
Molecular Mechanisms and Targeted Therapies of Advanced Basal Cell Carcinoma


1
Department of Dermatology, Nippon Medical School, Bunkyo-Ku, Tokyo 113-8602, Japan
2
Department of Dermatology, Nippon Medical School Chiba Hokusoh Hospital, Inzai 270-1694, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(19), 11968; https://doi.org/10.3390/ijms231911968
Submission received: 31 July 2022
/
Revised: 18 September 2022
/
Accepted: 19 September 2022
/
Published: 8 October 2022
(This article belongs to the Special Issue Molecular Advances in Skin Diseases 2.0)
Abstract
:Among human cutaneous malignancies, basal cell carcinoma is the most common. Solid advances in unveiling the molecular mechanisms of basal cell carcinoma have emerged in recent years. In Gorlin syndrome, which shows basal cell carcinoma predisposition, identification of the patched 1 gene (PTCH1) mutation was a dramatic breakthrough in understanding the carcinogenesis of basal cell carcinoma. PTCH1 plays a role in the hedgehog pathway, and dysregulations of this pathway are known to be crucial for the carcinogenesis of many types of cancers including sporadic as well as hereditary basal cell carcinoma. In this review, we summarize the clinical features, pathological features and hedgehog pathway as applied in basal cell carcinoma. Other crucial molecules, such as p53 and melanocortin-1 receptor are also discussed. Due to recent advances, therapeutic strategies based on the precise molecular mechanisms of basal cell carcinoma are emerging. Target therapies and biomarkers are also discussed.
1. Introduction
Basal cell carcinoma (BCC) is one of the most ordinary malignant tumors and its incidence continues to grow in many countries [1]. Moreover, BCCs are one of the most common cutaneous malignancies and account for approximately 80% to 90% of skin cancers in Caucasians [2,3]. Sunlight exposure is the most crucial risk factor, and radiation, age, and skin type [4] are also associated as possible causes. BCCs are defined as indolent growing tumors, which very rarely metastasize but can cause tissue destructions if they are not treated adequately [5]. Common metastasizing sites were regional lymph nodes (53%), lungs (33%), and bone (20%) according to a retrospective analysis of published metastatic BCC (mBCC) from 1981 to 2011, and the mean time between the occurrence of the primary tumor and signs of metastasis was about 9 years [6]. BCCs that show specific features can be classified as having a high metastatic potential. Concretely, tumors located in the midline of the face or ear, a tumor that has been present for a long period of duration, large tumor diameter size, and history of previous radiation exposure are such features [7,8]. Rare cases have reported BCC having intravascular invasion, showing higher metastatic rates than BCC cases without intravascular invasion. Systemic treatments are required for such mBCCs [9,10].
In this article, we review the clinical presentation and histologic features of BCCs. The molecular mechanisms that underpin BCCs and the systemic treatments based on the molecular mechanisms regulating the pathogenesis and development of BCCs are then described.
2. Clinical Presentations
Clinical manifestations may vary widely in BCCs, and BCC lesions usually grow slowly, are non-healing, and might show bleeding or ulceration [11,12,13,14].
2.1. Nodular BCC
The most frequent clinical subtype is nodular BCC [15]. The classical clinical description is that of a well-defined, pearly, shining, translucent and smooth nodule or papule with distinct, arborizing, and dilated blood vessels/telangiectasias often with ulcerations or erosions with rolled boundaries (Figure 1A). Nodular BCC may grow and crusting may occur over a depressed center. Hemorrhage with trivial trauma is frequent then the lesion sometimes may ulcerate, as known as rodent ulcer.
2.2. Superficial BCC
The second most frequent clinical subtype is superficial BCC, accounting for up to 15% of BCCs [15]. Clinical features appear as an annular, well-defined, thin plaque or patch. Superficial BCCs preferentially occur on the trunk and extremities, although other subtypes are usually seen on the head and neck [11,12,13,14,15].
2.3. Morpheic/Morpheaform/Sclerosing BCC
Morpheic BCC accounts for 5 to 10% of BCCs [15]. This type of BCCs are called morpheic or sclerosing because these show poorly defined lesions, scar-like features, infiltrative, or shining plaques, which might be flat or sometimes atrophic including telangiectasias, erosions, or small crusts (Figure 1B).
2.4. Pigmented BCC
Pigmented BCC appears as black nodules or papules sometimes with ulceration. Clinically, pigmented BCC can mimic melanoma and a histopathological examination is the only way to definitively diagnose BCC, to exclude the possibility of melanoma, and to decide on appropriate treatment [16]. Pigmented BCC is an unusual morphologic manifestation and has a low proportion of cases at 6.7% in Caucasians [17,18]. On the contrary, pigmented BCCs are the most frequent and typical clinical feature of Asian origin, especially in Japanese, and 88.3% of total BCCs in Japanese patients are pigmented [19].
3. Histopathology
The histopathology of BCC is generally distinguished by lobules of basaloid cells harboring large and hyperchromatic nuclei as well as scant cytoplasm, which are all accompanied by a fibromyxoid stroma and sometimes by tumor retraction spaces. The stroma surrounding BCC contains microvessel proliferations, linking to aggressive attitude [12,20].
Histopathologic subtypes of BCCs are classified according to the probability of the recurrence. BCCs with a low risk of recurrence are nodular, superficial, pigmented, infundibulocystic, and fibroepithelial, which show indolent attitudes [7]. In contrast, BCCs with a high risk of recurrence are morpheic/sclerosing, micronodular, infiltrating, basosquamous, and sarcomatous differentiated BCCs, which show aggressive histologic features [7]. Alternatively, BCCs were also suggested to be classified into ‘easy to treat’ and ‘difficult to treat’ [21,22,23]. We should bear in mind that clinical and histopathological classifications are distinct.
3.1. Nodular BCC
Islands of basaloid tumor cells or large nests with disorderly cell arrangements are observed in nodular BCCs [13]. Peripheral palisadings are usually seen. The tumor stroma with spindle cells is cleaving and is mucoid or myxoid, occasionally containing amyloid deposits (Figure 2A). The nests of tumor cells infiltrate deep into the dermis. Apoptosis also can be found in the center. According to the secondary findings such as keratotic, cystic/nodulocystic and adenoid BCCs, nodular BCC has several subtypes [12,20].
3.2. Superficial BCC
Superficial BCC appears as small lobules or islands containing basaloid tumor cells with peripheral palisading. Linking to the epidermis associated with a lichenoid or bandlike inflammatory cell infiltration within a myxoid stroma, the tumor localizes in the superficial dermis. Superficial BCCs can be multicentric and a mixed pattern tumor, with nodular, micronodular, or infiltrating subtype [12,13,20].
3.3. Morpheic/Sclerosing BCC
Morpheic/sclerosing BCC is characterized by very thin strands of tumor cells. These are buried in a collagenous stroma, however, tumor stroma cleftings are a rare phenomenon (Figure 2B). Morpheic/sclerosing BCC infiltrates deeply but differs from the infiltrating BCC by the stromal features. Infiltrating BCC lacks the collagenous stroma, which means sclerosing [12,20].
3.4. Micronodular BCC
Micronodular BCC is composed of dispersed micronodules of tumor cells. These tumor nests extend into the deeper dermis, and sometimes into the subcutaneous tissues. Satellite like arrangement of punctate nodules with irregular contours can be seen. Those are separated by normal dermal collagen and lined by a thin margin of stroma.
3.5. Infiltrating BCC
Infiltrating BCC is a subtype composed largely of chords of tumor cells which deeply infiltrate with angulated edges. This type of BCC shows a permeating or an irregular invasion pattern at the tumor periphery. Infiltrating BCC frequently overlaps with morpheic/sclerosing BCC and can be found with a nodular part [12,20].
3.6. Pigmented BCC
Pigmented BCC is a variant of superficial or nodular BCC, containing melanin pigment derived from activated melanocytes within the tumor nests, residing within the tumor cells or the melanophages [12,13,20,24]. In pigmented BCCs, tumor cell, as well as melanophages in the adjacent dermis, harbor pigment present within those cells in pigmented BCCs. This shows the reason for the black appearance of the tumor [25].
4. Molecular Mechanisms
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin Goltz syndrome, is an autosomal dominant disorder that manifests in the pits of the palms and soles, jaw keratocysts, various other tumors, and developmental abnormalities as well as multiple BCCs [26]. The candidate gene was narrowed down to chromosome 9q22.3 [27,28,29]. Subsequently, loss of heterozygosity in the region was proved to be quite crucial for the pathogenesis of sporadic BCCs as well as of NBCCS [30]. These data show that the gene might be a tumor suppressor. Thus, unveiling the molecular mechanisms of BCCs clearly relies on a careful analysis of NBCCS. Here we discuss the molecular mechanisms of BCCs; however, sporadic BCCs have several histopathological classifications as described above. What regulates the histological subtypes? Biomarkers might be of help and will be discussed later.
4.1. The Sonic Hedgehog Pathway
Molecular analysis revealed that the gene corresponds to a human homolog of Drosophila patched [31,32]. Now named patched 1 PTCH1, the gene encodes a twelve-pass transporter like transmembrane protein and a receptor of the hedgehog ligand (Sonic hedgehog (SHH), Desert hedgehog (DHH) and Indian hedgehog (IHH)). In vertebrate development, the hedgehog pathway is crucial [33]. The SHH ligand binds to the PTCH1 receptor, inhibiting it and allowing signaling through the hedgehog pathway (Figure 3). Mice overexpressing SHH show features of NBCCS and develop multiple BCCs [34,35]. The dysregulation of PTCH1, in other words, if its inhibitory functions are gone, is known to be crucial in the carcinogenesis of BCC.
