoun, M.; Trinchieri, G. Interleukin 12 Synergizes with B7/CD28 Interaction in Inducing Efficient Proliferation and Cytokine Production of Human T Cells. J. Exp. Med. 1994, 180, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Cogdill, A.P.; Andrews, M.C.; Wargo, J.A. Hallmarks of Response to Immune Checkpoint Blockade. Br. J. Cancer 2017, 117, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 Inhibits the Effector Phase of Tumor Rejection by T Cell Receptor (TCR) Transgenic CD8+ T Cells. Cancer Res. 2004, 64, 1140–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W.; Chen, L. Inhibitory B7-Family Molecules in the Tumour Microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 Improves Myeloid Dendritic Cell-Mediated Antitumor Immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Zou, L.; Rodriguez, P.; Zhu, G.; Wei, S.; Mottram, P.; Brumlik, M.; Cheng, P.; Curiel, T.; Myers, L.; et al. B7-H4 Expression Identifies a Novel Suppressive Macrophage Population in Human Ovarian Carcinoma. J. Exp. Med. 2006, 203, 871–881. [Google Scholar] [CrossRef]
- Wells, A.D. New Insights into the Molecular Basis of T Cell Anergy: Anergy Factors, Avoidance Sensors, and Epigenetic Imprinting. J. Immunol. 2009, 182, 7331–7341. [Google Scholar] [CrossRef]
- Soto-Nieves, N.; Puga, I.; Abe, B.T.; Bandyopadhyay, S.; Baine, I.; Rao, A.; Macian, F. Transcriptional Complexes Formed by NFAT Dimers Regulate the Induction of T Cell Tolerance. J. Exp. Med. 2009, 206, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.M.; Chunder, N.; Chen, C.; Umetsu, S.E.; Winandy, S.; Wells, A.D. Ikaros Enforces the Costimulatory Requirement for IL2 Gene Expression and Is Required for Anergy Induction in CD4 + T Lymphocytes. J. Immunol. 2007, 179, 7305–7315. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, S.; Duré, M.; Paroder, M.; Soto-Nieves, N.; Puga, I.; Macián, F. Interleukin 2 Gene Transcription Is Regulated by Ikaros-Induced Changes in Histone Acetylation in Anergic T Cells. Blood 2007, 109, 2878–2886. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T Cell Exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 Pathways to Reverse T Cell Exhaustion and Restore Anti-Tumor Immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/Galectin-9 Signaling Pathway Mediates T-Cell Dysfunction and Predicts Poor Prognosis in Patients with Hepatitis B Virus-Associated Hepatocellular Carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.A.L.; Tan, P.; Bradshaw, J.; Ledbetter, J.A.; Anasetti, C.; Damle, N.K. Coexpression and Functional Cooperation of CTLA-4 and CD28 on Activated T Lymphocytes. J. Exp. Med. 1992, 176, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Olive, D.; Kuchroo, V.; Zarour, H.M. CD8+ T Cells Specific for Tumor Antigens Can Be Rendered Dysfunctional by the Tumor Microenvironment through Upregulation of the Inhibitory Receptors BTLA and PD-1. Cancer Res. 2012, 72, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Derré, L.; Rivals, J.P.; Jandus, C.; Pastor, S.; Rimoldi, D.; Romero, P.; Michielin, O.; Olive, D.; Speiser, D.E. BTLA Mediates Inhibition of Human Tumor-Specific CD8+ T Cells That Can Be Partially Reversed by Vaccination. J. Clin. Investig. 2010, 120, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T Cell Exhaustion by Multiple Inhibitory Receptors during Chronic Viral Infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-Cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 Promotes T Cell Responses and Autoimmunity by Repressing Tim-3-Mediated Cell Death and Exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A Role for the Transcriptional Repressor Blimp-1 in CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [Green Version]
- Larsson, M.; Shankar, E.M.; Che, K.F.; Saeidi, A.; Ellegård, R.; Barathan, M.; Velu, V.; Kamarulzaman, A. Molecular Signatures of T-Cell Inhibition in HIV-1 Infection. Retrovirology 2013, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Adibzadeh, M.; Pohla, H.; Rehbein, A.; Pawelec, G. Long-Term Culture of Monoclonal Human T Lymphocytes: Models for Immunosenescence? Mech. Ageing Dev. 1995, 83, 171–183. [Google Scholar] [CrossRef]
- Effros, R.B. Replicative Senescence in the Immune System: Impact of the Hayflick Limit on T-Cell Function in the Elderly. Am. J. Hum. Genet. 1998, 62, 1003–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T Cell Anergy, Exhaustion, Senescence, and Stemness in the Tumor Microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Yang, O.O.; Daar, E.S.; Jamieson, B.D.; Balamurugan, A.; Smith, D.M.; Pitt, J.A.; Petropoulos, C.J.; Richman, D.D.; Little, S.J.; Brown, A.J.L. Human Immunodeficiency Virus Type 1 Clade B Superinfection: Evidence for Differential Immune Containment of Distinct Clade B Strains. J. Virol. 2005, 79, 860–868. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to Detect Senescence-Associated Beta-Galactosidase (SA-Βgal) Activity, a Biomarker of Senescent Cells in Culture and in Vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Appay, V.; Nixon, D.F.; Donahoe, S.M.; Gillespie, G.M.A.; Dong, T.; King, A.; Ogg, G.S.; Spiegel, H.M.L.; Conlon, C.; Spina, C.A.; et al. HIV-Specific CD8+ T Cells Produce Antiviral Cytokines but Are Impaired in Cytolytic Function. J. Exp. Med. 2000, 192, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Akbar, A.N.; Henson, S.M. Are Senescence and Exhaustion Intertwined or Unrelated Processes That Compromise Immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Larbi, A.; Fulop, T. From “Truly Naïve” to “Exhausted Senescent” T Cells: When Markers Predict Functionality. Cytom. Part A 2014, 85, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A Key Player in Cancer Development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Karandikar, N.J.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of CD57 Defines Replicative Senescence and Antigen-Induced Apoptotic Death of CD8+ T Cells. Blood 2003, 101, 2711–2720. [Google Scholar] [CrossRef]
- Heffner, M.; Fearon, D.T. Loss of T Cell Receptor-Induced Bmi-1 in the KLRG1+ Senescent CD8+ T Lymphocyte. Proc. Natl. Acad. Sci. USA 2007, 104, 13414–13419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voehringer, D.; Liang, H.-E.; Locksley, R.M. Homeostasis and Effector Function of Lymphopenia-Induced “Memory-Like” T Cells in Constitutively T Cell-Depleted Mice. J. Immunol. 2008, 180, 4742–4753. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 Expression Is Associated with Tumor Antigen-Specific CD8+ T Cell Dysfunction in Melanoma Patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.A.; et al. P53 Isoforms Regulate Aging- and Tumor-Associated Replicative Senescence in T Lymphocytes. J. Clin. Investig. 2013, 123, 5247–5257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Acevedo, J.A.; Kimbrough, E.M.O.; Lou, Y. Next Generation of Immune Checkpoint Inhibitors and Beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Co-Inhibitory Molecules of the B7-CD28 Family in the Control of T-Cell Immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Schildberg, F.A.; Klein, S.R.; Freeman, G.J.; Sharpe, A.H. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity 2016, 44, 955–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.v.; Brodie, D.W.; Gilbert, R.J.C.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The Interaction Properties of Costimulatory Molecules Revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Egen, J.G.; Kuhns, M.S.; Allison, J.P. CTLA-4: New Insights into Its Biological Function and Use in Tumor Immunotherapy. Nat. Immunol. 2002, 3, 611–618. [Google Scholar] [CrossRef]
- Parry, R.v.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.v.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [Green Version]
- Brunner-Weinzierl, M.C.; Rudd, C.E. CTLA-4 and PD-1 Control of T-Cell Motility and Migration: Implications for Tumor Immunotherapy. Front. Immunol. 2018, 9, 2737. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring Function in Exhausted CD8 T Cells during Chronic Viral Infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 Pathway in Tolerance and Autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 Expression in Human Cancers and Its Association with Clinical Outcomes. OncoTargets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef] [Green Version]
- Selenko-Gebauer, N.; Majdic, O.; Szekeres, A.; Höfler, G.; Guthann, E.; Korthäuer, U.; Zlabinger, G.; Steinberger, P.; Pickl, W.F.; Stockinger, H.; et al. B7-H1 (Programmed Death-1 Ligand) on Dendritic Cells Is Involved in the Induction and Maintenance of T Cell Anergy. J. Immunol. 2003, 170, 3637–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Crabill, G.A.; Pritchard, T.S.; McMiller, T.L.; Wei, P.; Pardoll, D.M.; Pan, F.; Topalian, S.L. Mechanisms Regulating PD-L1 Expression on Tumor and Immune Cells. J. ImmunoTher. Cancer 2019, 7, 305. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Loke, P.; Allison, J.P. PD-L1 and PD-L2 Are Differentially Regulated by Th1 and Th2 Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5336–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of Programmed Death 1 Ligands by Murine T Cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Serriari, N.-E.; Gondois-Rey, F.; Guillaume, Y.; Remmerswaal, E.B.M.; Pastor, S.; Messal, N.; Truneh, A.; Hirsch, I.; van Lier, R.A.W.; Olive, D. B and T Lymphocyte Attenuator Is Highly Expressed on CMV-Specific T Cells during Infection and Regulates Their Function. J. Immunol. 2010, 185, 3140–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedy, J.R.; Gavrieli, M.; Potter, K.G.; Hurchla, M.A.; Lindsley, R.C.; Hildner, K.; Scheu, S.; Pfeffer, K.; Ware, C.F.; Murphy, T.L.; et al. B and T Lymphocyte Attenuator Regulates T Cell Activation through Interaction with Herpesvirus Entry Mediator. Nat. Immunol. 2005, 6, 90–98. [Google Scholar] [CrossRef]
- Ning, Z.; Liu, K.; Xiong, H. Roles of BTLA in Immunity and Immune Disorders. Front. Immunol. 2021, 12, 654960. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, G.; Manick, B.; Hernandez, V.; Renelt, M.; Erickson, C.; Guan, J.; Singh, R.; Rollins, S.; Solorz, A.; et al. VSIG-3 as a Ligand of VISTA Inhibits Human T-Cell Function. Immunology 2019, 156, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a Novel Mouse Ig Superfamily Ligand That Negatively Regulates T Cell Responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Flies, D.B.; Han, X.; Higuchi, T.; Zheng, L.; Sun, J.; Ye, J.J.; Chen, L. Coinhibitory Receptor PD-1H Preferentially Suppresses CD4+ T Cell-Mediated Immunity. J. Clin. Investig. 2014, 124, 1966–1975. [Google Scholar] [CrossRef] [Green Version]
- Lines, J.L.; Sempere, L.F.; Broughton, T.; Wang, L.; Noelle, R. VISTA Is a Novel Broad-Spectrum Negative Checkpoint Regulator for Cancer Immunotherapy. Cancer Immunol. Res. 2014, 2, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Zong, L.; Mo, S.; Yu, S.; Zhou, Y.; Zhang, M.; Chen, J.; Xiang, Y. Expression of the Immune Checkpoint VISTA in Breast Cancer. Cancer Immunol. Immunother. 2020, 69, 1437–1446. [Google Scholar] [CrossRef]
- Mulati, K.; Hamanishi, J.; Matsumura, N.; Chamoto, K.; Mise, N.; Abiko, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Murakami, R.; et al. VISTA Expressed in Tumour Cells Regulates T Cell Function. Br. J. Cancer 2019, 120, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Villarroel-Espindola, F.; Yu, X.; Datar, I.; Mani, N.; Sanmamed, M.; Velcheti, V.; Syrigos, K.; Toki, M.; Zhao, H.; Chen, L.; et al. Spatially Resolved and Quantitative Analysis of Vista/Pd-1h as a Novel Immunotherapy Target in Human Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 1562–1573. [Google Scholar] [CrossRef] [Green Version]
- Zong, L.; Zhou, Y.; Zhang, M.; Chen, J.; Xiang, Y. VISTA Expression Is Associated with a Favorable Prognosis in Patients with High-Grade Serous Ovarian Cancer. Cancer Immunol. Immunother. 2020, 69, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ElTanbouly, M.A.; Croteau, W.; Noelle, R.J.; Lines, J.L. VISTA: A Novel Immunotherapy Target for Normalizing Innate and Adaptive Immunity. Semin. Immunol. 2019, 42, 101308. [Google Scholar] [CrossRef] [PubMed]
- Tagliamento, M.; Agostinetto, E.; Borea, R.; Brandão, M.; Poggio, F.; Addeo, A.; Lambertini, M. VISTA: A Promising Target for Cancer Immunotherapy? ImmunoTargets Ther. 2021, 10, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Podojil, J.R.; Miller, S.D. Potential Targeting of B7-H4 for the Treatment of Cancer. Immunol. Rev. 2017, 276, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a Checkpoint Molecule, as a Target for Cancer Immunotherapy. Int. J. Biol. Sci. 2020, 16, 1767–1773. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; Dekruyff, R.H. TIM Genes: A Family of Cell Surface Phosphatidylserine Receptors That Regulate Innate and Adaptive Immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.C. Tim-3: An Emerging Target in the Cancer Immunotherapy Landscape. Cancer Immunol. Res. 2014, 2, 393–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 Ligand Galectin-9 Negatively Regulates T Helper Type 1 Immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- DeKruyff, R.H.; Bu, X.; Ballesteros, A.; Santiago, C.; Chim, Y.-L.E.; Lee, H.-H.; Karisola, P.; Pichavant, M.; Kaplan, G.G.; Umetsu, D.T.; et al. T Cell/Transmembrane, Ig, and Mucin-3 Allelic Variants Differentially Recognize Phosphatidylserine and Mediate Phagocytosis of Apoptotic Cells. J. Immunol. 2010, 184, 1918–1930. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-Infiltrating DCs Suppress Nucleic Acid-Mediated Innate Immune Responses through Interactions between the Receptor TIM-3 and the Alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 Regulates TIM-3-Mediated Tolerance and Exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imaizumi, T.; Kumagai, M.; Sasaki, N.; Kurotaki, H.; Mori, F.; Seki, M.; Nishi, N.; Fujimoto, K.; Tanji, K.; Shibata, T.; et al. Interferon-Gamma Stimulates the Expression of Galectin-9 in Cultured Human Endothelial Cells. J. Leukoc. Biol. 2002, 72, 486–491. [Google Scholar] [CrossRef]
- Kobayashi, N.; Karisola, P.; Peña-Cruz, V.; Dorfman, D.M.; Jinushi, M.; Umetsu, S.E.; Butte, M.J.; Nagumo, H.; Chernova, I.; Zhu, B.; et al. TIM-1 and TIM-4 Glycoproteins Bind Phosphatidylserine and Mediate Uptake of Apoptotic Cells. Immunity 2007, 27, 927–940. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M.; Akiba, H.; Takeda, K.; Kojima, Y.; Hashiguchi, M.; Azuma, M.; Yagita, H.; Okumura, K. Tim-3 Mediates Phagocytosis of Apoptotic Cells and Cross-Presentation. Blood 2009, 113, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.F.; Liu, N.; Candolfi, M.; Xiong, W.; Assi, H.; Yagiz, K.; Edwards, M.R.; Michelsen, K.S.; Kroeger, K.M.; Liu, C.; et al. HMGB1 Mediates Endogenous TLR2 Activation and Brain Tumor Regression. PLoS Med. 2009, 6, e1000010. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 Proteins: Dual-Function Alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [Green Version]