Somatic mutations in smoothened (SMO), a seven-transmembrane protein immediately downstream of PTCH1, were activated in sporadic BCCs. Transgenic mice overexpressing mutant SMO developed skin abnormalities similar to bona-fide BCCs [36]. PTCH1 inhibits SMO function when the hedgehog ligand is missing [37]. PTCH-mediated repression of SMO is relieved on ligand binding. SMO transduces the signal to a SUFU (suppressor of fused)-GLI (glioma associated oncogene) complex residing in the cytoplasm. Then, it activates GLI transcription factors [38]. Mutated PTCH1, found in NBCCS, cannot inhibit SMO, in other words, SMO is constitutively active.
SMO is thought to be a target of pharmacological therapeutic strategy. Because SMO serves as an oncogenic molecule and has drug-binding pockets in its transmembrane helices [39,40]. Gain-of-function mutations in the SMO gene (L412F, S533N, W535L, and R562Q) activate the GLI transcription network [36,39,41]. In the drug-binding cavity, R400, D473 and E518 are key amino acid residues. Cyclopamine, which is a naturally occurring alkaloid and is found in the corn lily, serves as an SMO antagonist [42]. The acquired D473H SMO mutation causes resistance to vismodegib and sonidegib, which are engineered molecules from cyclopamine [39]. SMO serves as a proto-oncogene according to these findings.
In hereditary and in sporadic BCCs, mutations in several genes in the hedgehog pathway have been identified. Mutations in SMO as well as PTCH1 have been described in a subset of patients with sporadic BCC [36,43,44]. These mutations mostly consist of C to T substitutions at a dipyrimidine site, belonging to the so-called ‘ultraviolet (UV) signature’ mutations. PTCH2 shares structural similarities with PTCH1 and has a minor compensatory role in the hedgehog pathway. PTCH2 mutations were found in some cases of sporadic BCCs [45]. PTCH2 is overexpressed in both hereditary and sporadic BCCs, indicating that PTCH2 is a direct gene target of hedgehog signaling, and that PTCH1 may negatively regulate PTCH2 [46]. Loss of PTCH2 has been reported to contribute to enhanced tumorigenesis in PTCH1 haploinsufficient mice [47]. By sequestering GLI transcription factors in the nucleus and in the cytoplasm, SUFU is the main repressor of the mammalian hedgehog signaling pathway [48]. As a negative regulator of the hedgehog pathway that acts by sequestering all three hedgehog homologs with similar affinity to that of PTCH1 protein, the transmembrane hedgehog interacting protein (HIP) was identified [49]. HIP was dysregulated in the nodular type BCCs [50]. There are three GLI proteins, which are GLI1, GLI2 and GLI3. These are activated by SMO, and then can activate or repress hedgehog pathway target genes [39]. Overexpression of GLI proteins in mice has been proven to induce BCCs [51,52,53]. Moreover, hedgehog signaling has been shown to be crucial for carcinogenesis of BCC because conditionally GLI2-expressing mice show BCC regression when GLI2 expression is inactivated [53]. KIF7, a member of the kinesin-4 family, was identified as a GLI-interacting protein [54]. KIF7 binds to GLIs and regulates their degradation and stability. Then KIF7 controls GLI-mediated transcription [55]. KIF7 has SUFU-independent and -dependent regulatory functions. While KIF7 promotes hedgehog pathway activity through the dissociation of SUFU-GLI2 complex, it also represses the hedgehog target genes in the absence of SUFU. Inactivation of either KIF7 or SUFU alone in the epidermis cannot promote BCC carcinogenesis, although their simultaneous deletion can induce BCC. These results demonstrate the distinct and overlapping roles of SUFU and KIF7 in GLI2 regulation during tumorigenesis [56]. These studies suggest that overactivation of hedgehog signaling is necessary and perhaps sufficient for the development of BCCs.
4.2. Other Molecules
4.2.1. TP53
The TP53 gene is the most commonly mutated tumor suppressor in malignancy, and has been described as ‘guardian of the genome’ [57,58]. The gene TP53 encodes the protein p53; mutations of TP53 are found in a wide variety of tumors, and abrogation of this pathway is found in many human malignancies [59]. p53 senses genotoxic injury and causes arrested cell division, allowing DNA repairment prior to replication. p53 also induces apoptosis in order to eliminate potentially malignant cells if extensive DNA damage occurs. Mutations of TP53 are found in BCC tumors but not in the germ line, in 44% to 100% of the BCCs studied [60,61,62,63,64,65,66,67,68].
Li-Fraumeni syndrome harbors TP53 germ line mutation and shows a cancer predisposition [69]. Accordingly, BCC is not predisposed to by this syndrome, indicating that TP53 mutation is not necessary for BCC carcinogenesis. TP53 mutation may be a secondary event occurring after BCC carcinogenesis. On the contrary, p53 loss enhances BCC carcinogenesis if the hedgehog pathway is already activated in mice [70]. Currently, this discrepancy has not been fixed. The cell cycle checkpoint gene 14-3-3σ, which is under the regulation of p53, is silenced in BCC [71,72]. The protein encoded by 14-3-3σ is essential for keratinocyte senescence. Thus, loss of 14-3-3σ may contribute to the growth of BCC [73]. Overexpression of p53 in BCCs has been reported to be associated with aggressiveness [67,74,75,76,77,78]. Of those, necrosis was significantly associated with overexpression of p53 in BCCs [78]. p53 expression may have some role in determining the aggressiveness of BCCs.
4.2.2. Melanocortin 1 Receptor
Differences in the distribution and type of melanin produced in cutaneous melanocytes regulate wide variations of human hair and skin colors [79,80,81]. Melanin consists of two distinct types: one is the brown/black eumelanin, which is photoprotective. Another is the yellow/red pheomelanin, which is photosensitive. Individuals with red hair and light skin have a predominance of pheomelanin and/or a dysregulated ability to produce eumelanin. The red hair phenotype can be deeply associated with a single gene mutation which encodes the melanocortin 1 receptor (MC1R). Single gene mutations of MC1R are also responsible for freckling, and poor tanning response to UV as well as fair skin color.
MC1R, a membrane-anchored protein consisting of 317 amino acids, has 7 transmembrane spanning domains and is G-protein coupled receptor expressed on the surface of melanocytes [79,80]. In persons with a history of BCC, MC1R of red hair color mutations are overexpressed [82]. These mutants all bind α-melanocyte-stimulating hormone (α-MSH), which leads to production of melanin, but are unable to activate adenylate cyclase [83].
Several studies indicate the association of MC1R mutations with the risk of BCC, and show that a fair skin color in addition to MC1R mutations greatly increases this risk [82,84,85]. Melanocytes with loss-of-function MC1R mutations have an impaired ability to repair UV-induced DNA damage in vitro, however, their melanin content is not associated with this effect [86]. It is then possible that the same effect occurs in the carcinogenesis of BCCs. BCC risk in the two or more MC1R variants is reported to be increasing independently of skin type [81,85,87,88,89,90,91]. Interestingly, the frequency of P1315L mutation in BCCs is not associated with the MC1R genotype [89]. This indicates that the MC1R genotype is associated with the risk of BCC, independent of red hair characteristics.
5. Cell of Origin
The origin of BCC is still a controversial but intriguing matter due to conflicting evidences [92]. Several mouse models have been investigated so far to unveil the origin of BCC. Constitutively active SMO mutant in hair follicle bulge stem cells and in their transient amplifying progenies could not induce BCC. However, BCC arises from resident progenitor cells of the interfollicular epidermis and from the upper infundibulum when constitutively active SMO mutants are introduced [93]. Overexpression of GLI2 in stem cells of resting hair follicles induced nodular BCC. In contrast, overexpression of GLI2 in epidermis formed superficial BCCs [94]. BCC exclusively originated from keratin 15-expressing stem cells of the follicular bulge using fate tracking of X-ray induced BCCs in PTCH1+/- mice [95]. Loss of p53 enhanced BCC carcinogenesis from the bulge and produced BCCs from the interfollicular epidermis by enhancing SMO expression, at least in part, thus, loss of p53 may be a primary event in BCC formation through SMO upregulation.
Using BerEP4 immunohistochemistry, BCC originates from the basal cell of epidermis, located at the interfollicular epidermis and infundibulum of the hair follicle located along the basal layer, suggesting the notions that BCC originates from the basal cell layer of the interfollicular epidermis, not from the outer root sheath or of the hair follicles. These data indicate that BCCs may arise from basal keratinocytes of the interfollicular epidermis or of the hair follicle [96].
6. Therapy
The goal of treatment is the complete removal of BCCs and to avoid dysfunctions as well as disfigurements. Surgical removal is the treatment of choice considered as the traditional mainstay in most cases [7,21,97,98,99]. BCCs occur mainly in exposed areas including face and scalp, and such locations often make surgery difficult. Mohs micrographic surgery is the gold standard for aggressive or ‘difficult to treat’ BCCs, especially in difficult anatomic sites. Radiation therapy can be an alternative option for certain patients. For indolent BCCs, topical therapy with 5-fuorouracil or imiquimod is also approved and shows amazing results [100,101,102,103,104]. Although photodynamic therapy (PDT) has not been approved by the FDA, PDT has shown to be more effective for superficial BCCs than for nodular BCCs [105,106]. However, a subset of advanced BCC cases requires systemic therapy [7,21,97,98,99].