- Gray-Owen, S.D.; Blumberg, R.S. CEACAM1: Contact-Dependent Control of Immunity. Nat. Rev. Immunol. 2006, 6, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Cella, M.; Langen, H.; Friedlein, A.; Colonna, M. Activating Interactions in Human NK Cell Recognition: The Role of 2B4-CD48. Eur. J. Immunol. 1999, 29, 1676–1683. [Google Scholar] [CrossRef]
- Kammerer, R.; Stober, D.; Singer, B.B.; Öbrink, B.; Reimann, J. Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 on Murine Dendritic Cells Is a Potent Regulator of T Cell Stimulation. J. Immunol. 2001, 166, 6537–6544. [Google Scholar] [CrossRef] [Green Version]
- Horst, A.K.; Bickert, T.; Brewig, N.; Ludewig, P.; van Rooijen, N.; Schumacher, U.; Beauchemin, N.; Ito, W.D.; Fleischer, B.; Wagener, C.; et al. CEACAM1+ Myeloid Cells Control Angiogenesis in Inflammation. Blood 2009, 113, 6726–6736. [Google Scholar] [CrossRef]
- Coutelier, J.-P.; Godfraind, C.; Dveksler, G.S.; Wysocka, M.; Cardellichio, C.B.; Noël, H.; Holmes, K.V. B Lymphocyte and Macrophage Expression of Carcinoembryonic Antigen-related Adhesion Molecules That Serve as Receptors for Murine Coronavirus. Eur. J. Immunol. 1994, 24, 1383–1390. [Google Scholar] [CrossRef] [Green Version]
- Chihara, N.; Madi, A.; Kondo, T.; Zhang, H.; Acharya, N.; Singer, M.; Nyman, J.; Marjanovic, N.D.; Kowalczyk, M.S.; Wang, C.; et al. Induction and Transcriptional Regulation of the Co-Inhibitory Gene Module in T Cells. Nature 2018, 558, 454–459. [Google Scholar] [CrossRef] [Green Version]
- DeLong, J.H.; O’Hara Hall, A.; Rausch, M.; Moodley, D.; Perry, J.; Park, J.; Phan, A.T.; Beiting, D.P.; Kedl, R.M.; Hill, J.A.; et al. IL-27 and TCR Stimulation Promote T Cell Expression of Multiple Inhibitory Receptors. ImmunoHorizons 2019, 3, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, S.; Saito, H.; Ikeguchi, M. An Increased Number of PD-1+ and Tim-3+ CD8+ T Cells Is Involved in Immune Evasion in Gastric Cancer. Surg. Today 2016, 46, 1341–1347. [Google Scholar] [CrossRef]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a Promising Target for Cancer Immunotherapy. OncoTargets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a Novel Lymphocyte Activation Gene Closely Related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarour, H.M. Reversing T-Cell Dysfunction and Exhaustion in Cancer. Clin. Cancer Res. 2016, 22, 1856–1864. [Google Scholar] [CrossRef] [Green Version]
- Demeure, C.E.; Wolfers, J.; Martin-Garcia, N.; Gaulard, P.; Triebel, F. T Lymphocytes Infiltrating Various Tumour Types Express the MHC Class II Ligand Lymphocyte Activation Gene-3 (LAG-3): Role of LAG-3/MHC Class II Interactions in Cell-Cell Contacts. Eur. J. Cancer 2001, 37, 1709–1718. [Google Scholar] [CrossRef]
- Gandhi, M.K.; Lambley, E.; Duraiswamy, J.; Dua, U.; Smith, C.; Elliott, S.; Gill, D.; Marlton, P.; Seymour, J.; Khanna, R. Expression of LAG-3 by Tumor-Infiltrating Lymphocytes Is Coincident with the Suppression of Latent Membrane Antigen-Specific CD8+ T-Cell Function in Hodgkin Lymphoma Patients. Blood 2006, 108, 2280–2289. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schreiner, J.; Müller, P.; Herzig, P.; Roller, A.; Belousov, A.; Umana, P.; Pisa, P.; Klein, C.; Bacac, M.; et al. Progression of Lung Cancer Is Associated with Increased Dysfunction of T Cells Defined by Coexpression of Multiple Inhibitory Receptors. Cancer Immunol. Res. 2015, 3, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Workman, C.J.; Rice, D.S.; Dugger, K.J.; Kurschner, C.; Vignali, D.A.A. Phenotypic Analysis of the Murine CD4-Related Glycoprotein, CD223 (LAG-3). Eur. J. Immunol. 2002, 32, 2255–2263. [Google Scholar] [CrossRef]
- Huard, B.; Gaulard, P.; Faure, F.; Hercend, T.; Triebel, F. Cellular Expression and Tissue Distribution of the Human LAG-3-Encoded Protein, an MHC Class II Ligand. Immunogenetics 1994, 39, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8 T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin Expressed on Melanoma Cells Promotes Tumor Progression by Inhibiting Antitumor T-Cell Responses. Cancer Res. 2014, 74, 3418–3428. [Google Scholar] [CrossRef] [Green Version]
- Dumic, J.; Dabelic, S.; Flögel, M. Galectin-3: An Open-Ended Story. Biochim. Biophys. Acta-Gen. Subj. 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, Y.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e12. [Google Scholar] [CrossRef] [Green Version]
- Hannier, S.; Tournier, M.; Bismuth, G.; Triebel, F. CD3/TCR Complex-Associated Lymphocyte Activation Gene-3 Molecules Inhibit CD3/TCR Signaling. J. Immunol. 1998, 161, 4058–4065. [Google Scholar]
- Mathew, S.O.; Rao, K.K.; Kim, J.R.; Bambard, N.D.; Mathew, P.A. Functional Role of Human NK Cell Receptor 2B4 (CD244) Isoforms. Eur. J. Immunol. 2009, 39, 1632–1641. [Google Scholar] [CrossRef]
- Boles, K.S.; Stepp, S.E.; Bennett, M.; Kumar, V.; Mathew, P.A. 2B4 (CD244) and CS1: Novel Members of the CD2 Subset of the Immunoglobulin Superfamily Molecules Expressed on Natural Killer Cells and Other Leukocytes. Immunol. Rev. 2001, 181, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Schuhmachers, G.; Ariizumi, K.; Mathew, P.A.; Bennett, M.; Kumar, V.; Takashima, A. 2B4, a New Member of the Immunoglobulin Gene Superfamily, Is Expressed on Murine Dendritic Epidermal T Cells and Plays a Functional Role in Their Killing of Skin Tumors. J. Investig. Dermatol. 1995, 105, 592–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayos, J.; Wu, C.; Morra, M.; Wang, N.; Zhang, X.; Allen, D.; van Schaik, S.; Notarangelo, L.; Geha, R.; Roncarolo, M.G.; et al. The X-Linked Lymphoproliferative-Disease Gene Product SAP Regulates Signals Induced through the Co-Receptor SLAM. Nature 1998, 395, 462–469. [Google Scholar] [CrossRef]
- Morra, M.; Lu, J.; Poy, F.; Martin, M.; Sayos, J.; Calpe, S.; Gullo, C.; Howie, D.; Rietdijk, S.; Thompson, A.; et al. Structural Basis for the Interaction of the Free SH2 Domain EAT-2 with SLAM Receptors in Hematopoietic Cells. EMBO J. 2001, 20, 5840–5852. [Google Scholar] [CrossRef] [Green Version]
- Roncagalli, R.; Taylor, J.E.R.; Zhang, S.; Shi, X.; Chen, R.; Cruz-Munoz, M.E.; Yin, L.; Latour, S.; Veillette, A. Negative Regulation of Natural Killer Cell Function by EAT-2, a SAP-Related Adaptor. Nat. Immunol. 2005, 6, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Relouzat, F.; Roncagalli, R.; Aoukaty, A.; Tan, R.; Latour, S.; Veillette, A. Molecular Dissection of 2B4 Signaling: Implications for Signal Transduction by SLAM-Related Receptors. Mol. Cell. Biol. 2004, 24, 5144–5156. [Google Scholar] [CrossRef] [Green Version]
- Lichtenegger, F.S.; Schnorfeil, F.M.; Emmerig, K.; Neitz, J.S.; Beck, B.; Draenert, R.; Hiddemann, W.; Subklewe, M. Pseudo-Exhaustion of CD8+ T Cells in AML. Blood 2013, 122. [Google Scholar] [CrossRef]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of Tumor-Specific CD8+ T Cells in Metastases from Melanoma Patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [Green Version]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New Checkpoint Receptor Targets for Cancer Immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The Interaction of TIGIT with PVR and PVRL2 Inhibits Human NK Cell Cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Sho, M.; Nishiwada, S.; Yasuda, S.; Shimada, K.; Yamato, I.; Akahori, T.; Kinoshita, S.; Nagai, M.; Konishi, N.; Nakajima, Y. Clinical Significance of CD155 Expression in Human Pancreatic Cancer. Anticancer Res. 2015, 35, 2287–2297. [Google Scholar]
- Levin, S.D.; Taft, D.W.; Brandt, C.S.; Bucher, C.; Howard, E.D.; Chadwick, E.M.; Johnston, J.; Hammond, A.; Bontadelli, K.; Ardourel, D.; et al. Vstm3 Is a Member of the CD28 Family and an Important Modulator of T-Cell Function. Eur. J. Immunol. 2011, 41, 902–915. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.; Aoubala, M.; Jordier, F.; Isnardon, D.; Gomez, S.; Dubreuil, P. The Human Poliovirus Receptor Related 2 Protein Is a New Hematopoietic/Endothelial Homophilic Adhesion Molecule. Blood 1998, 92, 4602–4611. [Google Scholar] [CrossRef]
- Zhu, Y.; Paniccia, A.; Schulick, A.C.; Chen, W.; Koenig, M.R.; Byers, J.T.; Yao, S.; Bevers, S.; Edil, B.H. Identification of CD112R as a Novel Checkpoint for Human T Cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, H.; Ravens, I.; Papadogianni, G.; Bernhardt, G. Coming of Age: CD96 Emerges as Modulator of Immune Responses. Front. Immunol. 2018, 9, 1072. [Google Scholar] [CrossRef] [Green Version]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 Axis Regulates Human T Cell Function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef]
- Gomez-Lopez, N.; StLouis, D.; Lehr, M.A.; Sanchez-Rodriguez, E.N.; Arenas-Hernandez, M. Immune Cells in Term and Preterm Labor. Cell. Mol. Immunol. 2014, 11, 571–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara-Hanaoka, S.; Shibuya, K.; Onoda, Y.; Zhang, H.; Yamazaki, S.; Miyamoto, A.; Honda, S.I.; Lanier, L.L.; Shibuya, A. Functional Characterization of DNAM-1 (CD226) Interaction with Its Ligands PVR (CD155) and Nectin-2 (PRR-2/CD112). Int. Immunol. 2004, 16, 533–538. [Google Scholar] [CrossRef]
- Seth, S.; Maier, M.K.; Qiu, Q.; Ravens, I.; Kremmer, E.; Förster, R.; Bernhardt, G. The Murine Pan T Cell Marker CD96 Is an Adhesion Receptor for CD155 and Nectin-1. Biochem. Biophys. Res. Commun. 2007, 364, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8+ T Cell Effector Function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Zhu, L.; Schell, T.D.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; George, M.R.; Zeng, H.; Zheng, H. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin. Cancer Res. 2016, 22, 3057–3066. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 Impair Tumor Antigen-Specific CD8+ T Cells in Melanoma Patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Lee, W.J.; Lee, Y.J.; Choi, M.E.; Yun, K.A.; Won, C.H.; Lee, M.W.; Choi, J.H.; Chang, S.E. Expression of Lymphocyte-Activating Gene 3 and T-Cell Immunoreceptor with Immunoglobulin and ITIM Domains in Cutaneous Melanoma and Their Correlation with Programmed Cell Death 1 Expression in Tumor-Infiltrating Lymphocytes. J. Am. Acad. Dermatol. 2019, 81, 219–227. [Google Scholar] [CrossRef]
- He, W.; Zhang, H.; Han, F.; Chen, X.; Lin, R.; Wang, W.; Qiu, H.; Zhuang, Z.; Liao, Q.; Zhang, W.; et al. CD155T/TIGIT Signaling Regulates CD8+ T-Cell Metabolism and Promotes Tumor Progression in Human Gastric Cancer. Cancer Res. 2017, 77, 6375–6388. [Google Scholar] [CrossRef] [Green Version]
- Franke, W.W.; Heid, H.W.; Grund, C.; Winter, S.; Freudenstein, C.; Schmid, E.; Jarasch, E.D.; Keenan, T.W. Antibodies to the Major Insoluble Milk Fat Globule Membrane-Associated Protein: Specific Location in Apical Regions of Lactating Epithelial Cells. J. Cell Biol. 1981, 89, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefferl, A.; Schubart, A.; Storch, M.; Amini, A.; Mather, I.; Lassmann, H.; Linington, C. Butyrophilin, a Milk Protein, Modulates the Encephalitogenic T Cell Response to Myelin Oligodendrocyte Glycoprotein in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2000, 165, 2859–2865. [Google Scholar] [CrossRef] [Green Version]
- Ikemizu, S.; Gilbert, R.J.C.; Fennelly, J.A.; Collins, A.V.; Harlos, K.; Jones, E.Y.; Stuart, D.I.; Davis, S.J. Structure and Dimerization of a Soluble Form of B7-1. Immunity 2000, 12, 51–60. [Google Scholar] [CrossRef]
- Abeler-Dörner, L.; Swamy, M.; Williams, G.; Hayday, A.C.; Bas, A. Butyrophilins: An Emerging Family of Immune Regulators. Trends Immunol. 2012, 33, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Tazi-Ahnini, R.; Henry, J.; Offer, C.; Bouissou-Bouchouata, C.; Mather, I.H.; Pontarotti, P. Cloning, Localization, and Structure of New Members of the Butyrophilin Gene Family in the Juxta-Telomeric Region of the Major Histocompatibility Complex. Immunogenetics 1997, 47, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, M.; Tokarz-Deptula, B.; Deptula, W. Butyrophilins: An Important New Element of Resistance. Cent. Eur. J. Immunol. 2017, 42, 399–403. [Google Scholar] [CrossRef]
- Nguyen, T.; Liu, X.K.; Zhang, Y.; Dong, C. BTNL2, a Butyrophilin-Like Molecule That Functions to Inhibit T Cell Activation. J. Immunol. 2006, 176, 7354–7360. [Google Scholar] [CrossRef]
- Arnett, H.A.; Escobar, S.S.; Gonzalez-Suarez, E.; Budelsky, A.L.; Steffen, L.A.; Boiani, N.; Zhang, M.; Siu, G.; Brewer, A.W.; Viney, J.L. BTNL2, a Butyrophilin/B7-Like Molecule, Is a Negative Costimulatory Molecule Modulated in Intestinal Inflammation. J. Immunol. 2007, 178, 1523–1533. [Google Scholar] [CrossRef]
- Yamazaki, T.; Goya, I.; Graf, D.; Craig, S.; Martin-Orozco, N.; Dong, C. A Butyrophilin Family Member Critically Inhibits T Cell Activation. J. Immunol. 2010, 185, 5907–5914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.A.; Knezevic, B.R.; Ammann, J.U.; Rhodes, D.A.; Aw, D.; Palmer, D.B.; Mather, I.H.; Trowsdale, J. BTN1A1, the Mammary Gland Butyrophilin, and BTN2A2 Are Both Inhibitors of T Cell Activation. J. Immunol. 2010, 184, 3514–3525. [Google Scholar] [CrossRef] [PubMed]
- le Page, C.; Marineau, A.; Bonza, P.K.; Rahimi, K.; Cyr, L.; Labouba, I.; Madore, J.; Delvoye, N.; Mes-Masson, A.M.; Provencher, D.M.; et al. BTN3A2 Expression in Epithelial Ovarian Cancer Is Associated with Higher Tumor Infiltrating T Cells and a Better Prognosis. PLoS ONE 2012, 7, e38541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, K.K.; Mine, J.A.; Biswas, S.; Chaurio, R.A.; Perales-Puchalt, A.; Anadon, C.M.; Costich, T.L.; Harro, C.M.; Walrath, J.; Ming, Q.; et al. BTN3A1 Governs Antitumor Responses by Coordinating Aβ and Γδ T Cells. Science 2020, 369, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Klepsch, V.; Hermann-Kleiter, N.; Baier, G. Beyond CTLA-4 and PD-1: Orphan Nuclear Receptor NR2F6 as T Cell Signaling Switch and Emerging Target in Cancer Immunotherapy. Immunol. Lett. 2016, 178, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Ansa-Addo, E.A.; Huang, H.C.; Riesenberg, B.; Iamsawat, S.; Borucki, D.; Nelson, M.H.; Nam, J.H.; Chung, D.; Liu, B.; Li, Z.; et al. RNA Binding Protein Pcbp1 Is an Intracellular Immune Checkpoint for Shaping t Cell Responses in Cancer Immunity. Sci. Adv. 2020, 6, eaaz3865. [Google Scholar] [CrossRef]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; He, N.; Cai, C.; Cai, F.; Fan, P.; Zheng, Z.; Jin, X. HDAC3 Modulates Cancer Immunity via Increasing PD-L1 Expression in Pancreatic Cancer. Pancreatol. Off. J. Int. Assoc. Pancreatol. (IAP) 2019, 19, 383–389. [Google Scholar] [CrossRef]
- Casey, S.C.; Baylot, V.; Felsher, D.W. MYC: Master Regulator of Immune Privilege. Trends Immunol. 2017, 38, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Shen, Y.; Zhu, X.; Lv, R.; Li, S.; Zhang, Z.; Shi, Y.G.; Tan, L. ERα Is a Negative Regulator of PD-L1 Gene Transcription in Breast Cancer. Biochem. Biophys. Res. Commun. 2018, 505, 157–161. [Google Scholar] [CrossRef]