6.1. Conventional Chemotherapy
Historically, conventional cytotoxic chemotherapy in metastatic or locally aggressive BCCs has been used [107,108,109,110]. However, data are limited, primarily based on case reports, and no drug was approved for metastatic or locally aggressive BCCs by the US Food and Drug Administration (FDA).
Systemic platinum-based chemotherapy achieved an overall response rate (ORR) of up to 77% to 83%, with a complete response in up to 45% of treated lesions of locally aggressive BCCs or mBCCs, although the duration was several months [111,112]. Other case reports with platinum-based chemotherapy showed a low response rate (RR) (20-30%) and a short duration of response [113]. These reports consistently suggest that a platinum-based regimen should be considered for the systemic chemotherapy of BCCs.
In 2015, the term ‘locally advanced BCC’ (laBCC) was first used and refers to a complex clinical scenario in which the tumor was untreated for a long time or the tumor has had repetitive treatment failures as well as recurrences. The same term is also used to describe the presence of an extensive tissue destruction by BCC in the surrounding area that makes it quite difficult to treat by surgery or radiotherapy; in other words, only systemic drug therapy is prospecting [98].
6.2. FDA Approved Hedgehog Inhibitors
As mentioned above, the main driver of BCC carcinogenesis and progression is the constitutive activation and dysregulation of the hedgehog pathway. The hedgehog pathway is involved in many situations of fetal development, and is also strictly regulated after birth [114]. Hedgehog pathway reactivation, caused by several associated mutations in factors of the hedgehog pathway, induces uncontrolled proliferation of the malignant cells leading to tumor formation. NBCCS patients have germline mutations in PTCH1 and show BCC growth at a very early age [31]. Therefore, BCCs in NBCCS patients are currently mostly treated with hedgehog inhibitors (HHIs). Moreover, HHIs have determined a paradigmatic shift toward laBCCs or mBCCs [115]. As an HHI, SMO antagonist is promising and the first SMO antagonist is a naturally occurring alkaloid called cyclopamine, found in the corn lily [42]. However, poor oral bioavailability, acid sensitivity and some degrees of specificity constrict the clinical usage of cyclopamine [116,117]. Recently, several SMO inhibitors, including vismodegib (GDC-0449) and sonidegib (Erismodegib, NVP-LDE-225, LDE-225), have been engineered from cyclopamine and have gained success as targeted clinical cancer therapy [118] (Table 1).
6.2.1. Vismodegib
Vismodegib is the SMO antagonist approved by the FDA in 2012 based on a worldwide phase II study (ERIVANCE; NCT00833417) [119,120]. Vismodegib showed RR of 30% (laBCCs) and 43% (mBCCs) with a median response duration of 7.6 months [121]. The updated report showed RR of 48.5% (mBCC) and 60.3% (laBCC) at 39 month, and median response duration of 14.8 months (mBCC) and 26.2 months (laBCC) [119]. Most patients experienced adverse events (AEs) by vismodegib including muscle spasms, alopecia, taste loss, weight loss, decreased appetite, fatigue, nausea, or diarrhea [122,123]. BCC patients treated with vismodegib have an increased risk of developing cutaneous SCCs [124]. Squamous differentiation was observed in some BCC metastasis, and the activating SMO mutation c.1234C > T was found twice [124,125]. SMO mutations c.1234C > T provided resistance against vismodegib [126].
6.2.2. Sonidegib
Sonidegib is an orally administered SMO inhibitor which is structurally distinct from vismodegib. It was approved by the FDA in 2015 for the treatment of laBCC or recurrent BCC [127]. Approval was based on the results of the phase II BOLT trial (NCT01327053) [128,129]. Note that for mBCCs, sonidegib has not been approved. Patients were randomized to receive either 200 mg or 800 mg of sonidegib daily. Among those with laBCCs, the objective RR after 30 months were 38% (800 mg) and 43% (200 mg). In the patients with mBCCs, the objective RR were 17% (800 mg) and 15% (200 mg) [129]. Objective RR in the 200 mg group were sustained at 56.1% (laBCC) and 7.7% (mBCC) at 30-month follow-up [130]. The 200 mg dose also exhibited a lower rate of grade 3/4 AEs (31% vs. 56%) and AEs leading to drug discontinuation (22% vs. 36%) or dose reduction/interruption (32% vs. 60%) [130]. Most commonly reported AEs for sonidegib and vismodegib in the ERIVANCE [122] and BOLT [128] studies were muscle spasms, alopecia, and dysgeusia. Overall, time to onset of AEs was slightly later and less frequent with sonidegib.
6.3. Drug Resistance
Currently, no randomized controlled trial has compared vismodegib with sonidegib. Vismodegib appears to be the treatment of selection for mBCC, because vismodegib has FDA approval for mBCC and may have superior efficacy to sonidegib in treating mBCC [131]. The ORR with vismodegib and sonidegib was 47.6% at 21-month follow-up and 60.6% with 18-month follow-up, respectively. Treatment with vismodegib is known to show primary or secondary resistance in almost 20% of patients [132].
Again, D473 in the sixth transmembrane domain of SMO is one of the key amino acid residues having drug-binding cavity, to which vismodegib or sonidegib binds. Vismodegib and sonidegib are resistant to acquired D473H SMO mutation [39]. Approximately 50% of laBCCs initially show vismodegib resistance, while 21% of initial responders develop resistance later and experience disease progression or recurrence in a mean of 54.4 weeks [133]. This may be explained by the enrollment of only more aggressive mBCC [119]. The potency of hedgehog pathway inhibition by itraconazole was shown to be further enhanced when used in combination with HHI cyclopamine [134]. Therefore, inhibiting the hedgehog pathway through different mechanisms with a combination of itraconazole and HHI may overcome HHI resistance. Two patients with advanced BCC treated with itraconazole and vismodegib demonstrated disease control with acceptable toxicity profile [135]. A subset of patients under HHI shows progress of the disease due to primary resistance by the activation of noncanonical hedgehog pathway or additional signaling pathways [136]. Secondary HHI resistance is also observed due to additional mutations in the SMO drug-binding cavity [129,137]. Surprisingly, HHI treatment showed upregulation of MHC-I and -II and increased CD8+ T cells, which might enable an immune-related tumor response [138]. Thus, the combination of HHI with an immunotherapeutic agent is expected for synergic effects.
6.4. FDA Approved Immune Checkpoint Inhibitors
Immune checkpoints, including programmed death (PD)-1 and cytotoxic T cell lymphocyte-associated protein (CTLA)-4 receptors, are expressed on activated T cells [139]. PD-1 binds two ligands, the programmed death-ligand 1 (PD-L1) and programmed death-ligand 2 (PD-L2) on tumor cells [140]. CTLA-4 binds B7 (CD80/86) on antigen-presenting cells, which inhibits the immune cell activation. This may serve as a mechanism for inhibiting tumor infiltrating T cells, and has become a crucial therapeutic target for restoring immunity. Patients with advanced malignancies received significant benefit from studies of checkpoint inhibition with anti-CTLA-4 (ipilimumab) and anti-PD-1 (nivolumab and pembrolizumab) monoclonal antibodies. In fact, melanoma, a disease that is more likely to metastasize and become life-threatening as compared to BCC, was the first cancer to demonstrate clinical benefit from cytokines and immune checkpoint blockade [141].
Similar to melanoma, BCCs are generally characterized by UV damage, which translates into a high tumor mutational burden (TMB). Treatment with anti-PD-1 antibodies has shown a dramatic response in high TMB tumors [142]. Neoantigens, the putative targets of immune cells that recognize and eradicate neoplastic cells, is deeply associated with high TMB [141]. PD-L1 expression and TMB have been demonstrated to correlate with response to checkpoint inhibition [143]. BCCs have higher TMB with 47.3 median mutations/Mb than melanomas, which have TMB with 13.5 median mutations/Mb.
BCC was defined as a ‘cold tumor’, meaning that few immune cells can infiltrate tumor cells in BCC [14]. However, PD-L1 expression by tumor cells in BCCs ranges from 22% to 89.9%, while expression by tumor-infiltrating lymphocytes ranges from 82.0% to 94.9% [144,145,146]. Moreover, BCCs treated with checkpoint inhibition demonstrated greater PD-L1 expression in tumors (32% vs. 7%) and in TILs (47% vs. 18%), suggesting that treatment may induce PD-L1 expression, and that previously treated BCCs could possibly be more responsive to checkpoint inhibition [146].
Several case reports have been published of patients with mBCC or laBCC being treated with immune therapy, using ipilimumab, nivolumab or pembrolizumab, demonstrating meaningful and durable responses [147,148,149,150,151]. Thus, immune checkpoint inhibitors are promising toward laBCC and mBCC.