- You, L.; Wu, W.; Wang, X.; Fang, L.; Adam, V.; Nepovimova, E.; Wu, Q.; Kuca, K. The Role of Hypoxia-Inducible Factor 1 in Tumor Immune Evasion. Med. Res. Rev. 2021, 41, 1622–1643. [Google Scholar] [CrossRef]
- Venkatraman, S.; Meller, J.; Hongeng, S.; Tohtong, R.; Chutipongtanate, S. Transcriptional Regulation of Cancer Immune Checkpoints: Emerging Strategies for Immunotherapy. Vaccines 2020, 8, 735. [Google Scholar] [CrossRef]
- Mathas, S.; Hinz, M.; Anagnostopoulos, I.; Krappmann, D.; Lietz, A.; Jundt, F.; Bommert, K.; Mechta-Grigoriou, F.; Stein, H.; Dörken, B.; et al. Aberrantly Expressed C-Jun and JunB Are a Hallmark of Hodgkin Lymphoma Cells, Stimulate Proliferation and Synergize with NF-ΚB. EMBO J. 2002, 21, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 Activity and EBV Infection Induce PD-L1 in Hodgkin Lymphomas and Posttransplant Lymphoproliferative Disorders: Implications for Targeted Therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Hong, Y.W.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic Kinase NPM/ALK Induces through STAT3 Expression of Immunosuppressive Protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Chouaib, S. Targeting Hypoxia at the Forefront of Anticancer Immune Responses. OncoImmunology 2014, 3, e954463. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, A.A.; de la Roche, M.; Djamgoz, M.B.A.; Siddik, Z.H. Overview of the Oncogenic Signaling Pathways in Colorectal Cancer: Mechanistic Insights. Semin. Cancer Biol. 2019, 58, 65–79. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-MTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative Analysis Reveals Selective 9p24.1 Amplification, Increased PD-1 Ligand Expression, and Further Induction via JAK2 in Nodular Sclerosing Hodgkin Lymphoma and Primary Mediastinal Large B-Cell Lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiou, K.; Chen, L.; Berglund, M.; Ren, W.; De Miranda, N.F.C.C.; Lisboa, S.; Fangazio, M.; Zhu, S.; Hou, Y.; Wu, K.; et al. Genetic Basis of PD-L1 Overexpression in Diffuse Large B-Cell Lymphomas. Blood 2016, 127, 3026–3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, J.; Saito, M.; Tsuta, K.; Iwakawa, R.; Shiraishi, K.; Scheel, A.H.; Uchida, S.; Watanabe, S.I.; Nishikawa, R.; Noguchi, M.; et al. Genomic Amplification of CD274 (PD-L1) in Small-Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 1220–1226. [Google Scholar] [CrossRef] [Green Version]
- Vaeth, M.; Müller, G.; Stauss, D.; Dietz, L.; Klein-Hessling, S.; Serfling, E.; Lipp, M.; Berberich, I.; Berberich-Siebelt, F. Follicular Regulatory T Cells Control Humoral Autoimmunity via NFAT2-Regulated CXCR5 Expression. J. Exp. Med. 2014, 211, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.J.; Hu, J.K.; Pereira, R.M.; Crampton, J.S.; Togher, S.; Bild, N.; Crotty, S.; Rao, A. Cutting Edge: NFAT Transcription Factors Promote the Generation of Follicular Helper T Cells in Response to Acute Viral Infection. J. Immunol. 2016, 196, 2015–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oestreich, K.J.; Yoon, H.; Ahmed, R.; Boss, J.M. NFATc1 Regulates PD-1 Expression upon T Cell Activation. J. Immunol. 2008, 181, 4832–4839. [Google Scholar] [CrossRef] [PubMed]
- Tsytsykova, A.V.; Tsitsikov, E.N.; Geha, R.S. The CD40L Promoter Contains Nuclear Factor of Activated T Cells-Binding Motifs Which Require AP-1 Binding for Activation of Transcription. J. Biol. Chem. 1996, 271, 3763–3770. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, S.; Rao, A. NFAT1 and NFAT2 Are Positive Regulators of IL-4 Gene Transcription. Eur. J. Immunol. 2002, 32, 2971–2978. [Google Scholar] [CrossRef]
- Kim, H.P.; Korn, L.L.; Gamero, A.M.; Leonard, W.J. Calcium-Dependent Activation of Interleukin-21 Gene Expression in T Cells. J. Biol. Chem. 2005, 280, 25291–25297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, Y.; Lu, H.; Li, J.; Yan, X.; Xiao, M.; Hao, J.; Alekseev, A.; Khong, H.; Chen, T.; et al. Genome-Wide Analysis Identifies NR4A1 as a Key Mediator of T Cell Dysfunction. Nature 2019, 567, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.-W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A Transcription Factors Limit CAR T Cell Function in Solid Tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Macián, F.; García-Cózar, F.; Im, S.H.; Horton, H.F.; Byrne, M.C.; Rao, A. Transcriptional Mechanisms Underlying Lymphocyte Tolerance. Cell 2002, 109, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Scott-Browne, J.P.; López-Moyado, I.F.; Trifari, S.; Wong, V.; Chavez, L.; Rao, A.; Pereira, R.M. Dynamic Changes in Chromatin Accessibility Occur in CD8+ T Cells Responding to Viral Infection. Immunity 2016, 45, 1327–1340. [Google Scholar] [CrossRef] [Green Version]
- Mognol, G.P.; Spreafico, R.; Wong, V.; Scott-Browne, J.P.; Togher, S.; Hoffmann, A.; Hogan, P.G.; Rao, A.; Trifari, S. Exhaustion-Associated Regulatory Regions in CD8+ Tumor-Infiltrating T Cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2776–E2785. [Google Scholar] [CrossRef] [Green Version]
- O’Flaherty, E.; Kaye, J. TOX Defines a Conserved Subfamily of HMG-Box Proteins. BMC Genom. 2003, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX Transcriptionally and Epigenetically Programs CD8+ T Cell Exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX Reinforces the Phenotype and Longevity of Exhausted T Cells in Chronic Viral Infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX Is a Critical Regulator of Tumour-Specific T Cell Differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Yao, C.; Sun, H.-W.; Lacey, N.E.; Ji, Y.; Moseman, E.A.; Shih, H.-Y.; Heuston, E.F.; Kirby, M.; Anderson, S.; Cheng, J.; et al. Single-Cell RNA-Seq Reveals TOX as a Key Regulator of CD8+ T Cell Persistence in Chronic Infection. Nat. Immunol. 2019, 20, 890–901. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 Transcription Factors Cooperate with NR4A Transcription Factors to Impose CD8+ T Cell Exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Lin, W.; Tang, X.; Li, S.; Guo, L.; Lin, Y.; Kwok, H.F. The Roles of MicroRNAs in Regulating the Expression of PD-1/PD-L1 Immune Checkpoint. Int. J. Mol. Sci. 2017, 18, 2540. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Johnston, N.; Zheng, X.; Wang, H.; Zhang, X.; Gao, D.; Min, W. MiR-28 Modulates Exhaustive Differentiation of T Cells through Silencing Programmed Cell Death-1 and Regulating Cytokine Secretion. Oncotarget 2016, 7, 53735–53750. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Nie, W.; Jin, Y.; Zhuo, A.; Zang, Y.; Xiu, Q. B and T Lymphocyte Attenuator Is a Target of MiR-155 during Naive CD4+ T Cell Activation. Iran. J. Immunol. 2016, 13, 89–99. [Google Scholar]
- Zhang, Y.; Sun, E.; Li, X.; Zhang, M.; Tang, Z.; He, L.; Lv, K. MiR-155 Contributes to Df1-Induced Asthma by Increasing the Proliferative Response of Th Cells via CTLA-4 Downregulation. Cell. Immunol. 2017, 314, 1–9. [Google Scholar] [CrossRef]
- Kathuria, H.; Millien, G.; McNally, L.; Gower, A.C.; Tagne, J.B.; Cao, Y.; Ramirez, M.I. NKX2-1-AS1 Negatively Regulates CD274/PD-L1, Cell-Cell Interaction Genes, and Limits Human Lung Carcinoma Cell Migration. Sci. Rep. 2018, 8, 14418. [Google Scholar] [CrossRef] [PubMed]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Zha, Z.; Bucher, F.; Nejatfard, A.; Zheng, T.; Zhang, H.; Yea, K.; Lerner, R.A. Interferon-γ Is a Master Checkpoint Regulator of Cytokine-Induced Differentiation. Proc. Natl. Acad. Sci. USA 2017, 114, E6867–E6874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. IFN-γ from Lymphocytes Induces PD-L1 Expression and Promotes Progression of Ovarian Cancer. Br. J. Cancer 2015, 112, 1501–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movahedi, K.; Guilliams, M.; van den Bossche, J.; van den Bergh, R.; Gysemans, C.; Beschin, A.; de Baetselier, P.; van Ginderachter, J.A. Identification of Discrete Tumor-Induced Myeloid-Derived Suppressor Cell Subpopulations with Distinct T Cell Suppressive Activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef]
- Ibrahim, E.C.; Guerra, N.; Lacombe, M.J.T.; Angevin, E.; Chouaib, S.; Carosella, E.D.; Caignard, A.; Paul, P. Tumor-Specific up-Regulation of the Nonclassical Class I HLA-G Antigen Expression in Renal Carcinoma. Cancer Res. 2001, 61, 6838–6845. [Google Scholar]
- Van Horssen, R.; ten Hagen, T.L.M.; Eggermont, A.M.M. TNF-Alpha in Cancer Treatment: Molecular Insights, Antitumor Effects, and Clinical Utility. Oncologist 2006, 11, 397–408. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in Tumor Progression and Regression: A Review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef] [Green Version]
- Lu, X. Impact of IL-12 in Cancer. Curr. Cancer Drug Targets 2017, 17, 682–697. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in Cancer Immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [Green Version]
- Ranson, T.; Vosshenrich, C.A.J.; Corcuff, E.; Richard, O.; Müller, W.; di Santo, J.P. IL-15 Is an Essential Mediator of Peripheral NK-Cell Homeostasis. Blood 2003, 101, 4887–4893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillet, A.H.; Thèze, J.; Rose, T. Interleukin (IL)-2 and IL-15 Have Different Effects on Human Natural Killer Lymphocytes. Hum. Immunol. 2011, 72, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Stolfi, C.; Pallone, F.; Macdonald, T.T.; Monteleone, G. Interleukin-21 in Cancer Immunotherapy: Friend or Foe? Oncoimmunology 2012, 1, 351–354. [Google Scholar] [CrossRef] [Green Version]
- Yanaihara, N.; Hirata, Y.; Yamaguchi, N.; Noguchi, Y.; Saito, M.; Nagata, C.; Takakura, S.; Yamada, K.; Okamoto, A. Antitumor Effects of Interleukin-6 (IL-6)/Interleukin-6 Receptor (IL-6R) Signaling Pathway Inhibition in Clear Cell Carcinoma of the Ovary. Mol. Carcinog. 2016, 55, 832–841. [Google Scholar] [CrossRef]
- Beltraminelli, T.; de Palma, M. Biology and Therapeutic Targeting of Tumour-Associated Macrophages. J. Pathol. 2020, 250, 573–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borden, E.C. Interferons α and β in Cancer: Therapeutic Opportunities from New Insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef]
- Mantovani, A.; Barajon, I.; Garlanda, C. IL-1 and IL-1 Regulatory Pathways in Cancer Progression and Therapy. Immunol. Rev. 2018, 281, 57–61. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The Paradoxical Role of IL-10 in Immunity and Cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef]
- Oft, M. IL-10: Master Switch from Tumor-Promoting Inflammation to Antitumor Immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.E.; Shirota, H.; Kasahara, Y.; Saijo, K.; Ishioka, C. IL-4 Blockade Alters the Tumor Microenvironment and Augments the Response to Cancer Immunotherapy in a Mouse Model. Cancer Immunol. Immunother. CII 2017, 66, 1485–1496. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Leland, P.; Joshi, B.H.; Puri, R.K. Targeting of IL-4 and IL-13 Receptors for Cancer Therapy. Cytokine 2015, 75, 79–88. [Google Scholar] [CrossRef]
- Fabbi, M.; Carbotti, G.; Ferrini, S. Dual Roles of IL-27 in Cancer Biology and Immunotherapy. Mediat. Inflamm. 2017, 2017, 3958069. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Sakuishi, K.; Xiao, S.; Sun, Z.; Zaghouani, S.; Gu, G.; Wang, C.; Tan, D.J.; Wu, C.; Rangachari, M.; et al. An IL-27/NFIL3 Signalling Axis Drives Tim-3 and IL-10 Expression and T-Cell Dysfunction. Nat. Commun. 2015, 6, 6072. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Buchlis, G.; Kapoor, V.; Cheng, G.; Sun, J.; Singhal, S.; Crisanti, C.; Wang, L.C.S.; Heitjan, D.; Snyder, L.A.; et al. CCL2 Blockade Augments Cancer Immunotherapy. Cancer Res. 2010, 70, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in Innate Immunity and Inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef]
- Li, K.; Qu, S.; Chen, X.; Wu, Q.; Shi, M. Promising Targets for Cancer Immunotherapy: TLRs, RLRs, and STING-Mediated Innate Immune Pathways. Int. J. Mol. Sci. 2017, 18, 404. [Google Scholar] [CrossRef]
- Bai, L.; Li, W.; Zheng, W.; Xu, D.; Chen, N.; Cui, J. Promising Targets Based on Pattern Recognition Receptors for Cancer Immunotherapy. Pharmacol. Res. 2020, 159, 105017. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, D.H.; Mao, Y.; Mele, D.A. The Next Generation of Pattern Recognition Receptor Agonists: Improving Response Rates in Cancer Immunotherapy. Curr. Med. Chem. 2020, 27, 5654–5674. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A.; Medzhitov, R. Innate Immune Recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, A.A. Endogenous Ligands of Toll-like Receptors: Implications for Regulating Inflammatory and Immune Responses. Trends Immunol. 2002, 23, 509–512. [Google Scholar] [CrossRef]
- Hopkins, P.A.; Sriskandan, S. Mammalian Toll-like Receptors: To Immunity and Beyond. Clin. Exp. Immunol. 2005, 140, 395–407. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like Receptor Signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Beutler, B. Inferences, Questions and Possibilities in Toll-like Receptor Signalling. Nature 2004, 430, 257–263. [Google Scholar] [CrossRef]
- Liew, F.Y.; Xu, D.; Brint, E.K.; O’Neill, L.A.J. Negative Regulation of Toll-like Receptor-Mediated Immune Responses. Nat. Rev. Immunol. 2005, 5, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-Cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Saxena, M.; Yeretssian, G. NOD-like Receptors: Master Regulators of Inflammation and Cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef] [Green Version]
- Kent, A.; Blander, J.M. Nod-like Receptors: Key Molecular Switches in the Conundrum of Cancer. Front. Immunol. 2014, 5, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Kamiya, T.; Suabjakyong, P.; Tsuji, N.M. Targeting C-Type Lectin Receptors for Cancer Immunity. Front. Immunol. 2015, 6, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpisheh, V.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Ghalamfarsa, G.; Sabz, G.; Yousefi, M.; Yousefi, B.; Jadidi-Niaragh, F. Prostaglandin E2 as a Potent Therapeutic Target for Treatment of Colon Cancer. Prostaglandins Other Lipid Mediat. 2019, 144, 106338. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.P.; Smyth, E.M. COX-2 and PGE2-Dependent Immunomodulation in Breast Cancer. Prostaglandins Other Lipid Mediat. 2011, 96, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hida, T.; Yatabe, Y.; Achiwa, H.; Muramatsu, H.; Kozaki, K.; Nakamura, S.; Ogawa, M.; Mitsudomi, T.; Sugiura, T.; Takahashi, T. Increased Expression of Cyclooxygenase 2 Occurs Frequently in Human Lung Cancers, Specifically in Adenocarcinomas. Cancer Res. 1998, 58, 3761–3764. [Google Scholar] [PubMed]