Cemiplimab
The first clinical trial using checkpoint inhibitors for BCCs was performed. That trial was a non-randomized and open-label study of pembrolizumab with or without vismodegib for advanced BCCs (NCT02690948) [146]. The ORR at 18 weeks was 44 % (pembrolizumab monotherapy group) and 29% (pembrolizumab with vismodegib group). Pembolizumab was active against BCCs, but the dual therapy group did not show significant superiority compared to the pembolizumab monotherapy group. Cemiplimab (REGN2810) is a fully human hinge-stabilized IgG4 high-affinity anti-PD-1 antibody that potently blocks functional interaction between PD-1 and PD-L1 [152]. The pharmacological difference between cemiplimab and nivolumab was unclear [153]. Comparisons of pembrolizumab and cemiplimab have not been published so far. Cemiplimab was developed for treating metastatic and locally advanced cutaneous SCCs [154], which is candidate for neither radical surgery nor radiation. SCCs and BCCs share some common features, including the high UV-induced TMB, indicating a strong rationale for response to immunotherapy [155]. The first pivotal study performed on cemiplimab monotherapy for laBCC and mBCC patients escaping on HHIs was a worldwide, open-label, phase II, and single-arm trial (Study 1620; NCT03132636) [156,157,158]. ORR in the laBCC cohort was 31%, including 6% CR and 25% PR, at a median follow-up of 15.1 months. Moreover, ORR in the mBCC cohort was 21.4% at a median follow-up of 38.9 weeks. Pretreatment and posttreatment biopsies were assessed for the expression of PD-L1 and MHC-I as well as TMB. Unfortunately, these analyses were not associated with clinical efficacy. Based on these results, cemiplimab gained FDA and European Medicines Agency approval as a second line treatment option for BCC patients who are resistant, progressive or are intolerant to HHI first line treatment.
Currently, HHI therapy is a first-line option for laBCC or mBCC [7]. Cemiplimab is indicated and recommended as a first-line after HHIs’ treatments, either when the HHI therapy becomes intolerable, or when the BCC progresses despite HHI treatment, or when the BCC is stable but does not improve after nine consecutive months of HHI therapy [159].
7. Biomarkers
BCCs are classified into several subtypes in terms of the histopathological features described above [7,12,20]. Moreover, BCCs are also classified into indolent and aggressive, or ‘easy to treat’ and ‘difficult to treat’ [21,22,23]. This classification is crucial for the treatment of BCCs, and molecular basis biomarkers are helpful [7,12,20,160].
Paraoxonase-2 (PON2) is an intracellular enzyme having antioxidant roles in reducing intracellular oxidative stress [161,162]. PON2 enzyme expression was significantly increased in infiltrating BCCs compared with nodular BCCs [163]. Nicotinamide N-methyltransferase (NNMT) is a human cytosolic enzyme catalyzing the N-methylation of nicotinamide, pyrimidines and other structural analogs, and plays a crucial role in biotransformation [164,165,166]. NNMT was reported to be overexpressed in BCCs compared with intact margins, although was expressed relatively lower in infiltrating BCCs compared with nodular BCCs [167]. EZH2 is a histone methyltransferase of the polycomb repressive complex 2) and is associated with hedgehog pathway [168]. EZH2 expression was reported to correlate with Ki67 and with aggressive BCC subtypes, including morpheaform, infiltrative and micronodular [169]. miRNA are small single-stranded non-coding RNAs regulating posttranscriptional modification of related proteins, and are associated with the development of neoplasms [160,170]. Serum expression levels of miR-34a in BCC patients was significantly lower than in healthy controls and prognosis was poor when expression levels of miR-34a was low [170].
Again, PD-1 binds two ligands, the PD-L1 and PD-L2 on tumor cells [140]. PD-L1 expression is expected to be a biomarker for PD-1 inhibition therapy and was reported to be a positive predictor of pembrolizumab for cutaneous squamous cell carcinomas [171,172]. However, as for BCCs, PD-L1 expression was not associated with the response to cemiplimab [146,156,171].
8. Clinical Trials
8.1. XmAb20717 Monoclonal Bispecific PD-1 and CTLA-4 Antibody
Immune checkpoint inhibitor combination therapies have been quickly adopted to overcome resistance and have been shown to achieve greater ORR due to different modalities of action of CTLA-4 and PD-1 [173]. However, this success is also accompanied by a higher incidence of immune-related AEs [173]. Bispecific antibodies comprise at least two distinct antigen-binding sites and therefore have two different specificities, binding two different antigens or two different epitopes on the same antigen [174]. Targeted combination therapy with a bispecific antibody is an advantageous strategy for overcoming systemic responses by pairing tumor-associated antigens in the context of T cell engagement [175]. XmAb20717 is a humanized bispecific monoclonal antibody for PD-1 and CTLA-4. Its safety and tolerability profile are confirmed by a phase I open-label study (NCT03517488). Its anti-tumor activity is currently recruiting, in which BCCs are included (Table 2).
8.2. Anti LAG-3 Antibody
Lymphocyte activation gene 3 (LAG-3), also known as CD223, is a single-pass transmembrane glycoprotein and is an immune checkpoint receptor associated with tumor escape and decreased T cell effector function [176]. LAG-3 is expressed widely on different cells: T cell subpopulations including activated CD4+ helper T cells and cytotoxic CD8+ T cells [177,178]. T cell activation is a crucial condition for LAG-3 expression on T cell subpopulations [179]. MHC-II interacts with LAG-3, inhibiting T cell receptor (TCR) binding, thus hindering T cell activation and promoting an anergic T cell state [180]. It also regulates T cells differentiation toward regulatory T cells and favors regulatory T cell immune suppressive functions [181]. LAG-3 inhibition can induce reversal of the anergy and restore T cell antitumoral functions. In BCC patients, the soluble cell-free variants of CTLA-4, LAG-3, PD-1/PD-L1 and T-cell immunoglobulin and mucin domain-3 (TIM-3), which were prominent co-inhibitory immune checkpoints, were measured. Those of all five molecules were significantly upregulated [182]. These results show that LAG-3 could be a biomarker of immunotherapy as well as a candidate for immune checkpoint inhibition.
8.3. Combination of HHIs with Immune Checkpoint Inhibitors
Recent studies have shown that HHI-induced BCC regression leads to drastic changes in the tumor microenvironment, recruiting cytotoxic T cells, activating adaptive immunity, and stimulating dynamic changes in the cytokine/chemokine network [138]. These results suggested a synergic effect between HHI and checkpoint inhibition in advanced BCCs. Results from a phase III study for advanced melanomas assessing the clinical efficacy of the combination of relatlimab (anti LAG-3 antibody) plus nivolumab versus nivolumab monotherapy have been published recently [183]. Median progression-free survival was significantly longer in the combination treatment arm (10.1 months) versus nivolumab monotherapy (4.6 months), indicating that dual immune checkpoint inhibition is preferable. Similarly, for patients with advanced BCC, an open-label, phase II trial is investigating cemiplimab in combination with pulsed sonidegib (NCT04679480) and nivolumab alone or in combination with relatlimab or ipilimumab (NCT03521830).
8.4. Itraconazole
Vismodegib and sonidegib are derived from a naturally occurring alkaloid called cyclopamine. The activation of the hedgehog pathway is inhibited by binding to SMO receptor in the drug-binding pocket. Inevitable secondary drug resistance to vismodegib and sonidegib have been reported and were attributed to several mutations occurring in the drug-binding pocket of SMO [184]. Itraconazole is an FDA-approved, well-known and commonly used agent for the treatment of fungal infections. Itraconazole also inhibits SMO accumulation in the cilium by binding to a site on the SMO receptor different to that of cyclopamine, and efficiently shows SMO agonist effects [134]. Itraconazole inhibits 14-α-lanosterol demethylase, an enzyme involved in the cholesterol biosynthesis in mammals [134]. For SMO activity and hedgehog pathway function, cellular cholesterol is essential [185]. However, itraconazole-induced hedgehog pathway inhibition seems to be independent from cholesterol synthesis [134]. It has been shown that itraconazole does not bind SMO like vismodegib or sonidegib. The inhibitory effect on the hedgehog pathway by itraconazole is attributed to the downregulation of SMO in the cilium. Itraconazole efficiently inhibits medulloblastoma and BCC growth. Moreover, itraconazole shows synergistic inhibition effects if used with cyclopamine [134]. The combination of itraconazole and arsenic trioxide efficiently inhibits the proliferation of hedgehog-driven medulloblastoma in the context of GLI2 overexpression or in the case of vismodegib resistance due to SMO mutations. However, itraconazole could not inhibit entire SMO mutations associated with vismodegib-resistance [186]. Currently, itraconazole is under phase II clinical trials for the treatment of BCC [187]. Itraconazole reduced cell proliferation by 45%, hedgehog pathway activity by 65%, and reduced tumor volume by 24% [187].
8.5. Other Inhibitors of the Hedgehog Pathway
8.5.1. SMO Inhibitors
New drugs inhibiting the hedgehog pathway are under evaluation. Toxicity due to HHI is still a problem. Several SMO inhibitors other than vismodegib or sonidegib are currently being investigated in clinical trials.
8.5.2. Taladegib
Taladegib (LY2940680), a phthalazine-based SMO inhibitor, is one of the few successful examples that can inhibit both the wild-type and the D473H mutant SMO in clinical trials [188]. Taladegib binds to a thin pocket of SMO. It shows weak interaction with D473, and D473H mutation does not interfere with its binding to SMO, which contributes to its inhibitory activity towards SMO-D473H mutant [189]. In a phase I, multicenter, open-label study, 47 patients with laBCC or mBCC were treated with taladegib. Note that sonidegib was not efficacious in an investigational study of advanced BCC patients previously treated with vismodegib [190]. ORR was 46.8%. Responses were observed in patients previously treated with HHI therapy (35.4%) and in HHI therapy-naive (68.7%) patients [191].