- Krysan, K.; Merchant, F.H.; Zhu, L.; Dohadwala, M.; Luo, J.; Lin, Y.; Heuze-Vourc’h, N.; Põld, M.; Seligson, D.; Chia, D.; et al. COX-2-Dependent Stabilization of Survivin in Non-Small Cell Lung Cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 206–208. [Google Scholar] [CrossRef]
- Kamiyama, M.; Pozzi, A.; Yang, L.; DeBusk, L.M.; Breyer, R.M.; Lin, P.C. EP2, a Receptor for PGE2, Regulates Tumor Angiogenesis through Direct Effects on Endothelial Cell Motility and Survival. Oncogene 2006, 25, 7019–7028. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Soares, R.; Reis-Filho, J.S.; Leitão, D.; Amendoeira, I.; Schmitt, F.C. Cyclo-Oxygenase 2 Expression Is Associated with Angiogenesis and Lymph Node Metastasis in Human Breast Cancer. J. Clin. Pathol. 2002, 55, 429–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Lv, Y.; Yan, D. Inhibition of Ep3 Attenuates Migration and Promotes Apoptosis of Non-Small Cell Lung Cancer Cells via Suppression of TGF-β/Smad Signaling. Oncol. Lett. 2018, 16, 5645–5654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, F.G.; Gorden, D.L.; Matta, P.; Shi, Q.; Matrisian, L.M.; DuBois, R.N. Role of β-Arrestin 1 in the Metastatic Progression of Colorectal Cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 1492–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of CDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Peng, P.; Loschko, J.; Feng, L.; Pham, P.; Cui, W.; Lee, K.P.; Krug, A.B.; Jiang, A. Plasmacytoid Dendritic Cells Cross-Prime Naive CD8 T Cells by Transferring Antigen to Conventional Dendritic Cells through Exosomes. Proc. Natl. Acad. Sci. USA 2020, 117, 23730–23741. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 Promotes Tumor Progression by Inducing Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive Feedback between PGE2 and COX2 Redirects the Differentiation of Human Dendritic Cells toward Stable Myeloid-Derived Suppressor Cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef]
- Rong, Y.; Yuan, C.H.; Qu, Z.; Zhou, H.; Guan, Q.; Yang, N.; Leng, X.H.; Bu, L.; Wu, K.; Wang, F.B. Doxorubicin Resistant Cancer Cells Activate Myeloid-Derived Suppressor Cells by Releasing PGE2. Sci. Rep. 2016, 6, 23824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Ubreva, J.; Català-Moll, F.; Obermajer, N.; Álvarez-Errico, D.; Ramirez, R.N.; Company, C.; Vento-Tormo, R.; Moreno-Bueno, G.; Edwards, R.P.; Mortazavi, A.; et al. Prostaglandin E2 Leads to the Acquisition of DNMT3A-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017, 21, 154–167. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Poschke, I.; Wennerberg, E.; de Coaña, Y.P.; Brage, S.E.; Schultz, I.; Hansson, J.; Masucci, G.; Lundqvist, A.; Kiessling, R. Melanoma-Educated CD14+ Cells Acquire a Myeloid-Derived Suppressor Cell Phenotype through COX-2-Dependent Mechanisms. Cancer Res. 2013, 73, 3877–3887. [Google Scholar] [CrossRef] [Green Version]
- Eruslanov, E.; Kaliberov, S.; Daurkin, I.; Kaliberova, L.; Buchsbaum, D.; Vieweg, J.; Kusmartsev, S. Altered Expression of 15-Hydroxyprostaglandin Dehydrogenase in Tumor-Infiltrated CD11b Myeloid Cells: A Mechanism for Immune Evasion in Cancer. J. Immunol. 2009, 182, 7548–7557. [Google Scholar] [CrossRef] [Green Version]
- Oshima, H.; Hioki, K.; Popivanova, B.K.; Oguma, K.; van Rooijen, N.; Ishikawa, T.; Oshima, M. Prostaglandin E2 2 Signaling and Bacterial Infection Recruit Tumor-Promoting Macrophages to Mouse Gastric Tumors. Gastroenterology 2011, 140, 596–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Yang, S.C.; Zhu, L.; Reckamp, K.; Gardner, B.; Baratelli, F.; Huang, M.; Batra, R.K.; Dubinett, S.M. Tumor Cyclooxygenase-2/Prostaglandin E2-Dependent Promotion of FOXP3 Expression and CD4+CD25+ T Regulatory Cell Activities in Lung Cancer. Cancer Res. 2005, 65, 5211–5220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonavita, E.; Bromley, C.P.; Jonsson, G.; Pelly, V.S.; Sahoo, S.; Walwyn-Brown, K.; Mensurado, S.; Moeini, A.; Flanagan, E.; Bell, C.R.; et al. Antagonistic Inflammatory Phenotypes Dictate Tumor Fate and Response to Immune Checkpoint Blockade. Immunity 2020, 53, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Singer, K.; Cheng, W.C.; Kreutz, M.; Ho, P.C.; Siska, P.J. Immunometabolism in Cancer at a Glance. DMM Dis. Models Mech. 2018, 11, dmm034272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; vander Heiden, M.G.; Muir, A. Quantification of Microenvironmental Metabolites in Murine Cancers Reveals Determinants of Tumor Nutrient Availability. Elife 2019, 8, e44235. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Thompson, C.B. Metabolic Regulation of Cell Growth and Proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T Cell Metabolism Drives Immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.C.; Bihuniak, J.D.; MacIntyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Bantug, G.R.; Galluzzi, L.; Kroemer, G.; Hess, C. The Spectrum of T Cell Metabolism in Health and Disease. Nat. Rev. Immunol. 2018, 18, 19–34. [Google Scholar] [CrossRef]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Sukumar, M.; Roychoudhuri, R.; Restifo, N.P. Nutrient Competition: A New Axis of Tumor Immunosuppression. Cell 2015, 162, 1206–1208. [Google Scholar] [CrossRef] [Green Version]
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer Mediates Effector T Cell Dysfunction by Targeting MicroRNAs and EZH2 via Glycolysis Restriction. Nat. Immunol. 2016, 17, 95–103. [Google Scholar] [CrossRef]
- Peng, X.; Chen, Z.; Farshidfar, F.; Xu, X.; Lorenzi, P.L.; Wang, Y.; Cheng, F.; Tan, L.; Mojumdar, K.; Du, D.; et al. Molecular Characterization and Clinical Relevance of Metabolic Expression Subtypes in Human Cancers. Cell Rep. 2018, 23, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-Tumor Activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.K.; Park, H.J.; MacLeod, M.; Chandler, P.; Munn, D.H.; Mellor, A.L. Tryptophan Deprivation Sensitizes Activated T Cells to Apoptosis Prior to Cell Division. Immunology 2002, 107, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T Cell Apoptosis by Tryptophan Catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Venkateswaran, N.; Lafita-Navarro, M.C.; Hao, Y.H.; Kilgore, J.A.; Perez-Castro, L.; Braverman, J.; Borenstein-Auerbach, N.; Kim, M.; Lesner, N.P.; Mishra, P.; et al. MYC Promotes Tryptophan Uptake and Metabolism by the Kynurenine Pathway in Colon Cancer. Genes Dev. 2019, 33, 1236–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.; Yue, L.; Shi, J.; Shao, M.; Wu, T. Role of IDO and TDO in Cancers and Related Diseases and the Therapeutic Implications. J. Cancer 2019, 10, 2771–2782. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Wick, W.; van den Eynde, B.J. Tryptophan Catabolism in Cancer: Beyond IDO and Tryptophan Depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [Green Version]
- Schramme, F.; Crosignani, S.; Frederix, K.; Hoffmann, D.; Pilotte, L.; Stroobant, V.; Preillon, J.; Driessens, G.; van den Eynde, B.J. Inhibition of Tryptophan-Dioxygenase Activity Increases the Antitumor Efficacy of Immune Checkpoint Inhibitors. Cancer Immunol. Res. 2020, 8, 32–45. [Google Scholar] [CrossRef]
- Clever, D.; Roychoudhuri, R.; Constantinides, M.G.; Askenase, M.H.; Sukumar, M.; Klebanoff, C.A.; Eil, R.L.; Hickman, H.D.; Yu, Z.; Pan, J.H.; et al. Oxygen Sensing by T Cells Establishes an Immunologically Tolerant Metastatic Niche. Cell 2016, 166, 1117–1131. [Google Scholar] [CrossRef] [Green Version]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer Acidity: An Ultimate Frontier of Tumor Immune Escape and a Novel Target of Immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Eil, R.; Vodnala, S.K.; Clever, D.; Klebanoff, C.A.; Sukumar, M.; Pan, J.H.; Palmer, D.C.; Gros, A.; Yamamoto, T.N.; Patel, S.J.; et al. Ionic Immune Suppression within the Tumour Microenvironment Limits T Cell Effector Function. Nature 2016, 537, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Vodnala, S.K.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.H.; Lee, P.H.; Shin, M.H.; Patel, S.J.; Yu, Z.; et al. T Cell Stemness and Dysfunction in Tumors Are Triggered by a Common Mechanism. Science 2019, 363, eaau0135. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xie, J.; Wu, G.; Shen, J.; Collins, R.; Chen, W.; Kang, X.; Luo, M.; Zou, Y.; Huang, L.J.-S.; et al. Fasting Selectively Blocks Development of Acute Lymphoblastic Leukemia via Leptin-Receptor Upregulation. Nat. Med. 2017, 23, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Rao, S.; Enot, D.P.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Jacquelot, N.; Yamazaki, T.; et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 2016, 30, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987. [Google Scholar] [CrossRef]
- Leone, R.D.; Emens, L.A. Targeting Adenosine for Cancer Immunotherapy. J. ImmunoTher. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Xu, X.; Qiao, M.; Li, X.; Zhao, C.; Zhou, F.; Gao, G.; Wu, F.; Chen, X.; Su, C.; et al. Comprehensive Evaluation of NT5E/CD73 Expression and Its Prognostic Significance in Distinct Types of Cancers. BMC Cancer 2018, 18, 267. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Lo, Y.C.; Powell, J.D. A2aR Antagonists: Next Generation Checkpoint Blockade for Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic Landscape of the Tumor Microenvironment at Single Cell Resolution. Nat. Commun. 2019, 10, 3763. [Google Scholar] [CrossRef]
- Stoll, G.; Kremer, M.; Bloy, N.; Joseph, A.; Castedo, M.; Meurice, G.; Klein, C.; Galluzzi, L.; Michels, J.; Kroemer, G. Metabolic Enzymes Expressed by Cancer Cells Impact the Immune Infiltrate. OncoImmunology 2019, 8, e1571389. [Google Scholar] [CrossRef] [Green Version]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbatino, F.; Liguori, L.; Polcaro, G.; Salvato, I.; Caramori, G.; Salzano, F.A.; Casolaro, V.; Stellato, C.; Col, J.D.; Pepe, S. Role of Human Leukocyte Antigen System as A Predictive Biomarker for Checkpoint-Based Immunotherapy in Cancer Patients. Int. J. Mol. Sci. 2020, 21, 7295. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Kambayashi, T.; Laufer, T.M. Atypical MHC Class II-Expressing Antigen-Presenting Cells: Can Anything Replace a Dendritic Cell? Nat. Rev. Immunol. 2014, 14, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, M.L.; Cook, R.S.; Johnson, D.B.; Balko, J.M. Biological Consequences of MHC-II Expression by Tumor Cells in Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2392–2402. [Google Scholar] [CrossRef]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative Tumour-Specific Antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef]
- Gubin, M.M.; Artyomov, M.N.; Mardis, E.R.; Schreiber, R.D. Tumor Neoantigens: Building a Framework for Personalized Cancer Immunotherapy. J. Clin. Investig. 2015, 125, 3413–3421. [Google Scholar] [CrossRef]
- McFarlane, R.J.; Feichtinger, J.; Larcombe, L. Cancer Germline Gene Activation: Friend or Foe? Cell Cycle 2014, 13, 2151–2152. [Google Scholar] [CrossRef] [Green Version]
- Dersh, D.; Hollý, J.; Yewdell, J.W. A Few Good Peptides: MHC Class I-Based Cancer Immunosurveillance and Immunoevasion. Nat. Rev. Immunol. 2020, 21, 116–128. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N. Cancer Immune Escape: MHC Expression in Primary Tumours versus Metastases. Immunology 2019, 158, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef] [PubMed]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Cabrera, T.; Aptsiauri, N. “Hard” and “Soft” Lesions Underlying the HLA Class I Alterations in Cancer Cells: Implications for Immunotherapy. Int. J. Cancer 2010, 127, 249–256. [Google Scholar] [CrossRef]
- Jongsma, M.L.M.; Guarda, G.; Spaapen, R.M. The Regulatory Network behind MHC Class I Expression. Mol. Immunol. 2019, 113, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Kärre, K. Natural Killer Cell Recognition of Missing Self. Nat. Immunol. 2008, 9, 477–480. [Google Scholar] [CrossRef]
- Ljunggren, H.-G.; Kärre, K. In Search of the ‘Missing Self’: MHC Molecules and NK Cell Recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Anfossi, N.; André, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK Cell Education by Inhibitory Receptors for MHC Class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef]
- Judge, S.J.; Murphy, W.J.; Canter, R.J. Characterizing the Dysfunctional NK Cell: Assessing the Clinical Relevance of Exhaustion, Anergy, and Senescence. Front. Cell. Infect. Microbiol. 2020, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Ardolino, M.; Azimi, C.S.; Iannello, A.; Trevino, T.N.; Horan, L.; Zhang, L.; Deng, W.; Ring, A.M.; Fischer, S.; Garcia, K.C.; et al. Cytokine Therapy Reverses NK Cell Anergy in MHC-Deficient Tumors. J. Clin. Investig. 2014, 124, 4781–4794. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.; Jeon, I.; Kim, B.-S.; Park, M.; Bae, E.-A.; Song, B.; Koh, C.-H.; Shin, K.-S.; Kim, I.-K.; Choi, K.; et al. IL-21-Mediated Reversal of NK Cell Exhaustion Facilitates Anti-Tumour Immunity in MHC Class I-Deficient Tumours. Nat. Commun. 2017, 8, 15776. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The Urgent Need to Recover MHC Class I in Cancers for Effective Immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Galat, V.; Galat4, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK Cell-Based Cancer Immunotherapy: From Basic Biology to Clinical Development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A MAb Is a Checkpoint Inhibitor That Promotes Anti-Tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef] [Green Version]
- Creelan, B.C.; Antonia, S.J. The NKG2A Immune Checkpoint—A New Direction in Cancer Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 277–278. [Google Scholar] [CrossRef] [PubMed]
- Bastidas-Legarda, L.Y.; Khakoo, S.I. Conserved and Variable Natural Killer Cell Receptors: Diverse Approaches to Viral Infections. Immunology 2019, 156, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Snyder, A. Adding to the Checkpoint Blockade Armamentarium. Nat. Med. 2019, 25, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A-HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef]
- Gallegos, C.E.; Michelin, S.; Dubner, D.; Carosella, E.D. Immunomodulation of Classical and Non-Classical HLA Molecules by Ionizing Radiation. Cell. Immunol. 2016, 303, 16–23. [Google Scholar] [CrossRef]
- Lin, A.; Zhang, X.; Ruan, Y.Y.; Wang, Q.; Zhou, W.J.; Yan, W.H. HLA-F Expression Is a Prognostic Factor in Patients with Non-Small-Cell Lung Cancer. Lung Cancer 2011, 74, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, A.; Zhang, J.G.; Bao, W.G.; Xu, D.P.; Ruan, Y.Y.; Yan, W.H. Alteration of HLA-F and HLA I Antigen Expression in the Tumor Is Associated with Survival in Patients with Esophageal Squamous Cell Carcinoma. Int. J. Cancer 2013, 132, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Human Leukocyte Antigen (HLA)-E and HLA-F Expression in Gastric Cancer—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/25862890/ (accessed on 12 November 2021).