9. Conclusions
We reviewed the studies regarding the clinical presentation and histologic features of BCCs. We then described the molecular mechanisms that underpin BCCs and the systemic treatments based on the molecular mechanisms underlying the pathogenesis and development BCCs. The discovery of hedgehog signaling in BCCs is quite striking and targeted therapies have rapidly emerged. However, differences between nodular BCCs and morpheic BCCs, which are quite distinct, have not yet been unveiled. Drug resistances are also a big obstacle. Future investigations are eagerly awaited.
Author Contributions
T.H. is the main author in manuscript drafting; N.K. revised the bibliography and updated the figure; H.S. made critical revisions to the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Number 19K07720.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AE | Adverse event |
| α-MSH | α-melanocyte-stimulating hormone |
| BCC | Basal cell carcinoma |
| CTLA-4 | Cytotoxic T cell lymphocyte associated protein-4 |
| DHH | Desert hedgehog |
| FDA | Food and Drug Administration |
| GLI | Glioma associated oncogene |
| HHI | Hedgehog inhibitor |
| HIP | Hedgehog interacting protein |
| IHH | Indian hedgehog |
| laBCC | Locally advanced basal cell carcinoma |
| LAG-3 | Lymphocyte activation gene 3 |
| mBCC | Metastatic basal cell carcinoma |
| MC1R | Melanocortin 1 receptor |
| NBCCS | Nevoid basal cell carcinoma syndrome |
| NNMT | Nicotinamide N-methyltransferase |
| ORR | Overall response rate |
| PD-1 | Programmed death-1 |
| PD-L1 | Programmed death-ligand 1 |
| PD-L2 | Programmed death-ligand 2 |
| PDT | Photodynamic therapy |
| PON2 | Paraoxonase-2 |
| PTCH1 | Patched 1 |
| RR | Response rate |
| SHH | Sonic hedgehog |
| SMO | Smoothened |
| SUFU | Suppressor of fused |
| TCR | T cell receptor |
| TIM-3 | T-cell immunoglobulin and mucin domain-3 |
| TMB | Tumor mutational burden |
| UV | Ultraviolet |
References
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Hogue, L.; Harvey, V.M. Basal Cell Carcinoma, Squamous Cell Carcinoma, and Cutaneous Melanoma in Skin of Color Patients. Dermatol. Clin. 2019, 37, 519–526. [Google Scholar] [CrossRef]
- Vílchez-Márquez, F.; Borregón-Nofuentes, P.; Barchino-Ortiz, L.; Ruíz-De-Casas, A.; Palacios-Álvarez, I.; Soria-Rivas, A.; Descalzo-Gallego, M.; García-Doval, I.; Ríos-Buceta, L.; Redondo-Bellón, P. Carcinoma basocelular cutáneo: Diagnóstico y tratamiento en atención especializada dermatológica. Guía De Práctica Clínica De La AEDV 2020, 111, 291–299. [Google Scholar] [CrossRef]
- Fania, L.; Didona, D.; Morese, R.; Campana, I.; Coco, V.; Di Pietro, F.R.; Ricci, F.; Pallotta, S.; Candi, E.; Abeni, D.; et al. Basal Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2020, 8, 449. [Google Scholar] [CrossRef] [PubMed]
- Sobanko, J.F.; Lynm, C.; Rosenbach, M. Basal Cell Carcinoma. JAMA Dermatol. 2013, 149, 766. [Google Scholar] [CrossRef] [Green Version]
- Wysong, A.; Aasi, S.Z.; Tang, J.Y. Update on Metastatic Basal Cell Carcinoma: A Summary of Published Cases From 1981 Through 2011. JAMA Dermatol. 2013, 149, 615–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCCN. NCCN Guidelines Basal Cell Skin Cancer Version 2. 2022. Available online: https://www.nccn.org/professionals/physician_gls/pdf/nmsc.pdf (accessed on 31 May 2022).
- Piva de Freitas, P.; Senna, C.G.; Tabai, M.; Chone, C.T.; Altemani, A. Metastatic Basal Cell Carcinoma: A Rare Manifestation of a Common Disease. Case Rep. Med. 2017, 2017, 8929745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poignet, B.; Gardrat, S.; Dendale, R.; Lemaitre, S.; Rouic, L.L.-L.; Desjardins, L.; Cassoux, N.; Gabriel, C.L. Basal cell carcinomas of the eyelid: Results of an initial surgical management. J. Français D’ophtalmologie 2019, 42, 1094–1099. [Google Scholar] [CrossRef]
- Muzumdar, S.; Stewart, C.L.; Feng, H. Rare case of a basal cell carcinoma with intravascular invasion. Int. J. Women’s Dermatol. 2020, 6, 334–335. [Google Scholar] [CrossRef] [PubMed]
- Wade, T.R.; Ackerman, A.B. The Many Faces of Basal-Cell Carcinoma. J. Dermatol. Surg. Oncol. 1978, 4, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.C.; Lee, E.; Hibler, B.P.; Barker, C.A.; Mori, S.; Cordova, M.; Nehal, K.S.; Rossi, A.M. Basal cell carcinoma. J. Am. Acad. Dermatol. 2018, 80, 303–317. [Google Scholar] [CrossRef] [Green Version]
- Niculet, E.; Craescu, M.; Rebegea, L.; Bobeica, C.; Nastase, F.; Lupasteanu, G.; Stan, D.J.; Chioncel, V.; Anghel, L.; Lungu, M.; et al. Basal cell carcinoma: Comprehensive clinical and histopathological aspects, novel imaging tools and therapeutic approaches (Review). Exp. Ther. Med. 2021, 23, 10982. [Google Scholar] [CrossRef] [PubMed]
- Marzuka, A.G.; Book, S.E. Basal Cell Carcinoma: Pathogenesis, Epidemiology, Clinical Features, Diagnosis, Histopathology, and Management. Yale J. Boil. Med. 2015, 88, 167–179. [Google Scholar]
- Scrivener, Y.; Grosshans, E.; Cribier, B. Variations of basal cell carcinomas according to gender, age, location and histopathological subtype. Br. J. Dermatol. 2002, 147, 41–47. [Google Scholar] [CrossRef]
- Demierre, M.-F.; Chung, C.; Miller, D.R.; Geller, A.C. Early Detection of Thick Melanomas in the United States. Arch. Dermatol. 2005, 141, 745–750. [Google Scholar] [CrossRef] [Green Version]
- Demırtaşoǧlu, M.; Lebe, B.; Kuşku, E.; Akarsu, S.; Ozkan, S.; Ilknur, T. Evaluation of dermoscopic and histopathologic features and their correlations in pigmented basal cell carcinomas. J. Eur. Acad. Dermatol. Venereol. 2006, 20, 916–920. [Google Scholar] [CrossRef]
- Maloney, M.E.; Jones, D.B.; Sexton, F.M. Pigmented basal cell carcinoma: Investigation of 70 cases. J. Am. Acad. Dermatol. 1992, 27, 74–78. [Google Scholar] [CrossRef]
- Takenouchi, T.; Takatsuka, S. Long-term prognosis after surgical excision of basal cell carcinoma: A single institutional study in Japan. J. Dermatol. 2013, 40, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.; Epstein, E.J.; Kossard, S.; McKenzie, C.; Patel, R.; Patterson, J. Basal cell carcinoma. In WHO Classification of Skin Tumors, 4th ed.; Elder, D., Massi, D., Scolyer, R., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 26–34. [Google Scholar]
- Peris, K.; Fargnoli, M.C.; Garbe, C.; Kaufmann, R.; Bastholt, L.; Seguin, N.B.; Bataille, V.; Marmol, V.D.; Dummer, R.; Harwood, C.A.; et al. Diagnosis and treatment of basal cell carcinoma: European consensus–based interdisciplinary guidelines. Eur. J. Cancer 2019, 118, 10–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grob, J.; Gaudy-Marqueste, C.; Guminski, A.; Malvehy, J.; Basset-Seguin, N.; Bertrand, B.; Fernandez-Penas, P.; Kaufmann, R.; Zalaudek, I.; Fargnoli, M.; et al. Position statement on classification of basal cell carcinomas. Part 2: EADO proposal for new operational staging system adapted to basal cell carcinomas. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 2149–2153. [Google Scholar] [CrossRef] [PubMed]
- Grob, J.; Guminski, A.; Malvehy, J.; Basset-Seguin, N.; Bertrand, B.; Fernandez-Penas, P.; Kaufmann, R.; Zalaudek, I.; Gaudy-Marqueste, C.; Fargnoli, M.; et al. Position statement on classification of basal cell carcinomas. Part 1: Unsupervised clustering of experts as a way to build an operational classification of advanced basal cell carcinoma based on pattern recognition. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 1949–1956. [Google Scholar] [CrossRef]
- Abudu, B.; Cohen, P.R. Pigmented Basal Cell Carcinoma Masquerading as a Melanoma. Cureus 2019, 11, e4369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deppe, R.; Pullmann, H.; Steigleder, G.K. Dopa-positive cells and melanin in basal cell epithelioma (BCE). Arch. Dermatol. Res. 1976, 256, 79–83. [Google Scholar] [CrossRef]
- Gorlin, R.J.; Goltz, R.W. Multiple Nevoid Basal-Cell Epithelioma, Jaw Cysts and Bifid Rib. N. Engl. J. Med. 1960, 262, 908–912. [Google Scholar] [CrossRef]