- Harada, A.; Ishigami, S.; Kijima, Y.; Nakajo, A.; Arigami, T.; Kurahara, H.; Kita, Y.; Yoshinaka, H.; Natsugoe, S. Clinical Implication of Human Leukocyte Antigen (HLA)-F Expression in Breast Cancer. Pathol. Int. 2015, 65, 569–574. [Google Scholar] [CrossRef]
- Apps, R.; Murphy, S.P.; Fernando, R.; Gardner, L.; Ahad, T.; Moffett, A. Human Leucocyte Antigen (HLA) Expression of Primary Trophoblast Cells and Placental Cell Lines, Determined Using Single Antigen Beads to Characterize Allotype Specificities of Anti-HLA Antibodies. Immunology 2009, 127, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Loustau, M.; Anna, F.; Dréan, R.; Lecomte, M.; Langlade-Demoyen, P.; Caumartin, J. HLA-G Neo-Expression on Tumors. Front. Immunol. 2020, 11, 1685. [Google Scholar] [CrossRef] [PubMed]
- Pazmany, L.; Mandelboim, O.; Valés-Gómez, M.; Davis, D.M.; Reyburn, H.T.; Strominger, J.L. Protection from Natural Killer Cell-Mediated Lysis by HLA-G Expression on Target Cells. Science 1996, 274, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhou, Y.; Wei, H. Roles of HLA-G in the Maternal-Fetal Immune Microenvironment. Front. Immunol. 2020, 11, 592010. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.M.R.; Meissner, T.B.; Tilburgs, T.; Strominger, J.L. HLA-G: At the Interface of Maternal-Fetal Tolerance. Trends Immunol. 2017, 38, 272–286. [Google Scholar] [CrossRef]
- De Kruijf, E.M.; Sajet, A.; van Nes, J.G.H.; Natanov, R.; Putter, H.; Smit, V.T.H.B.M.; Liefers, G.J.; van den Elsen, P.J.; van de Velde, C.J.H.; Kuppen, P.J.K. HLA-E and HLA-G Expression in Classical HLA Class I-Negative Tumors Is of Prognostic Value for Clinical Outcome of Early Breast Cancer Patients. J. Immunol. 2010, 185, 7452–7459. [Google Scholar] [CrossRef] [Green Version]
- Zeestraten, E.C.M.; Reimers, M.S.; Saadatmand, S.; Dekker, J.W.T.; Liefers, G.J.; van den Elsen, P.J.; van de Velde, C.J.H.; Kuppen, P.J.K. Combined Analysis of HLA Class I, HLA-E and HLA-G Predicts Prognosis in Colon Cancer Patients. Br. J. Cancer 2014, 110, 459–468. [Google Scholar] [CrossRef]
- Yie, S.-M.; Yang, H.; Ye, S.-R.; Li, K.; Dong, D.-D.; Lin, X. mei Expression of Human Leucocyte Antigen G (HLA-G) Is Associated with Prognosis in Non-Small Cell Lung Cancer. Lung Cancer 2007, 58, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Xiao, J.; Li, W.; Li, S.; Xie, B.; He, J.; Liu, C. Prognostic and Clinicopathological Value of Human Leukocyte Antigen G in Gastrointestinal Cancers: A Meta-Analysis. Front. Oncol. 2021, 11, 642902. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Yan, W.-H. Heterogeneity of HLA-G Expression in Cancers: Facing the Challenges. Front. Immunol. 2018, 9, 2164. [Google Scholar] [CrossRef]
- Lin, A.; Yan, W.H. Human Leukocyte Antigen-G (HLA-G) Expression in Cancers: Roles in Immune Evasion, Metastasis and Target for Therapy. Mol. Med. 2015, 21, 782–791. [Google Scholar] [CrossRef]
- Carosella, E.D.; Rouas-Freiss, N.; Roux, D.T.L.; Moreau, P.; LeMaoult, J. HLA-G: An Immune Checkpoint Molecule. Adv. Immunol. 2015, 127, 33–144. [Google Scholar] [CrossRef]
- Krijgsman, D.; Roelands, J.; Hendrickx, W.; Bedognetti, D.; Kuppen, P.J.K. HLA-G: A New Immune Checkpoint in Cancer? Int. J. Mol. Sci. 2020, 21, 4528. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Yan, W.H. Intercellular transfer of HLA‐G: Its potential in cancer immunology. Clin. Transl. Immunol. 2019, 8, e1077. [Google Scholar] [CrossRef] [Green Version]
- Carosella, E.D.; Gregori, S.; Roux, D.T.-L. HLA-G/LILRBs: A Cancer Immunotherapy Challenge. Trends Cancer 2021, 7, 389–392. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Yan, Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer 2020, 6, 580–592. [Google Scholar] [CrossRef]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the Epigenetic Regulation of Antitumour Immunity. Nat. Rev. Drug Discov. 2020, 19, 776–800. [Google Scholar] [CrossRef]
- Gjetting, T.; Gad, M.; Fröhlich, C.; Lindsted, T.; Melander, M.C.; Bhatia, V.K.; Grandal, M.M.; Dietrich, N.; Uhlenbrock, F.; Galler, G.R.; et al. Sym021, a Promising Anti-PD1 Clinical Candidate Antibody Derived from a New Chicken Antibody Discovery Platform. In MAbs; Taylor & Francis: New York, NY, USA, 2019; Volume 11, pp. 666–680. [Google Scholar] [CrossRef]
- Attermann, A.; Bjerregaard, A.-M.; Saini, S.; Grønbæk, K.; Hadrup, S. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann. Oncol. 2018, 29, 2183–2191. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic Dendritic Cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [Green Version]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic Cells Induce Peripheral T Cell Unresponsiveness under Steady State Conditions in Vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Heeger, P.S.; Lalli, P.N.; Lin, F.; Valujskikh, A.; Liu, J.; Muqim, N.; Xu, Y.; Medof, M.E. Decay-Accelerating Factor Modulates Induction of T Cell Immunity. J. Exp. Med. 2005, 201, 1523–1530. [Google Scholar] [CrossRef] [Green Version]
- Eisenbarth, S.C.; Piggott, D.A.; Huleatt, J.W.; Visintin, I.; Herrick, C.A.; Bottomly, K. Lipopolysaccharide-Enhanced, Toll-like Receptor 4-Dependent T Helper Cell Type 2 Responses to Inhaled Antigen. J. Exp. Med. 2002, 196, 1645–1651. [Google Scholar] [CrossRef] [Green Version]
- Wakkach, A.; Fournier, N.; Brun, V.; Breittmayer, J.P.; Cottrez, F.; Groux, H. Characterization of Dendritic Cells That Induce Tolerance and T Regulatory 1 Cell Differentiation in Vivo. Immunity 2003, 18, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Ochando, J.C.; Homma, C.; Yang, Y.; Hidalgo, A.; Garin, A.; Tacke, F.; Angeli, V.; Li, Y.; Boros, P.; Ding, Y.; et al. Alloantigen-Presenting Plasmacytoid Dendritic Cells Mediate Tolerance to Vascularized Grafts. Nat. Immunol. 2006, 7, 652–662. [Google Scholar] [CrossRef]
- Döhler, A.; Schneider, T.; Eckert, I.; Ribechini, E.; Andreas, N.; Riemann, M.; Reizis, B.; Weih, F.; Lutz, M.B. RelB+ Steady-State Migratory Dendritic Cells Control the Peripheral Pool of the Natural Foxp3+ Regulatory T Cells. Front. Immunol. 2017, 8, 726. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.W.; Tjota, M.Y.; Clay, B.S.; vander Lugt, B.; Bandukwala, H.S.; Hrusch, C.L.; Decker, D.C.; Blaine, K.M.; Fixsen, B.R.; Singh, H.; et al. Transcription Factor IRF4 Drives Dendritic Cells to Promote Th2 Differentiation. Nat. Commun. 2013, 4, 2990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Søndergaard, J.N.; van Heeringen, S.J.; Looman, M.W.G.; Tang, C.; Triantis, V.; Louche, P.; Janssen-Megens, E.M.; Sieuwerts, A.M.; Martens, J.W.M.; Logie, C.; et al. Dendritic Cells Actively Limit Interleukin-10 Production under Inflammatory Conditions via DC-SCRIPT and Dual-Specificity Phosphatase 4. Front. Immunol. 2018, 9, 1420. [Google Scholar] [CrossRef] [PubMed]
- Hontelez, S.; Ansems, M.; Karthaus, N.; Zuidscherwoude, M.; Looman, M.W.; Triantis, V.; Adema, G.J. Dendritic Cell-Specific Transcript: Dendritic Cell Marker and Regulator of TLR-Induced Cytokine Production. J. Immunol. 2012, 189, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, A.L.; Munn, D.H. IDO Expression by Dendritic Cells: Tolerance and Tryptophan Catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Lee, J.R.; Jhaver, K.G.; Johnson, T.S.; Keskin, D.B.; Marshall, B.; Chandler, P.; Antonia, S.J.; Burgess, R.; et al. Potential Regulatory Function of Human Dendritic Cells Expressing Indoleamine 2,3-Dioxygenase. Science 2002, 297, 1867–1870. [Google Scholar] [CrossRef]
- Molinier-Frenkel, V.; Castellano, F. Immunosuppressive Enzymes in the Tumor Microenvironment. FEBS Lett. 2017, 591, 3135–3157. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; von Knebel Doeberitz, N.; Oezen, I.; Wick, W.; Ochs, K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front. Immunol. 2015, 6, 673. [Google Scholar] [CrossRef]
- Rizeq, B.; Zakaria, Z.; Ouhtit, A. Towards Understanding the Mechanisms of Actions of Carcinoembryonic Antigen-Related Cell Adhesion Molecule 6 in Cancer Progression. Cancer Sci. 2018, 109, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.; Mahadevan, D. Emerging Role and Targeting of Carcinoembryonic Antigen-Related Cell Adhesion Molecule 6 (CEACAM6) in Human Malignancies. Clin. Cancer Drugs 2015, 2, 100–111. [Google Scholar] [CrossRef]
- Calinescu, A.; Turcu, G.; Nedelcu, R.I.; Brinzea, A.; Hodorogea, A.; Antohe, M.; Diaconu, C.; Bleotu, C.; Pirici, D.; Jilaveanu, L.B.; et al. On the Dual Role of Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 (CEACAM1) in Human Malignancies. J. Immunol. Res. 2018, 2018, 7169081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual-García, M.; Bonfill-Teixidor, E.; Planas-Rigol, E.; Rubio-Perez, C.; Iurlaro, R.; Arias, A.; Cuartas, I.; Sala-Hojman, A.; Escudero, L.; Martínez-Ricarte, F.; et al. LIF Regulates CXCL9 in Tumor-Associated Macrophages and Prevents CD8+ T Cell Tumor-Infiltration Impairing Anti-PD1 Therapy. Nat. Commun. 2019, 10, 2416. [Google Scholar] [CrossRef] [Green Version]
- Weiskopf, K. Cancer Immunotherapy Targeting the CD47/SIRPα Axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar] [CrossRef]
- Tong, B.; Wang, M. CD47 Is a Novel Potent Immunotherapy Target in Human Malignancies: Current Studies and Future Promises. Future Oncol. 2018, 14, 2179–2188. [Google Scholar] [CrossRef]
- Koncina, E.; Roth, L.; Gonthier, B.; Bagnard, D. Role of Semaphorins during Axon Growth and Guidance. Adv. Exp. Med. Biol. 2007, 621, 50–64. [Google Scholar] [CrossRef]
- Chen, L.H.; Cuang, E.Y. Importance of Semaphorins in Cancer Immunity. Transl. Lung Cancer Res. 2019, 8, S468–S470. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.E.; Jonason, A.S.; Bussler, H.; Torno, S.; Veeraraghavan, J.; Reilly, C.; Doherty, M.A.; Seils, J.; Winter, L.A.; Mallow, C.; et al. Antibody Blockade of Semaphorin 4D Promotes Immune Infiltration into Tumor and Enhances Response to Other Immunomodulatory Therapies. Cancer Immunol. Res. 2015, 3, 698–701. [Google Scholar] [CrossRef] [Green Version]
- McClellan, J.L.; Mark Davis, J.; Steiner, J.L.; Enos, R.T.; Jung, S.H.; Carson, J.A.; Pena, M.M.; Carnevale, K.A.; Berger, F.G.; Angela Murphy, E. Linking Tumor-Associated Macrophages, Inflammation, and Intestinal Tumorigenesis: Role of MCP-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1087–G1095. [Google Scholar] [CrossRef] [Green Version]
- Kzhyshkowska, J.; Gratchev, A.; Goerdt, S. Stabilin-1, a Homeostatic Scavenger Receptor with Multiple Functions. J. Cell. Mol. Med. 2006, 10, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Viitala, M.; Virtakoivu, R.; Tadayon, S.; Rannikko, J.; Jalkanen, S.; Hollmen, M. Immunotherapeutic Blockade of Macrophage Clever-1 Reactivates the CD8 + T-Cell Response against Immunosuppressive Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3289–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, G. Biology of the TAM Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef]
- Zhu, C.; Wei, Y.; Wei, X. AXL Receptor Tyrosine Kinase as a Promising Anti-Cancer Approach: Functions, Molecular Mechanisms and Clinical Applications. Mol. Cancer 2019, 18, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera, T.A.; Giaccia, A.J. Molecular Pathways: Oncologic Pathways and Their Role in T-Cell Exclusion and Immune Evasion-A New Role for the AXL Receptor Tyrosine Kinase. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2928–2933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayoub, A.S.; Brekken, R.A. TIMs, TAMs, and PS- Antibody Targeting: Implications for Cancer Immunotherapy. Cell Commun. Signal. CCS 2020, 18, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Huang, X.; Lynn, K.D.; Thorpe, P.E. Phosphatidylserine-Targeting Antibody Induces M1 Macrophage Polarization and Promotes Myeloid-Derived Suppressor Cell Differentiation. Cancer Immunol. Res. 2013, 1, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Pennock, G.K.; Chow, L.Q.M. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. Oncologist 2015, 20, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Ghobrial, I.M. Immunotherapy for Hematological Malignancies. J. Life Sci. 2019, 1, 46. [Google Scholar] [CrossRef]
- Joshi, S.; Durden, D.L. Combinatorial Approach to Improve Cancer Immunotherapy: Rational Drug Design Strategy to Simultaneously Hit Multiple Targets to Kill Tumor Cells and to Activate the Immune System. J. Oncol. 2019, 2019, 5245034. [Google Scholar] [CrossRef] [Green Version]