- Gailani, M.R.; Bale, S.J.; Leffell, D.J.; DiGiovanna, J.J.; Peck, G.L.; Poliak, S.; Drum, M.A.; Pastakia, B.; McBride, O.W.; Kase, R.; et al. Developmental defects in gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell 1992, 69, 111–117. [Google Scholar] [CrossRef]
- Farndon, P.; Del Mastro, R.; Kilpatrick, M.; Evans, D. Location of gene for Gorlin syndrome. Lancet 1992, 339, 581–582. [Google Scholar] [CrossRef]
- Reis, A.; Küster, W.; Gebel, E.; Fuhrmann, W.; Groth, W.; Kuklik, M.; Wegner, R.; Linss, G.; Hamm, H.; Wolff, G.; et al. Localisation of gene for the naevoid basal-cell carcinoma syndrome. Lancet 1992, 339, 617. [Google Scholar] [CrossRef]
- Gailani, M.R.; Ståhle-Bäckdahl, M.; Leffell, D.J.; Glyn, M.; Zaphiropoulos, P.; Undén, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; Toftgård, R. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet. 1996, 14, 78–81. [Google Scholar] [CrossRef]
- Hahn, H.; Wicking, C.; Zaphiropoulos, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the Human Homolog of Drosophila patched in the Nevoid Basal Cell Carcinoma Syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H.; et al. Human Homolog of patched, a Candidate Gene for the Basal Cell Nevus Syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [Green Version]
- Stone, D.M.; Hynes, A.M.; Armanini, M.P.; Swanson, T.A.; Gu, Q.; Johnson, R.L.; Scott, M.P.; Pennica, D.; Goddard, A.; Phillips, H.S.; et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996, 384, 129–134. [Google Scholar] [CrossRef]
- Fan, H.; Oro, A.E.; Scott, M.P.; Khavari, P.A. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nat. Med. 1997, 3, 788–792. [Google Scholar] [CrossRef]
- Oro, A.E.; Higgins, K.M.; Hu, Z.; Bonifas, J.M.; Epstein, E.H.; Scott, M.P. Basal Cell Carcinomas in Mice Overexpressing Sonic Hedgehog. Science 1997, 276, 817–821. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Murone, M.; Luoh, S.-M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.-W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002, 418, 892–896. [Google Scholar] [CrossRef]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. Genomic testing, tumor microenvironment and targeted therapy of Hedgehog-related human cancers. Clin. Sci. 2019, 133, 953–970. [Google Scholar] [CrossRef]
- Nguyen, N.M.; Cho, J. Hedgehog Pathway Inhibitors as Targeted Cancer Therapy and Strategies to Overcome Drug Resistance. Int. J. Mol. Sci. 2022, 23, 1733. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-Mediated Inhibition of Target Tissue Response to Shh Signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Köhler, B.; Schönicke, A.; Scharwächter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Smyth, I.; Narang, M.A.; Evans, T.; Heimann, C.; Nakamura, Y.; Chenevix-Trench, G.; Pietsch, T.; Wicking, C.; Wainwright, B.J. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Hum. Mol. Genet. 1999, 8, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Zaphiropoulos, P.G.; Unden, A.B.; Rahnama, F.; Hollingsworth, R.E.; Toftgard, R. PTCH2, a novel human patched gene, un-dergoing alternative splicing and up-regulated in basal cell carcinomas. Cancer Res. 1999, 59, 787–792. [Google Scholar]
- Lee, Y.; Miller, H.L.; Russell, H.R.; Boyd, K.; Curran, T.; McKinnon, P.J. Patched2 Modulates Tumorigenesis in Patched1 Heterozygous Mice. Cancer Res. 2006, 66, 6964–6971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.R.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P. Mammalian Suppressor-of-Fused modulates nuclear–cytoplasmic shuttling of GLI-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef]
- Chuang, P.-T.; McMahon, A.P. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 1999, 397, 617–621. [Google Scholar] [CrossRef]
- Tojo, M.; Kiyosawa, H.; Iwatsuki, K.; Kaneko, F. Expression of a sonic hedgehog signal transducer, hedgehog-interacting protein, by human basal cell carcinoma. Br. J. Dermatol. 2002, 146, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Grachtchouk, M.; Mo, R.; Yu, S.; Zhang, X.; Sasaki, H.; Hui, C.-C.; Dlugosz, A.A. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat. Genet. 2000, 24, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Undèn, A.B.; Krause, D.; Malmqwist, U.; Raza, K.; Zaphiropoulos, P.G.; Toftgård, R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. USA 2000, 97, 3438–3443. [Google Scholar] [CrossRef]
- Hutchin, M.E.; Kariapper, M.S.; Grachtchouk, M.; Wang, A.; Wei, L.; Cummings, D.; Liu, J.; Michael, L.E.; Glick, A.; Dlugosz, A.A. Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: Conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev. 2004, 19, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Varjosalo, M.; Li, S.-P.; Taipale, J. Divergence of Hedgehog Signal Transduction Mechanism between Drosophila and Mammals. Dev. Cell 2006, 10, 177–186. [Google Scholar] [CrossRef]
- Cheung, H.O.-L.; Zhang, X.; Ribeiro, A.; Mo, R.; Makino, S.; Puviindran, V.; Law, K.K.L.; Briscoe, J.; Hui, C.-C. The Kinesin Protein Kif7 Is a Critical Regulator of Gli Transcription Factors in Mammalian Hedgehog Signaling. Sci. Signal. 2009, 2, ra29. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Nieuwenhuis, E.; Nien, W.; Zhang, X.; Zhang, J.; Puviindran, V.; Wainwright, B.; Kim, P.C.W.; Hui, C.-C. Kif7 regulates Gli2 through Sufu-dependent and -independent functions during skin development and tumorigenesis. Development 2012, 139, 4152–4161. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zwaan, E.S.; Haass, N.K. Genetics of basal cell carcinoma. Australas. J. Dermatol. 2009, 51, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Piipponen, M.; Riihilä, P.; Nissinen, L.; Kähäri, V.-M. The Role of p53 in Progression of Cutaneous Squamous Cell Carcinoma. Cancers 2021, 13, 4507. [Google Scholar] [CrossRef]
- Rady, P.; Scinicariello, F.; Wagner, R.F.; Tyring, S.K. p53 mutations in basal cell carcinomas. Cancer Res. 1992, 52, 3804–3806. [Google Scholar] [PubMed]
- Ziegler, A.; Leffell, D.J.; Kunala, S.; Sharma, H.W.; Gailani, M.; Simon, A.J.; Halperin, A.J.; Baden, H.P.; Shapiro, E.P.; Bale, E.A. Mutation hotspots due to sunlight in the p53 gene of nonmelanoma skin cancers. Proc. Natl. Acad. Sci. USA 1993, 90, 4216–4220. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B. p53 function and dysfunction. Cell 1992, 70, 523–526. [Google Scholar] [CrossRef]
- Levine, A.J. The p53 tumor suppressor gene and gene product. Princess Takamatsu Symp. 1989, 20, 221–230. [Google Scholar] [PubMed]
- Van Der Riet, P.; Karp, D.; Farmer, E.; Wei, Q.; Grossman, L.; Tokino, K.; Ruppert, J.M.; Sidransky, D. Progression of basal cell carcinoma through loss of chromosome 9q and inactivation of a single p53 allele. Cancer Res. 1994, 54, 25–27. [Google Scholar]
- Rosenstein, B.S.; Phelps, R.G.; Weinstock, M.A.; Bernstein, J.L.; Gordon, M.L.; Rudikoff, D.; Kantor, I.; Shelton, R.; Lebwohl, M.G. p53 mutations in basal cell carcinomas arising in routine users of sunscreens. Photochem. Photobiol. 1999, 70, 798–806. [Google Scholar] [CrossRef]
- Pontén, F.; Williams, C.; Ling, G.; Ahmadian, A.; Nistér, M.; Lundeberg, J.; Pontén, J.; Uhlén, M. Genomic analysis of single cells from human basal cell cancer using laser-assisted capture microscopy. Mutat. Res. Res. Genom. 1997, 382, 45–55. [Google Scholar] [CrossRef]
- Bolshakov, S.; Walker, C.M.; Strom, S.S.; Selvan, M.S.; Clayman, G.L.; El-Naggar, A.; Lippman, S.M.; Kripke, M.L.; Ananthaswamy, H.N. p53 mutations in human aggressive and nonaggressive basal and squamous cell carcinomas. Clin. Cancer Res. 2003, 9. [Google Scholar]
- Molès, J.-P.; Moyret, C.; Guillot, B.; Jeanteur, P.; Guilhou, J.J.; Theillet, C.; Basset-Sèguin, N. p53 gene mutations in human epithelial skin cancers. Oncogene 1993, 8, 228–334. [Google Scholar]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ Line p53 Mutations in a Familial Syndrome of Breast Cancer, Sarcomas, and Other Neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Falcicchio, M.; Ward, J.A.; Macip, S.; Doveston, R.G. Regulation of p53 by the 14-3-3 protein interaction network: New opportunities for drug discovery in cancer. Cell Death Discov. 2020, 6, 126. [Google Scholar] [CrossRef] [PubMed]
- Molin, S.C.; Grgic, M.; Ruzicka, T.; Herzinger, T. Silencing of the cell cycle checkpoint gene 14-3-3σ in basal cell carcinomas correlates with reduced expression of IKK-α. J. Eur. Acad. Dermatol. Venereol. 2013, 28, 1113–1116. [Google Scholar] [CrossRef] [PubMed]
- Lodygin, D.; Yazdi, A.S.; Sander, A.C.; Herzinger, T.; Hermeking, H. Analysis of 14-3-3σ expression in hyperproliferative skin diseases reveals selective loss associated with CpG-methylation in basal cell carcinoma. Oncogene 2003, 22, 5519–5524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef]
- Enache, A.O.; Stepan, A.E.; Mărgăritescu, C.; Pătraşcu, V.; Ciurea, R.N.; Simionescu, C.E.; Camen, A. Immunoexpression of p53 and COX-2 in basal cell carcinoma. Rom. J. Morphol. Embryol. 2018, 59, 1115–1120. [Google Scholar]
- Ansarin, H.; Daliri, M.; Soltani-Arabshahi, R. Expression of p53 in aggressive and non-aggressive histologic variants of basal cell carcinoma. Eur. J. Dermatol. 2006, 16, 543–547. [Google Scholar]
- Auepemkiate, S.; Boonyaphiphat, P.; Thongsuksai, P. p53 expression related to the aggressive infiltrative histopathological feature of basal cell carcinoma. Histopathology 2002, 40, 568–573. [Google Scholar] [CrossRef]
- Koseoglu, R.D.; Sezer, E.; Eyibilen, A.; Aladag, I.; Etikan, I. Expressions of p53, cyclinD1 and histopathological features in basal cell carcinomas. J. Cutan. Pathol. 2009, 36, 958–965. [Google Scholar] [CrossRef]
- Castanheira, A.; Vieira, M.J.; Pinto, M.; Dias, C.; Prada, L.; Macedo, S.; Fernandes, M.S.; Vieira, F.; Soares, P.; Mota, A.; et al. TERTp mutations and p53 expression in head and neck cutaneous basal cell carcinomas with different aggressive features. Sci. Rep. 2021, 11, 10395. [Google Scholar] [CrossRef]
- Rouzaud, F.; Costin, G.; Yamaguchi, Y.; Valencia, J.C.; Berens, W.F.; Chen, K.G.; Hoashi, T.; Böhm, M.; Abdel-Malek, Z.A.; Hearing, V.J. Regulation of constitutive and UVR-induced skin pigmentation by melanocortin 1 receptor isoforms. FASEB J. 2006, 20, 1927–1929. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Hearing, V.J. Physiological factors that regulate skin pigmentation. BioFactors 2009, 35, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manganelli, M.; Guida, S.; Ferretta, A.; Pellacani, G.; Porcelli, L.; Azzariti, A.; Guida, G. Behind the Scene: Exploiting MC1R in Skin Cancer Risk and Prevention. Genes 2021, 12, 1093. [Google Scholar] [CrossRef]
- Box, N.; Chen, W.; Sturm, R.; Duffy, D.; Irving, R.E.; Russell, A.; Griffyths, L.R.; Parsons, P.; Green, A.C. Melanocortin-1 Receptor Genotype is a Risk Factor for Basal and Squamous Cell Carcinoma. J. Investig. Dermatol. 2001, 116, 224–229. [Google Scholar] [CrossRef]
- Frändberg, P.-A.; Doufexis, M.; Kapas, S.; Chhajlani, V. Amino acid residues in third intracellular loop of melanocortin 1 receptor are involved in G-protein coupling. IUBMB Life 1998, 46, 913–922. [Google Scholar] [CrossRef]
- Han, J.; Kraft, P.; Colditz, G.; Wong, J.; Hunter, D.J. Melanocortin 1 receptor variants and skin cancer risk. Int. J. Cancer 2006, 119, 1976–1984. [Google Scholar] [CrossRef]
- Scherer, D.; Bermejo, J.L.; Rudnai, P.; Gurzau, E.; Koppova, K.; Hemminki, K.; Kumar, R. MC1R variants associated susceptibility to basal cell carcinoma of skin: Interaction with host factors and XRCC3 polymorphism. Int. J. Cancer 2007, 122, 1787–1793. [Google Scholar] [CrossRef]
- Hauser, J.E.; Kadekaro, A.L.; Kavanagh, R.J.; Wakamatsu, K.; Terzieva, S.; Schwemberger, S.; Babcock, G.; Rao, M.B.; Ito, S.; Abdel-Malek, Z.A. Melanin content and MC1R function independently affect UVR-induced DNA damage in cultured human melanocytes. Pigment. Cell Res. 2006, 19, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Dessinioti, C.; Sypsa, V.; Kypreou, K.; Dimisianos, G.; Kodela, E.; Nikolaou, V.; Antoniou, C.; Stratigos, A.J. A case-control study of MC1R variants in Greek patients with basal cell carcinoma: Increased risk independently of pigmentary characteristics. Exp. Dermatol. 2015, 24, 476–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastiaens, M.T.; Ter Huurne, J.A.C.; Kielich, C.; Gruis, N.A.; Westendorp, R.G.; Vermeer, B.J.; Bavinck, J.N.B. Melanocortin-1 Receptor Gene Variants Determine the Risk of Nonmelanoma Skin Cancer Independently of Fair Skin and Red Hair. Am. J. Hum. Genet. 2001, 68, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Liboutet, M.; Portela, M.; Delestaing, G.; Vilmer, C.; Dupin, N.; Gorin, I.; Saiag, P.; Lebbé, C.; Kerob, D.; Dubertret, L.; et al. MC1R and PTCH Gene Polymorphism in French Patients with Basal Cell Carcinomas. J. Investig. Dermatol. 2006, 126, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.M.; Cartmel, B.; Molinaro, A.M.; Gordon, P.B.; Leffell, D.J.; Bale, A.E.; Mayne, S.T. Host Phenotype Characteristics and MC1R in Relation to Early-Onset Basal Cell Carcinoma. J. Investig. Dermatol. 2012, 132, 1272–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliabue, E.; Fargnoli, M.C.; Gandini, S.; Maisonneuve, P.; Cornelius, A.L.; Kayser, M.; Nijsten, T.; Han, J.; Kumar, R.; Gruis, A.N.; et al. MC1R gene variants and non-melanoma skin cancer: A pooled-analysis from the M-SKIP project. Br. J. Cancer 2015, 113, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josiah, A.; Twilley, D.; Pillai, S.; Ray, S.; Lall, N. Pathogenesis of Keratinocyte Carcinomas and the Therapeutic Potential of Medicinal Plants and Phytochemicals. Molecules 2021, 26, 1979. [Google Scholar] [CrossRef]
- Youssef, K.K.; Van Keymeulen, A.; Lapouge, G.; Beck, B.H.; Michaux, C.; Achouri, Y.; Sotiropoulou, P.A.; Blanpain, C. Identification of the cell lineage at the origin of basal cell carcinoma. Nature 2010, 12, 299–305. [Google Scholar] [CrossRef]
- Grachtchouk, M.; Pero, J.; Yang, S.H.; Ermilov, A.N.; Michael, L.E.; Wang, A.; Wilbert, D.; Patel, R.M.; Ferris, J.; Diener, J.; et al. Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. J. Clin. Investig. 2011, 121, 1768–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.Y.; Wang, J.; Mancianti, M.-L.; Epstein, E.H. Basal Cell Carcinomas Arise from Hair Follicle Stem Cells in Ptch1+/− Mice. Cancer Cell 2011, 19, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.T.; Ghaznawie, M.; Heenan, P.J.; Dosan, R. Basal Cell Carcinoma Arises from Interfollicular Layer of Epidermis. J. Oncol. 2018, 2018, 3098940. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.S.; Kozlow, J.H.; Mittal, B.; Moyer, J.; Olencki, T.; Rodgers, P.; Bichakjian, C.; Armstrong, A.; Baum, C.; Bordeaux, J.S.; et al. Guidelines of care for the management of basal cell carcinoma. J. Am. Acad. Dermatol. 2018, 78, 540–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dika, E.; Scarfì, F.; Ferracin, M.; Broseghini, E.; Marcelli, E.; Bortolani, B.; Campione, E.; Riefolo, M.; Ricci, C.; Lambertini, M. Basal Cell Carcinoma: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 5572. [Google Scholar] [CrossRef] [PubMed]
- Cives, M.; Mannavola, F.; Lospalluti, L.; Sergi, M.C.; Cazzato, G.; Filoni, E.; Cavallo, F.; Giudice, G.; Stucci, L.S.; Porta, C.; et al. Non-Melanoma Skin Cancers: Biological and Clinical Features. Int. J. Mol. Sci. 2020, 21, 5394. [Google Scholar] [CrossRef] [PubMed]
- Geisse, J.K.; Rich, P.; Pandya, A.; Gross, K.; Andres, K.; Ginkel, A.; Owens, M. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: A double-blind, randomized, vehicle-controlled study*. J. Am. Acad. Dermatol. 2002, 47, 390–398. [Google Scholar] [CrossRef] [Green Version]
- Bath-Hextall, F.; Ozolins, M.; Armstrong, S.J.; Colver, G.B.; Perkins, W.; Miller, P.S.J.; Williams, H.C. Surgical excision versus imiquimod 5% cream for nodular and superficial basal-cell carcinoma (SINS): A multicentre, non-inferiority, randomised controlled trial. Lancet Oncol. 2013, 15, 96–105. [Google Scholar] [CrossRef]