- Eggermont, A.M.M.; Chiarion-Sileni, V.; Grob, J.J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant Ipilimumab versus Placebo after Complete Resection of High-Risk Stage III Melanoma (EORTC 18071): A Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2015, 16, 522–530. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in Patients with Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-Line Nivolumab plus Ipilimumab Combined with Two Cycles of Chemotherapy in Patients with Non-Small-Cell Lung Cancer (CheckMate 9LA): An International, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-Line Nivolumab plus Ipilimumab in Unresectable Malignant Pleural Mesothelioma (CheckMate 743): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab with or without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.A.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III Randomized Clinical Trial Comparing Tremelimumab with Standard-of-Care Chemotherapy in Patients with Advanced Melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Sharma, P.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. CheckMate 025 Randomized Phase 3 Study: Outcomes by Key Baseline Factors and Prior Therapy for Nivolumab Versus Everolimus in Advanced Renal Cell Carcinoma. Eur. Urol. 2017, 72, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B.; et al. Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus Chemotherapy in Patients with Advanced Melanoma Who Progressed after Anti-CTLA-4 Treatment (CheckMate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab Alone and Nivolumab plus Ipilimumab in Recurrent Small-Cell Lung Cancer (CheckMate 032): A Multicentre, Open-Label, Phase 1/2 Trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Borghaei, H.; Gettinger, S.; Vokes, E.E.; Chow, L.Q.M.; Burgio, M.A.; de Castro Carpeno, J.; Pluzanski, A.; Arrietac, O.; Frontera, O.A.; Chiari, R.; et al. Five-Year Outcomes from the Randomized, Phase Iii Trials Checkmate 017 and 057: Nivolumab versus Docetaxel in Previously Treated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2021, 39, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.-Y.; Breder, V.V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Results of KEYNOTE-240: Phase 3 Study of Pembrolizumab (Pembro) vs Best Supportive Care (BSC) for Second Line Therapy in Advanced Hepatocellular Carcinoma (HCC). J. Clin. Oncol. 2019, 37, 4004. [Google Scholar] [CrossRef]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus Investigator-Choice Chemotherapy for Ipilimumab-Refractory Melanoma (KEYNOTE-002): A Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.C.; Ros, W.; Delord, J.P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab Alone or with Chemotherapy versus Cetuximab with Chemotherapy for Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-048): A Randomised, Open-Label, Phase 3 Study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Kojima, T.; Shah, M.A.; Muro, K.; Francois, E.; Adenis, A.; Hsu, C.H.; Doi, T.; Moriwaki, T.; Kim, S.B.; Lee, S.H.; et al. Randomized Phase III KEYNOTE-181 Study of Pembrolizumab Versus Chemotherapy in Advanced Esophageal Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 4138–4148. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Kojima, T.; Hochhauser, D.; Enzinger, P.; Raimbourg, J.; Hollebecque, A.; Lordick, F.; Kim, S.B.; Tajika, M.; Kim, H.T.; et al. Efficacy and Safety of Pembrolizumab for Heavily Pretreated Patients with Advanced, Metastatic Adenocarcinoma or Squamous Cell Carcinoma of the Esophagus: The Phase 2 KEYNOTE-180 Study. JAMA Oncol. 2019, 5, 546–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus Docetaxel for Previously Treated, PD-L1-Positive, Advanced Non-Small-Cell Lung Cancer (KEYNOTE-010): A Randomised Controlled Trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.K.; Wu, Y.L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus Chemotherapy for Previously Untreated, PD-L1-Expressing, Locally Advanced or Metastatic Non-Small-Cell Lung Cancer (KEYNOTE-042): A Randomised, Open-Label, Controlled, Phase 3 Trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; de Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Ott, P.A.; Elez, E.; Hiret, S.; Kim, D.W.; Morosky, A.; Saraf, S.; Piperdi, B.; Mehnert, J.M. Pembrolizumab in Patients with Extensive-Stage Small-Cell Lung Cancer: Results From the Phase Ib KEYNOTE-028 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3823–3829. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet. Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- O’Malley, D.; Marabelle, A.; de Jesus-Acosta, A.; Piha-Paul, S.A.; Arkhipov, A.; Longo, F.; Motola-Kuba, D.; Shapira-Frommer, R.; Geva, R.; Rimel, B.J.; et al. Pembrolizumab in Patients with MSI-H Advanced Endometrial Cancer from the KEYNOTE-158 Study. Ann. Oncol. 2019, 30, v425–v426. [Google Scholar] [CrossRef]
- Oaknin, A.; Tinker, A.V.; Gilbert, L.; Samouëlian, V.; Mathews, C.; Brown, J.; Barretina-Ginesta, M.P.; Moreno, V.; Gravina, A.; Abdeddaim, C.; et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients with Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020, 6, 1766–1772. [Google Scholar] [CrossRef]
- Kasherman, L.; Ahrari, S.; Lheureux, S. Dostarlimab in the Treatment of Recurrent or Primary Advanced Endometrial Cancer. Future Oncol. 2021, 17, 877–892. [Google Scholar] [CrossRef]
- Keam, S.J. Toripalimab: First Global Approval. Drugs 2019, 79, 573–578. [Google Scholar] [CrossRef]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1–Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D.; Arija, J.Á.A.; Bamias, A.; Davis, I.D.; de Santis, M.; Kikuchi, E.; Garcia-del-Muro, X.; de Giorgi, U.; Mencinger, M.; Izumi, K.; et al. Atezolizumab with or without Chemotherapy in Metastatic Urothelial Cancer (IMvigor130): A Multicentre, Randomised, Placebo-Controlled Phase 3 Trial. Lancet 2020, 395, 1547–1557. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus Docetaxel in Patients with Previously Treated Non-Small-Cell Lung Cancer (OAK): A Phase 3, Open-Label, Multicentre Randomised Controlled Trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [Green Version]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.U.; Grischke, E.M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A Randomised Phase II Study Investigating Durvalumab in Addition to an Anthracycline Taxane-Based Neoadjuvant Therapy in Early Triple-Negative Breast Cancer: Clinical Results and Biomarker Analysis of GeparNuevo Study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Lavaud, P.; Hamilou, Z.; Loriot, Y.; Massard, C. Durvalumab in Urothelial Cancers. Expert Rev. Anticancer. Ther. 2018, 18, 311–318. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Russell, J.; Lebbé, C.; Chmielowski, B.; Gambichler, T.; Grob, J.J.; Kiecker, F.; Rabinowits, G.; Terheyden, P.; Zwiener, I.; et al. Efficacy and Safety of First-Line Avelumab Treatment in Patients with Stage IV Metastatic Merkel Cell Carcinoma a Preplanned Interim Analysis of a Clinical Trial. JAMA Oncol. 2018, 4, e180077. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Motzer, R.J.; Rini, B.I.; Haanen, J.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Gravis-Mescam, G.; Uemura, M.; Lee, J.L.; et al. Updated Efficacy Results from the JAVELIN Renal 101 Trial: First-Line Avelumab plus Axitinib versus Sunitinib in Patients with Advanced Renal Cell Carcinoma. Ann. Oncol. 2020, 31, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Huang, X.; Chen, X.; Liu, J.; Wu, C.; Pu, Q.; Wang, Y.; Kang, X.; Zhou, L. Characterization of a Novel Anti-Human Lymphocyte Activation Gene 3 (LAG-3) Antibody for Cancer Immunotherapy. In MAbs; Taylor & Francis: Abingdon, UK, 2019; Volume 11, pp. 1139–1148. [Google Scholar] [CrossRef]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and Efficacy of Combination Treatment with Anti-LAG-3 and Anti-PD-1 Monoclonal Antibodies in Glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Harding, J.J.; Moreno, V.; Bang, Y.-J.; Hong, M.H.; Patnaik, A.; Trigo, J.; Szpurka, A.M.; Yamamoto, N.; Doi, T.; Fu, S.; et al. Blocking TIM-3 in Treatment-Refractory Advanced Solid Tumors: A Phase Ia/b Study of LY3321367 with or without an Anti-PD-L1 Antibody. Clin. Cancer Res. 2021, 27, 2168–2178. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Shimizu, T.; Doi, T.; Hodi, F.S.; Rottey, S.; Aftimos, P.G.; Tina Liu, Z.; Velez de Mendizabal, N.; Szpurka, A.M.; Piao, Y.; et al. A Phase Ia/b Study of TIM-3/PD-L1 Bispecific Antibody in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2019, 37, TPS2654. [Google Scholar] [CrossRef]
- Mesa, R.A.; Kiladjian, J.J.; Catalano, J.V.; Devos, T.; Egyed, M.; Hellmann, A.; McLornan, D.; Shimoda, K.; Winton, E.F.; Deng, W.; et al. Simplify-1: A Phase III Randomized Trial of Momelotinib versus Ruxolitinib in Janus Kinase Inhibitor–Naïve Patients with Myelofibrosis. J. Clin. Oncol. 2017, 35, 3844–3850. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Spira, A.; Kelly, K.; Humeniuk, R.; Kawashima, J.; Kong, S.; Koczywas, M. Phase 1B Study of Momelotinib Combined with Trametinib in Metastatic, Kirsten Rat Sarcoma Viral Oncogene Homolog-Mutated Non–Small-Cell Lung Cancer After Platinum-Based Chemotherapy Treatment Failure. Clin. Lung Cancer 2018, 19, e853–e859. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.J.; Cervantes, F.; Harrison, C.N.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-Term Survival in Patients Treated with Ruxolitinib for Myelofibrosis: COMFORT-I and-II Pooled Analyses. J. Hematol. Oncol. 2017, 10, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Bubnoff, N.; Ihorst, G.; Grishina, O.; Röthling, N.; Bertz, H.; Duyster, J.; Finke, J.; Zeiser, R. Ruxolitinib in GvHD (RIG) Study: A Multicenter, Randomized Phase 2 Trial to Determine the Response Rate of Ruxolitinib and Best Available Treatment (BAT) versus BAT in Steroid-Refractory Acute Graft-versus-Host Disease (AGvHD) (NCT02396628). BMC Cancer 2018, 18, 1132. [Google Scholar] [CrossRef]
- Ishikawa, C.; Senba, M.; Mori, N. Anti-Adult T-Cell Leukemia/Lymphoma Activity of Cerdulatinib, a Dual SYK/JAK Kinase Inhibitor. Int. J. Oncol. 2018, 53, 1681–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamlin, P.A.; Flinn, I.W.; Wagner-Johnston, N.; Burger, J.A.; Coffey, G.P.; Conley, P.B.; Michelson, G.; Leeds, J.M.; Der, K.; Kim, Y.; et al. Efficacy and Safety of the Dual SYK/JAK Inhibitor Cerdulatinib in Patients with Relapsed or Refractory B-Cell Malignancies: Results of a Phase I Study. Am. J. Hematol. 2019, 94, E90–E93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdeja, J.; Palandri, F.; Baer, M.R.; Quick, D.; Kiladjian, J.J.; Martinelli, G.; Verma, A.; Hamid, O.; Walgren, R.; Pitou, C.; et al. Phase 2 Study of Gandotinib (LY2784544) in Patients with Myeloproliferative Neoplasms. Leuk. Res. 2018, 71, 82–88. [Google Scholar] [CrossRef]
- Knapper, S.; Russell, N.; Gilkes, A.; Hills, R.K.; Gale, R.E.; Cavenagh, J.D.; Jones, G.; Kjeldsen, L.; Grunwald, M.R.; Thomas, I.; et al. A Randomized Assessment of Adding the Kinase Inhibitor Lestaurtinib to First-Line Chemotherapy for FLT3-Mutated AML. Blood 2017, 129, 1143–1154. [Google Scholar] [CrossRef]
- Hexner, E.O.; Mascarenhas, J.; Prchal, J.; Roboz, G.J.; Baer, M.R.; Ritchie, E.K.; Leibowitz, D.; Demakos, E.P.; Miller, C.; Siuty, J.; et al. Phase I Dose Escalation Study of Lestaurtinib in Patients with Myelofibrosis. Leuk. Lymphoma 2015, 56, 2543–2551. [Google Scholar] [CrossRef] [Green Version]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients with Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2018, 4, 652–659. [Google Scholar] [CrossRef]
- Schmidinger, M.; Hejna, M.; Zielinski, C.C. Aldesleukin in Advanced Renal Cell Carcinoma. Expert Rev. Anticancer. Ther. 2004, 4, 957–980. [Google Scholar] [CrossRef]
- Diab, A.; Tannir, N.M.; Bentebibel, S.E.; Hwu, P.; Papadimitrakopoulou, V.; Haymaker, C.; Kluger, H.M.; Gettinger, S.N.; Sznol, M.; Tykodi, S.S.; et al. Bempegaldesleukin (NKTR-214) plus Nivolumab in Patients with Advanced Solid Tumors: Phase I Dose-Escalation Study of Safety, Effi Cacy, and Immune Activation (PIVOT-02). Cancer Discov. 2020, 10, 1158–1173. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus Gemcitabine vs. Gemcitabine for First-Line Treatment of Patients with Unresectable Pancreatic Cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [Green Version]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-B1 Receptor Type i Inhibitor) and Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Ramanathan, R.; Choudry, H.; Jones, H.; Girgis, M.; Gooding, W.; Kalinski, P.; Bartlett, D.L. Phase II Trial of Adjuvant Dendritic Cell Vaccine in Combination with Celecoxib, Interferon-α, and Rintatolimod in Patients Undergoing Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy for Peritoneal Metastases. Ann. Surg. Oncol. 2021, 28, 4637–4646. [Google Scholar] [CrossRef] [PubMed]
- Herbert, W.C. Imiquimod and the Treatment of Cutaneous T-Cell Proliferative Diseases: At the Threshold. Skinmed 2003, 2, 273–274. [Google Scholar]
- Martínez-González, M.C.; Verea-Hernando, M.M.; Yebra-Pimentel, M.T.; del Pozo, J.; Mazaira, M.; Fonseca, E. Imiquimod in Mycosis Fungoides. Eur. J. Dermatol. 2008, 18, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Ramelyte, E.; Dummer, R.; Guenova, E. Investigative Drugs for the Treatment of Cutaneous T-Cell Lymphomas (CTCL): An Update. Expert Opin. Investig. Drugs 2019, 28, 799–809. [Google Scholar] [CrossRef]
- Rook, A.H.; Gelfand, J.C.; Wysocka, M.; Troxel, A.B.; Benoit, B.; Surber, C.; Elenitsas, R.; Buchanan, M.A.; Leahy, D.S.; Watanabe, R.; et al. Topical Resiquimod Can Induce Disease Regression and Enhance T-Cell Effector Functions in Cutaneous T-Cell Lymphoma. Blood 2015, 126, 1452–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, N.H.; Logan, T.F.; Hodi, F.S.; McDermott, D.; Melero, I.; Hamid, O.; Schmidt, H.; Robert, C.; Chiarion-Sileni, V.; Ascierto, P.A.; et al. Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin. Cancer Res. 2017, 23, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Flinn, I.; Taylor, M.H.; Sikic, B.I.; Brody, J.; Nemunaitis, J.; Feldman, A.; Hawthorne, T.R.; Rawls, T.; Keler, T.; et al. Safety and Activity of Varlilumab, a Novel and First-in-Class Agonist Anti-CD27 Antibody, for Hematologic Malignancies. Blood Adv. 2020, 4, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Infante, J.R.; Ansell, S.M.; Nemunaitis, J.J.; Weiss, G.R.; Villalobos, V.M.; Sikic, B.I.; Taylor, M.H.; Northfelt, D.W.; Carson, W.E.; et al. Safety and Activity of Varlilumab, a Novel and First-in-Class Agonist Anti-CD27 Antibody, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 2028–2036. [Google Scholar] [CrossRef] [Green Version]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, J.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J.; Anderson, L.D.; et al. Daratumumab, Lenalidomide, Bortezomib, and Dexamethasone for Transplant-Eligible Newly Diagnosed Multiple Myeloma: The GRIFFIN Trial. Blood 2020, 136, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Platzbecker, U.; Ades, L. How We Manage Adults with Myelodysplastic Syndrome. Br. J. Haematol. 2020, 189, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.P.; Trneny, M.; Izutsu, K.; Fowler, N.H.; Hong, X.; Zhu, J.; Zhang, H.; Offner, F.; Scheliga, A.; Nowakowski, G.S.; et al. AUGMENT: A Phase III Study of Lenalidomide plus Rituximab versus Placebo plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2019, 37, 1188–1199. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon Is a Direct Protein Target for Immunomodulatory and Antiproliferative Activities of Lenalidomide and Pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Rajkumar, S.V. Drug Therapy: Multiple Myeloma. N. Engl. J. Med. 2004, 351, 1860–1873. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef]



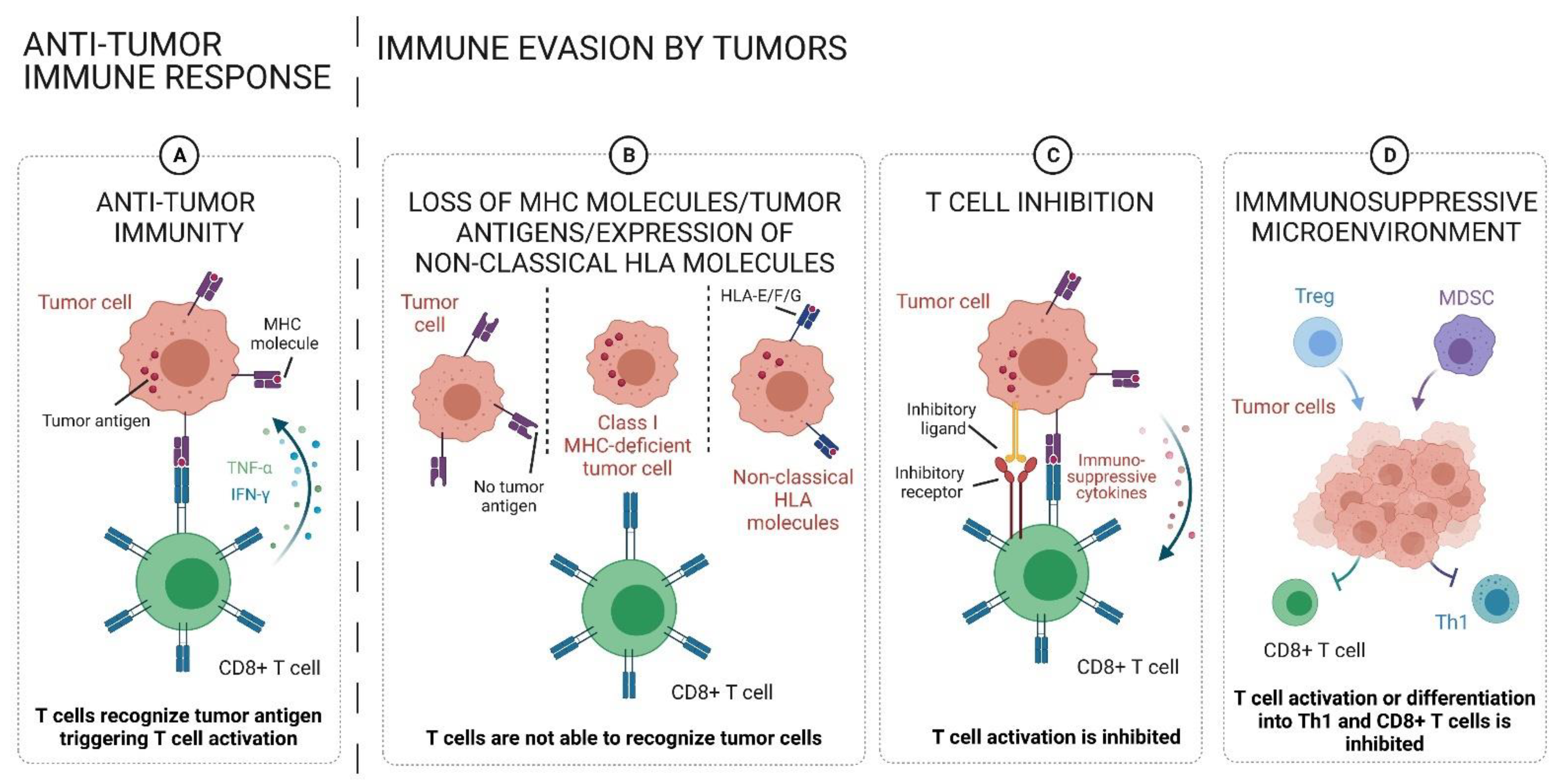{kind=link}
{kind=link}
| Cell Type | Marker | Production | Function | Reference |
|---|---|---|---|---|
| Macrophages | CD14+ CD16+ CD64+ CD68+ CD80+ (M1) CD206+ (M2) | M1: IL-12 IL-23 TNF-α M2: IL-10 TGF-β | Macrophages derive from monocytes. They play an important role in host defense against pathogens, stimulation of the adaptive immune system mainly by their function as antigen-presenting cells (APCs), and tissue remodeling. By Th1 cytokines (IFN-γ and/or lipopolysaccharide (LPS)) macrophages differentiate into M1 phenotype and produce pro-inflammatory cytokines. In contrast, macrophages activated by Th2 cytokines (IL-4, IL-10, IL-2) possess the M2 phenotype producing anti-inflammatory factors. Malignant conditions: Tumor-associated macrophages (TAM) can contribute to tumor cell proliferation, invasion, and metastasis, as well as angiogenesis and suppression of T cell-mediated anti-tumor immune responses. TAMs can adopt their differentiation stage in a wide range between M1 and M2 expressing both markers. | [1,6] |
| Neutrophils | CD11b+ CD15+ CD16+ CD62L+ CD66b+ | N1: ICAM1 TNF-α N2: VEGF MMP9 | Neutrophils are essential effector cells of the innate immune system. They are the first responders in infection, injury, and defense against pathogens. Malignant conditions: Tumor-associated neutrophils (TANs) can exhibit anti-tumor properties as N1 TANs—mediating cytotoxicity—or pro-tumoral effects as N2 TANs—secreting angiogenesis and invasion promoting factors. | [6] |
| Eosinophils | Siglec8+ CD193+ CD11b+ CD14- CD62L+ | TNF-α granzyme IL-18 | Eosinophils are crucial for the control of parasitic infections, bacterial and viral pathogens. Besides, these cells play a central role in inflammation and allergic processes. Malignant conditions: Eosinophils can possess anti-tumorigenic or tumor-promoting functions in different types of tumors. Their different function is mediated by the secretion of anti-tumorigenic or pro-tumorigenic molecules depending on the milieu. | [7] |
| Mast cells | CD117+ CD203+ | VEGF FGF-2 | Mast cells represent another important myeloid component of the immune system that contributes to the innate and the acquired immune responses. Malignant conditions: Tumor-associated mast cells (TAMCs) can possess tumor-promoting functions mediated by the secretion of growth- and angiogenesis-promoting factors. In contrast, in some types of cancer TAMCs induce tumor cell apoptosis by IL-4 and TNF-α. | [6] |
| Myeloid-derived suppressor cells (MDSCs) | CD11b+ CD33+ CD14+ CD15+ CD16+ HLA-DR- | NO ROS iNOS Arginase1 PD-L1 MMP9 | MDSCs compromise a heterogeneous immature immune cell population derived from the myeloid compartment. This cell population plays an essential role in the negative regulation of immune responses. Malignant conditions: MDSCs can be induced by GM-CSF, VEGF, and IL-6, which are mainly produced by tumor cells. They can modulate the inflammatory microenvironment via depletion of amino acids and/or via expression of immune inhibitory ligands to inhibit T cell effector function | [6] |
| DCs | HLA-DR+ lineage− | IFNs | DCs are the central coordinator of immune response and play a central role in immunity. Their main functions are endocytosis, antigen presentation, and IFN production. Malignant conditions: DCs can play a key role in inducing and maintaining anti-tumor immunity, but in the TME their antigen-presenting function may be inefficient. DCs can differentiate into immunosuppressive regulatory DCs, which limit the T cell activity. | [2] |
| NK cells | CD3- CD56+ | GM-CSF IL-5 IL-8 IL-10 IL-13 CCL2 CCL3 CCL4 CCL5 CXCL10 | NK cells belong to the family of innate lymphoid cells with both cytotoxicity and cytokine-producing effector functions. These cells also possess the ability to discriminate target cells, i.e., virus-infected or malignant cells, from healthy cells. This function is based on various cell surfaces consisting of numerous activating and inhibitory receptors. Activating NK cell receptors detect ligands, such as the stress-induced self ligands, infectious non-self ligands, and/or toll-like receptor (TLR), resulting in IFN-γ production and cytotoxicity. Tolerance to self-ligands is mediated by the interaction of the inhibitory receptors and MHC class I molecule. Furthermore, NK cells express the low-affinity Fc receptor CD16, enabling them to exert antibody-dependent cellular cytotoxicity (ADCC). Additionally, NK cells also play a major role in the orchestration of adaptive immune responses by IL secretion. Cytotoxic human NK cells are defined as CD56dimCD16hi, while immunomodulatory and cytokine-producing NK cells are defined as CD56brightCD16lo. Malignant conditions: NK cells can directly cause tumor cell lysis; regulate T cell-mediated anti-tumor immune responses by IL secretion and are implicated in the ADCC. | [3] |
| Cell Type | Marker | Production | Function | Reference |
|---|---|---|---|---|
| T cells | CD3+ | various cytokines | T cells express the TCR complex, which consists of two variable regions—the α- and the β-chains (αβTCR)—in the vast majority of human T cells. The smaller T cell subset—γδ-T cells—just express γ- and δ-chains. Reactive conditions: T cells can recognize foreign or “non-self” material presented as peptides bound to MHC class I or II molecules at the cell surface. Therefore, these cells play an essential role in immune response (bacterial and viral infection via MHC-mediated antigen presentation and tissue/cell graft rejection caused by MHC mismatches). Malignant conditions: T cells can be crucial in anti-tumor immunity initiated by recognition of tumor-specific antigens presented by MHC molecules. | [1,6] |
| CD8+ T cells | CD3+ CD8+ | IL-2 IL-12 Type I IFN granzymes perforin | Reactive conditions: CD8+ T cells mediate immune reactions against pathogens such as viruses and bacteria leading to lysis of infected cells. Malignant conditions: CD8+ T cells can mediate the anti-tumor immune response by recognition of tumor-specific antigens presented by MHC class I molecules leading to tumor cell killing. | [1,6] |
| CD4+ T cells | CD3+ CD4+ | Various cytokines | Reactive conditions: CD4+ T cells are activated by antigens through APCs. These can differentiate into T helper 1 (Th1), T helper 2 (Th2), T helper 9 (Th9), T helper 17 (Th17) cells, follicular helper T cells (Tfh), and regulatory T cells (Tregs). Malignant conditions: CD4+ T cells are less investigated in cancer immunity compared to CD8+ T cells. However, tumor-promoting or anti-tumor immunity was reported for this T cell subtype. | [1,6] |
| Th1 | CD3+ CD4+ STAT4+ T-bet+ | IL-2 IL-12 IFN-γ TNF-α | Reactive conditions: Th1 cells secrete IFN-γ to activate macrophages and CD8+ T cells. These cells play an essential role in immunity against intracellular pathogens. Malignant conditions: Th1 cells can promote anti-tumor immunity by activating cytotoxic CD8+ T cells, macrophages, and other APCs. | [1,6] |
| Th2 | CD3+ CD4+ Gata3+ | IL-4 IL-5 IL-10IL-13 | Reactive conditions: Th2 cells lead to humoral immune responses, typically against extracellular antigens mediated by effector immune cells, including B cells, eosinophils, basophils, and mast cells as well as CD8+ T cells. Malignant conditions: Th2 cells can produce cytokines to downregulate anti-tumor CD8+ T cell-mediated immunity, thereby contributing to tumor growth. This function is particularly mediated by IL-10, which causes inhibition of the DC-mediated antigen presentation and activation of immunosuppressive Tregs. However, Th2-mediated immune responses, such as IL-4 production and activation of eosinophils, can decrease tumor growth. | [1,6] |
| Th9 | CD3+ CD4+ IRF-4+ | IL-9 IL-21 IL-10 | Reactive conditions: Th9 cells are involved in various pathophysiological conditions of immune response, such as allergic reactions, inflammation and elimination of extracellular pathogens. Malignant conditions: Th9 cells can play a key role in inducing CD8+ T cell-mediated anti-tumor immune responses. Besides, Th9 cells can induce innate cells such as DCs, mast cells, and NK cells to promote a robust anti-tumor immune response. | [1,6] |
| Th17 | CD3+ CD4+ RORγt+ | IL-17A IL-17F IL-21 IL-22 CCL20 | Reactive conditions: Th17 cells are implicated in immune responses toward bacteria and fungi by recruitment of neutrophils and macrophages. Malignant conditions: Th17 cells and Th17-derived cytokines, such as IL-17, can exhibit anti-tumor and tumor-promoting activity by shaping the TME. | [1,6] |
| Tregs | CD3+ CD4+ CD25+ FOXP3+ | TGF-β IL-2 GITR9 PD-L1 CTLA-4 TIGIT GARP | Reactive conditions: Tregs are specialized to suppress abnormal immune responses to both self and foreign antigens in order to maintain immune homeostasis by inhibiting T cell proliferation and the production of anti-inflammatory cytokines. Malignant conditions: Tregs can suppress anti-tumor immunity, thus promoting tumor development and progression. | [1,6] |
| γδ- T cells | CD3+ TCRγδ+ | IFN-γ | Reactive conditions: γδ-T cells can recognize a broad range of antigens without any presentation via MHC molecules. They can attack target cells directly through their cytotoxic activity or indirectly through the activation of other immune cells. These cells are involved in pathogen clearance, inflammation, and tissue homeostasis. Malignant conditions: γδ-T cells can possess tumor-suppressing function mediated by their own cytolytic properties or activation of other immune cells. However, the tumor-promoting effect, which is mainly mediated by other effector cells, has also been observed in different types of cancers. | [1] |
| NK-T cells | CD3 CD56 CD4+/− CD8+/− | IFN-γ TNF IL-4 IL-10 IL-13 IL-2 | Reactive conditions: NK-T cells rapidly produce large numbers of immunomodulatory cytokines when they are activated and, thus, modulate immune responses against infectious agents, autoantigens, tissue grafts, and allergens. Malignant conditions: NK-T cells can stimulate T and NK cells to eliminate tumor cells during early tumor development. However, in an overstimulated state, these cells can become anergic and differentiate into immunosuppressive NK-T cell subsets, thereby facilitating tumor progression and immune escape. | [1,6] |
| Cell Type | Marker | Production | Function | Reference |
|---|---|---|---|---|
| B cells | CD19+ CD20+ | various cytokines | B cells are essential players of humoral immunity through antibody (Ab) production. They recognize antigens by the B cell receptor (BCR) composed of membrane-bound antibody. B cells are divided into (1) B1 B cells, mainly found in the peritoneal and pleural cavities; (2) B2 or FO B cells, which are located in lymph nodes; and (3) marginal zone B cells, which are in the marginal sinus of the spleen. The different subsets are activated in a T cell-dependent or -independent way. Reactive conditions: B cell activation begins when the B cell binds to an antigen via its BCR. This activation causes proliferation and differentiation into plasma B cells (PCs), which produce and release antibodies. Malignant conditions: B cells can be implicated in the presentation of tumor-specific antigens to T cells, the production of tumor-specific antibodies and immune regulation as Bregs. | [1,6] |
| Ab-producing B cells | CD19+ CD20+ | tumor-specific IgG and IgA | Reactive conditions: B cells can secrete Abs, and thereby start a specific immune response in viral and/or bacterial infections and autoimmune diseases. Malignant conditions: B cell-mediated Ab production can lead to the killing of tumor cells through the complement cascade activation, phagocytosis by macrophages, and activation of tumor-killing activity of NK cells. | [1,6] |
| B cells as APCs | CD19+ CD20+ CD21+ CD23+ CD27− IgG1+ CD40+ CD80+ CD86+ MHC class II+ | IL-2 IL-6 CCL3 CCL4 ICAM1 GM-CSF | Reactive conditions: B cells can recognize antigen in inflammatory processes in a T cell-independent manner, and they possess the function to present these antigens via their MHC class II surface molecule and thus induce T cell responses. Malignant conditions: B cells can be found nearby T cells in several types of cancers, including when DCs are absent. These cells act as APCs to CD4+ T cells. There exist two forms of this type of cell, causing either anti-tumor immune responses or immunosuppressive intratumoral conditions: (1) the activated B cells (CD69+, HLA-DR+, CD27+, CD21+), possessing Th1 T cell activating function (anti-tumor immune responses); and (3) the exhausted B cells (CD69+, HLA-DR+, CD27−, CD21−), leading to the generation of Tregs (immunosuppressive condition). | [1,6] |
| Bregs | CD19+ CD21+ CD24+ CD25+ FOXP3+ | IL-10 IL-35 TGF-β | Reactive conditions: Bregs are mainly implicated in mediating immune tolerance in autoimmune diseases, including systemic lupus erythematosus (SLE) and multiple sclerosis. Malignant conditions: Bregs possess a tumor-promoting function. They can suppress CD4+ T cell proliferation and induce forkhead box P3 (FOXP3) expression in Tregs mediated by IL-10 and TGF-β. Bregs can also suppress CD8+ T cells in their effector function by IL-10. Furthermore, they cause immune inhibitory receptors PD-L1 on cancer cells through IL-10, IL-35, and TGF-β secretion. | [1,6] |
| Cytokine | Function | Reference |
|---|---|---|
| anti-tumorigenic effects—immune activating | ||
| TNF-α | Enhanced T cell activation Enhanced T cell survival Suppression of Tregs | [202] |
| IFN-γ | Enhanced antigen presentation by induction of MHC class I Promotion of Th1 together with inhibition of Th2 response, and activation of macrophages | [203] |
| IL-33 | Local increasement of CD8+ T cells and NK cells Increased type I immune response (increased expression of IFN-γ, IL-12 and granzyme B) | [204] |
| IL-36 | Enhanced effector function of CD8+ T cells, NK T cells, and γδ T cells | [204] |
| IL-12 | Enhanced cellular cytotoxicity Enhanced IFN-γ production and differentiation of naïve T cells towards Th1 cells | [205] |
| IL-2 | Expansion of CD8+ T cells by IL-2 Acquisition of effector and memory functions of CD8+ T cells | [206] |
| IL-18 | Activation of CD4+ T cells and/or NK immune responses | [204] |
| IL-15 | Homeostasis and activation of NK cells Expansion and activation of memory T cells | [207,208] |
| IL-21 | Regulation of lymphoid cell, NK cells and myeloid cells | [209] |
| IL-1 | T cell activation Proliferation of B cells | [204] |
| IL-6 | Inhibition of Tregs differentiation Stimulation of Tfh differentiation Production of IL-21 Promotion of differentiation of B cells into IgA secreting plasma cells Involvement in CD28-independent T cell activation | [210] |
| pro-tumorigenic effects—immunosuppressive function | ||
| CSF-1 | TAM recruitment and differentiation onto an M2 phenotype | [211] |
| IFN-γ | Upregulation of indoleamine-pyrrole 2,3-dioxygenase (IDO) and HLA-G, PD-L1 and other immunoregulatory molecules by the JAK/STAT pathway Induction of MDSCs | [203] |
| IL-18 | Regulation of PD-1 by conventional NK cells | [204] |
| Type I IFN (IFN-α and-β) | Increased expression of TIM-3 and IL-10 Upregulation of CD80 and CD86 | [212] |
| IL-1 | Suppression of immune reactions in the TME Recruitment of MDSC and M2 macrophages | [213] |
| IL-8 | Attraction of TAMs, neutrophils, and MSDCs causing suppression of anti-tumor immune responses | [214] |
| IL-10 | Inhibition of cytotoxic effector functions of T cells Interfering with T cell priming Supporting activity of dendritic cells and macrophages | [215,216] |
| IL-4 | Stimulating factors for antigen presenting capacities Promotion of macrophage activation Attraction of M2 macrophages and MDSCs | [217,218] |
| IL-13 | Stimulating factors for antigen presenting capacities Promotion of the macrophage activation Attraction of M2 macrophages and MDSC | [218] |
| pro-tumorigenic effects—immunosuppressive function | ||
| IL-27 | Increased expression of immune inhibitory molecules mediated through the transcription factors c-Mac and Prdm1 The key factor for the maximal effector cell expression of PD-L1, LAG-3, CTLA-4, and TIGIT | [219,220] |
| IL-33 | Increased number of immunosuppressive immune cells and innate lymphoid cells | [204] |
| CCL2 | Activation of Tregs and inhibition of T cell effector function | [221] |
| TGF-β | Inhibition of CD8+, CD4+, NK cells proliferation and cytotoxicity Polarization of macrophages and neutrophils towards a suppressive phenotype Together with FOXP3 driver of Tregs differentiation Together with IL-6 and IL-21 driver of Th17 differentiation | [222] |
| Target | Drugs | Malignancies | Effect on Immune Response | Phase | Reference * |
|---|---|---|---|---|---|
| CTLA-4 | Ipilimumab | melanoma NSCLC RCC MSI-H/dMMR CRC HCC MPM MDS AML MPN | anti-CTLA-4 IgG1 mAB | Approved: melanoma NSCLC RCC CRC HCC MPM Phase 1: MDS AML Phase 3: MPN | [380,381,382,383,384,385,386] |
| Tremelimumab | melanoma MSTO NSCLC mPDAC PPC mKC MM aHCC | anti-CTLA-4 mAB | Phase 3: melanoma MSTO Phase 2: SCLC HCC mPDAC Phase 1: PPC MM pilot study: mKC | NCT02558894 NCT02485990 NCT02626130 NCT02716805 NCT02519348 Combination with other drugs for many types of cancer (Phase I/II) [387,388] | |
| PD-1 | Nivolumab | melanoma SCLC mNSCLC aRCC urMPM, rc/mHNSCC HCC previously treated with sorafenib previously treated a/mTCC, rMSI-H/dMMR mCRC r/r cHL aESCC | anti-PD-1 IgG4 mAB | Approved | [382,383,386,389,390,391,392,393,394,395,396] |
| PD-1 | Pembrolizumab | NCLCS melanoma r/r cHL locally a/m TCC rc/m HNSCC SCLC RCC advanced cervical cancer a/m ESCC ur/ m MSI-H or dMMR CRC | anti-PD-1 mAB | Approved: NCLCS melanoma r/r cHL locally a/m TCC HNSCC Locally a/m ESCC Approved in US:RCC SCLS MSI-H or dMMR CRCadvanced cervial cancer | [397,398,399,400,401,402,403,404,405,406,407,408,409,410,411,412] |
| Cemiplimab | aCSCC rc Stage III-IV HNSCC before surgery resNSCLC HCC high risk or locally advanced hormone receptor positive HER2 negative or triple-negative breast cancer mHSPC | anti-PD-1 IgG4 mAB | Appoved: CSCC Phase 2: HNSCC high risk or locally advanced hormone receptor positive HER2 negative or triple-negative breast cancer mHSPC Clinical Trial: resNSCLC HCC | NCT03565783 NCT03916627 NCT04243616 NCT03951831 | |
| Spartalizumab (PDR001) | melanoma metastatic tumors with high PD-1 expression | anti-PD-1 IgG4 mAB | Phase 3: melanoma Phase 2: metastatic tumors with high PD-1 expression | NCT04802876 | |
| Tislelizumab | Rc or a ur/m ESCC rcHCC r/r cHL r/r DLBCL MSI-H/dMMR solid tumors | anti-PD-1 IgG4 mAB | Phase 3: ESCC r/r DLBCLPhase 2: rcHCC r/r cHL MSI-H/dMMR solid tumors | NCT04271956 NCT04663035 NCT04615143 NCT04318080 NCT04799314 NCT04732494 NCT03736889 | |
| Dostarlimab (TSR-042) | aEC a/m CCS LACC melanoma CC NSCLC ovarian neoplasms | anti-PD-1 IgG4 mAB | Approved: aEC: Phase 2: melanoma CCS LACC CC NSCLC ovarian neoplasms | [413,414] NCT04274023 NCT03833479 NCT02715284 NCT04139902 NCT04068753 NCT04655976 NCT03955471 NCT04679064 | |
| PD-1 | Sym021 | advanced solid tumors lymphomas | anti-PD-1 IgG1 mAB | Phase 1 | NCT03311412 NCT04641871 [344] |
| Camrelizumab | advanced solid tumors NSCLC rcPCNSL r/r cHL | anti-PD-1 IgG4 mAB | Approved in China: r/r cHL Phase 2: resNSCLC PCNSL Phase 1–3: HCC advanced solid tumors | NCT04510610 NCT04342936 [345] NCT04338620 NCT04070040 NCT04564313 NCT04490421 | |
| Toripalimab | melanoma ESCC aNSCLC, r/r DLBCL advanced solid tumors | anti-PD-1 IgG4 mAB | Approved in China: melanoma Phase 3: ESCC Phase 2: r/r DLBCL aNSCLC, ESCC Phase 1/2: DLBCL advanced solid tumors | [415] NCT03829969 NCT04425824 NCT04613804 NCT03811379 NCT04058470 NCT04284488 | |
| PD-L1 | Atezolizumab | ES-SCLC NSCLC a/m TCC | anti-PD-L1 IgG1 mAB | Approved | [416,417,418] |
| Durvalumab | ES-SCLC NSCLC BC TCC PC DLBCL FL CLL | anti-PD-L1 IgG1 mAB | Approved: ES-SCLC Phase 2: BC TCC PC Phase 1/2: DLBCL FL CLL | [409,419,420,421] NCT02401048 NCT02733042 | |
| Avelumab | MCC RCC TCC HL DLBCL | anti-PD-L1 IgG1 AB | Approved: MCC RCC Phase 2: HL Phase 1: DLBCL | [422,423,424,425] NCT03617666 NCT03244176 | |
| LAG3 | REGN 3767 | DLBCL advanced solid tumors | anti-LAG-3 mAB | Phase 1 | NCT04566978 NCT03005782 NCT04706715 [426] |
| Relatlimab (BMS-986016) | advanced solid tumors melanoma | anti-LAG-3 IgG4 mAB | Phase 1/2: Other Phase 3: melanoma | NCT04080804 NCT03724968 NCT01968109 [427] NCT03470922 | |
| LAG3 | Sym022 | advanced solid tumors lymphomas | anti-LAG-3 IgG1 mAB | Phase 1 | NCT03489369 NCT04641871 NCT03311412 |
| TIM-3 | MBG453 | MDS AML | anti-TIM-3 IgG4 mAB | Phase 2/3 | NCT04823624 NCT04150029 NCT04266301 |
| LY3321367 | solid tumors | anti-TIM-3 mAB | Phase 1 | NCT03099109 [428] | |
| BGB-A425 | advanced solid tumors | anti-TIM-3 IgG1 mAB | Phase 1 | NCT03744468 | |
| TSR-022/ Cobolimab | advanced solid tumors | anti-TIM-3 mAB | Phase 1/2 | NCT03680508 NCT04139902 | |
| Sym023 | advanced solid tumors lymphomas | anti-TIM-3 IgG1 mAB | Phase 1 | NCT03489343 NCT04641871 NCT03311412 | |
| INCAGN02390 | advanced solid tumors | anti-TIM-3 IgG1 mAB | Phase 1/2 | NCT03652077 NCT04370704 | |
| bispecific ab (anti-PD-1/TIM3) | RO7121661 | advanced solid tumors | Bispecific antibody against PD-1 and TIM3 | Phase 1 | NCT03708328 |
| LY3415244 | advanced solid tumors | Bispecific antibody against PD-1 and TIM3 | Phase 1 | [429] NCT03752177 | |
| JAK | Momelotinib | MF NSCLC | JAK1/2 inhibitor | Phase 3: MF Phase 1: NSCLC | [430,431] |
| Ruxolitinib | MF PC | JAK2 inhibitor | Approved: MF Phase 3: PC | [432,433] NCT02119663 | |
| Cerdulatinib | PTCL aggressive B-NHL | SYK/JAK inhibitor | Phase 1 | [434,435] | |
| Gandotinib | MPN | JAK2/STAT 3 inhibitor | Phase 2 | [436] NCT01594723 | |
| Lestaurtinib | AML MF | JAK2, FLT3 and TrkA Inhibitor | Phase 2: AML Phase 1: MF | NCT00469859 [437,438] | |
| Pacritinib | MF CRC | JAK2/FLT3 inhibitor | Phase 3: MF Phase 2: CRC | [439] NCT02277093 | |
| IL-2 | Aldesleukin | mRCC | IL-2 agonist | Approved: mRCC | [440] |
| Bempegaldesleukin | advanced solid tumors | IL-2 pathway agonist | Phase 1/3 | [441] NCT04410445 NCT04540705 | |
| IL-1 and IL-1R3 (IL-1RAP) | Canakinumab (ACZ885) | Early-stage NSCLC | anti-IL-1R3 IgG1 mAB | Phase 2 | NCT03968419 |
| IL-1 and IL-1R3 (IL-1RAP) | CAN04 | a/m NSCLC a/m CRC a/m BC a/m PDAC | anti-IL-1R3 IgG1 mAB | Phase 1 | NCT03267316 |
| IL-8 | BMS-986253 | advanced solid tumors | anti-IL-8 IgG1 mAB | Phase 1/2 | NCT02536469 NCT03400332 |
| Interleukines | TGF-β/Galunisertib | advanced solid tumors | TGF-β Receptor inhibitor | Phase 2 | [442,443] NCT02452008 |
| CCL2/CCR2 | PF-04136309 | mPDAC | CCR2 antagonist | Phase 1 | NCT02732938 |
| TLR3 | Rintatolimod | advanced solid tumors | Agonist of TLR3 | Phase 2 | NCT04119830 NCT03899987 NCT03734692 [444] |
| Poly-ICLC | PC MM melanomaHCC BC FL B-NHL NSCLC RCC TCC | Agonist of TLR3 | Phase 1/2 | NCT01079741 NCT02834052 NCT01976585 NCT02643303 NCT02452775 NCT02834052 NCT02061449 | |
| TRL4 | GLA-SE (GL100) | melanoma FL | Agonist of TLR4 | Phase 1/2 | NCT02320305 NCT02501473 NCT04364230 |
| TLR5 | Entolimod | CRC SCC advanced solid tumors | Agonist of TRL5 | Phase 1/2 | NCT01527136 NCT01728480 NCT02715882 |
| Mobilan | PRC | Agonist of TRL5 | Phase 1/2 | NCT02654938 NCT02844699 | |
| TLR7/8 | Imiqumod | melanoma various skin cancer CIN PRC BC BCC CL | Agonist of TLR7/8 | Approved | [445,446] |
| Resiquimod | CL melanoma | Agonist of TLR7/8 | Approved | [447,448] | |
| TLR9 | CpG7909 | CLL B-NHL ESCC | Agonist of TLR9 | Phase 1/2 | NCT00185965 NCT00669292 |
| TLR2, 4, 9, NLR NOD2 | Bacillus Calmette-Guerin (BCG) | BLC | Agonist of TLR2, 4, 9, NLR NOD2 | Phase 2/3 | NCT03022825 |
| 4-1BB (CD137) | Urelumab | advanced solid tumors r/r B-NHL melanoma | anti-4-1BB mAB | Phase 2: melanoma Phase 1: Other | NCT01471210 NCT01775631 NCT02534506 NCT00612664 [449] |
| CD27 | Varlilumab | hematologic malignancies advanced solid tumors | Anti-CD27 IgG1 mAB | Phase 1 | [450,451] |
| CD47/SIRP | Hu5F9-G4 (5F9) | advanced solid tumors r/r B-NHL | anti-CD47 IgG4 mAB | Phase 1: advanced solid tumors Phase 1/2 r/r NHL | NCT02953509 NCT02216409 |
| ALX148 | advanced solid tumors rf B-NHL | blocking SIRPα fusion protein | Phase 1 | NCT03013218 | |
| RRx-001 | advanced solid tumors lymphomas | molecule that targets CD27/SIRP | Phase 1 | NCT02518958 | |
| CD73 | CPI-006/ Mupadolimab | advanced solid tumors | anti-CD73 mAB | Phase 1 | NCT03454451 |
| A2aR | EOS100850 | advanced solid tumors | A2AR antagonist | Phase 1 | NCT02740985 |
| AB928/ Etrumadenant | advanced solid tumors | A2AR antagonist | Phase 1 | NCT02740985 | |
| NKG2A | Monalizumab | rc/m HNSCC | anti-NKG2A mAb | Phase 2 | NCT03088059 NCT02643550 |
| LIF | MSC-1 | advanced solid tumors | anti-LIF IgG1 mAB | Phase 1 | NCT03490669 |
| CSF-1 (M-CSF)/CSF-1R | Lacnotuzumab (MCS110) | advanced malignancies | anti-M-CSF IgG1 mAB | Phase 1/2 | NCT02807844 |
| LY3022855 | mBC and mCRPC | anti-M-CSF IgG1 mAB | Phase 1 | NCT02265536 | |
| SNDX-6352 | advanced solid tumors | anti-M-CSF IgG4 mAB | Phase 1 | NCT03238027 | |
| Emactuzumab (RG7155) | advanced solid tumor | anti-CSF1R IgG1 mAB | Phase 1 | NCT01494688 | |
| Pexidartinib (PLX3397) | advanced solid tumors a/m PDAC a/m CRC | inhibitor of tyrosine kinase activity of CSF-1R | Phase 1 | NCT01525602 NCT02777710 NCT02734433 | |
| SEMA4D | Pepinemab (VX15/2503) | aNSCLC | anti-SEMA4D IgG4 mAB | Phase 1/2 | NCT03268057 |
| CLEVER-1 | FP-1305 | advanced solid tumors | anti-CLEVER-1 IgG4 mAB | Phase 1/2 | NCT03733990 |
| Axl | Enapotamab vedotin (EnaV) | advanced solid tumors | AXL targeted Antibody-Drug Conjugate (ADC) | Phase 1 | NCT02988817 |
| Phosphatidylserine | Bavituximab | a/un HCC | anti-Phosphatidylserine IgG3 mAB | Phase 2 | NCT01264705 |
| Imids | Lenalidomide | MM MDS with 5q- indolent lymphoma | inhibitor of ubiquitin E3 ligase cereblon | Approved | [452,453,454,455,456] |
| Imids | Thalidomide | MM | inhibitor of ubiquitin E3 ligase cereblon | Approved | [456,457] |
| Pomalidomide | MM | inhibitor of ubiquitin E3 ligase cereblon | Approved | [456,458] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pansy, K.; Uhl, B.; Krstic, J.; Szmyra, M.; Fechter, K.; Santiso, A.; Thüminger, L.; Greinix, H.; Kargl, J.; Prochazka, K.; et al. Immune Regulatory Processes of the Tumor Microenvironment under Malignant Conditions. Int. J. Mol. Sci. 2021, 22, 13311. https://doi.org/10.3390/ijms222413311
Pansy K, Uhl B, Krstic J, Szmyra M, Fechter K, Santiso A, Thüminger L, Greinix H, Kargl J, Prochazka K, et al. Immune Regulatory Processes of the Tumor Microenvironment under Malignant Conditions. International Journal of Molecular Sciences. 2021; 22(24):13311. https://doi.org/10.3390/ijms222413311
Chicago/Turabian StylePansy, Katrin, Barbara Uhl, Jelena Krstic, Marta Szmyra, Karoline Fechter, Ana Santiso, Lea Thüminger, Hildegard Greinix, Julia Kargl, Katharina Prochazka, and et al. 2021. "Immune Regulatory Processes of the Tumor Microenvironment under Malignant Conditions" International Journal of Molecular Sciences 22, no. 24: 13311. https://doi.org/10.3390/ijms222413311