- Thomas, D.; Zalcberg, J.R. 5-Fluorouracil: A Pharmacological Paradigm in the Use of Cytotoxics. Clin. Exp. Pharmacol. Physiol. 1998, 25, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Schön, M. Imiquimod: Mode of action. Br. J. Dermatol. 2007, 157, 8–13. [Google Scholar] [CrossRef]
- Kim, D.P.; Kus, K.J.; Ruiz, E. Basal Cell Carcinoma Review. Hematol. Clin. North Am. 2018, 33, 13–24. [Google Scholar] [CrossRef]
- Mosterd, K.; Thissen, M.; Nelemans, P.; Janssen, R.; Broekhof, K.; Neumann, H.; Steijlen, P.; Kuijpers, D.; Kelleners-Smeets, N. Fractionated 5-aminolaevulinic acid-photodynamic therapy vs. surgical excision in the treatment of nodular basal cell carcinoma: Results of a randomized controlled trial. Br. J. Dermatol. 2008, 159, 864–870. [Google Scholar] [CrossRef]
- Fantini, F.; Greco, A.; Del Giovane, C.; Cesinaro, A.M.; Venturini, M.; Zane, C.; Surrenti, T.; Peris, K.; Calzavara-Pinton, P. Photodynamic therapy for basal cell carcinoma: Clinical and pathological determinants of response. J. Eur. Acad. Dermatol. Venereol. 2010, 25, 896–901. [Google Scholar] [CrossRef]
- Woods, R.L.; Stewart, J.F. Metastatic basal cell carcinoma: Report of a case responding to chemotherapy. Postgrad. Med. J. 1980, 56, 272–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthrie, T.H., Jr.; McElveen, L.J.; Porubsky, E.S.; Harmon, J.D. Cisplatin and doxorubicin. An effective chemotherapy com-bination in the treatment of advanced basal cell and squamous carcinoma of the skin. Cancer 1985, 55, 1629–1632. [Google Scholar] [CrossRef]
- Jefford, M.; Kiffer, J.D.; Somers, G.; Daniel, F.J.; Davis, I.D. Metastatic basal cell carcinoma: Rapid symptomatic response to Cisplatin and Paclitaxel. ANZ J. Surg. 2004, 74, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; Watkin, W.G.; Mehta, U.K.; Brockstein, B.E. Metastatic Basal Cell Carcinoma: Complete Response to Chemotherapy and Associated Pure Red Cell Aplasia. Cancer Investig. 2006, 24, 396–400. [Google Scholar] [CrossRef]
- Moeholt, K.; Aagaard, H.; Pfeiffer, P.; Hansen, O. Platinum-Based Cytotoxic Therapy in Basal Cell Carcinoma a review of the literature. Acta Oncol. 1996, 35, 677–682. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, P.; Hansen, O.; Rose, C. Systemic cytotoxic therapy of basal cell carcinoma: A review of the literature. Eur. J. Cancer Clin. Oncol. 1990, 26, 73–77. [Google Scholar] [CrossRef]
- Peris, K.; Tambone, S.; Kostaki, D.; Varrassi, E.; Fargnoli, M.C. Treatments of advanced basal cell carcinoma: A review of the literature. G. Ital. Di Dermatol. E Venereol. 2014, 151, 77–86. [Google Scholar]
- Armas-López, L.; Zúñiga, J.; Arrieta, O.; Ávila-Moreno, F. The Hedgehog-GLI pathway in embryonic development and cancer: Implications for pulmonary oncology therapy. Oncotarget 2017, 8, 60684–60703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peris, K.; Licitra, L.; Ascierto, A.P.; Corvò, R.; Simonacci, M.; Picciotto, F.; Gualdi, G.; Pellacani, G.; Santoro, A. Identifying locally advanced basal cell carcinoma eligible for treatment with vismodegib: An expert panel consensus. Futur. Oncol. 2015, 11, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.D.; Isaacs, A.; Holderbaum, L.; Tatard, V.; Dahmane, N. Design and Synthesis of Inhibitors of Hedgehog Signaling Based on the Alkaloid Cyclopamine. Org. Lett. 2009, 11, 2824–2827. [Google Scholar] [CrossRef]
- Lin, T.L.; Matsui, W. Hedgehog pathway as a drug target: Smoothened inhibitors in development. Onco Targets Ther. 2012, 5, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Hedberg, M.L.; Berry, C.T.; Moshiri, A.S.; Xiang, Y.; Yeh, C.J.; Attilasoy, C.; Capell, B.C.; Seykora, J.T. Molecular Mechanisms of Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 3478. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: Final update of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef] [Green Version]
- Kilgour, J.; Jia, J.; Sarin, K. Review of the Molecular Genetics of Basal Cell Carcinoma; Inherited Susceptibility, Somatic Mutations, and Targeted Therapeutics. Cancers 2021, 13, 3870. [Google Scholar] [CrossRef]
- Axelson, M.; Liu, K.; Jiang, X.; He, K.; Wang, J.; Zhao, H.; Kufrin, D.; Palmby, T.; Dong, Z.; Russell, A.M.; et al. U.S. Food and Drug Administration Approval: Vismodegib for Recurrent, Locally Advanced, or Metastatic Basal Cell Carcinoma. Clin. Cancer Res. 2013, 19, 2289–2293. [Google Scholar] [CrossRef] [Green Version]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and Safety of Vismodegib in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [Green Version]
- Basset-Séguin, N.; Hauschild, A.; Kunstfeld, R.; Grob, J.; Dréno, B.; Mortier, L.; Ascierto, P.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma: Primary analysis of STEVIE, an international, open-label trial. Eur. J. Cancer 2017, 86, 334–348. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.V.; Chang, J.; Li, S.; Henry, A.S.; Wood, D.J.; Chang, A.L.S. Increased Risk of Cutaneous Squamous Cell Carcinoma After Vismodegib Therapy for Basal Cell Carcinoma. JAMA Dermatol. 2016, 152, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Khamaysi, Z.; Bochner, R.; Indelman, M.; Magal, L.; Avitan-Hersh, E.; Sarig, O.; Sprecher, E.; Bergman, R. Segmental basal cell naevus syndrome caused by an activating mutation in smoothened. Br. J. Dermatol. 2016, 175, 178–181. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic Analysis of Smoothened Inhibitor Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Casey, D.; Demko, S.; Shord, S.; Zhao, H.; Chen, H.; He, K.; Putman, A.; Helms, W.S.; Keegan, P.; Pazdur, R. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 2377–2381. [Google Scholar] [CrossRef] [Green Version]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef]
- Xie, P.; Lefrançois, P. Efficacy, safety, and comparison of sonic hedgehog inhibitors in basal cell carcinomas: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2018, 79, 1089–1100.e17. [Google Scholar] [CrossRef]
- Chen, L.; Aria, A.B.; Silapunt, S.; Lee, H.-H.; Migden, M.R. Treatment of advanced basal cell carcinoma with sonidegib: Perspective from the 30-month update of the BOLT trial. Futur. Oncol. 2018, 14, 515–525. [Google Scholar] [CrossRef]
- Odom, D.; Mladsi, D.; Purser, M.; Kaye, J.A.; Palaka, E.; Charter, A.; Jensen, J.A.; Sellami, D. A Matching-Adjusted Indirect Comparison of Sonidegib and Vismodegib in Advanced Basal Cell Carcinoma. J. Ski. Cancer 2017, 2017, 6121760. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.; Chang, A.L.S.; Oro, A.E. Hedgehog pathway inhibition and the race against tumor evolution. J. Cell Biol. 2012, 199, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.L.S.; Oro, A.E. Initial Assessment of Tumor Regrowth After Vismodegib in Advanced Basal Cell Carcinoma. Arch. Dermatol. 2012, 148, 1324–1325. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a Commonly Used Antifungal that Inhibits Hedgehog Pathway Activity and Cancer Growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramelyte, E.; Restivo, G.; Imhof, L.; Nägeli, M.; Dummer, R. How to break resistance to hedgehog inhibitors in advanced basal cell carcinoma? Br. J. Dermatol. 2020, 184, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF–MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, A.; Dreier, J.; Cheng, P.F.; Nägeli, M.; Lehmann, H.; Felderer, L.; Frew, I.J.; Matsushita, S.; Levesque, M.P.; Dummer, R. Hedgehog Pathway Inhibitors Promote Adaptive Immune Responses in Basal Cell Carcinoma. Clin. Cancer Res. 2015, 21, 1289–1297. [Google Scholar] [CrossRef] [Green Version]
- Stonesifer, C.J.; Djavid, A.R.; Grimes, J.M.; Khaleel, A.E.; Soliman, Y.S.; Maisel-Campbell, A.; Garcia-Saleem, T.J.; Geskin, L.J.; Carvajal, R.D. Immune Checkpoint Inhibition in Non-Melanoma Skin Cancer: A Review of Current Evidence. Front. Oncol. 2021, 11, 734354. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Ralli, M.; Botticelli, A.; Visconti, I.C.; Angeletti, D.; Fiore, M.; Marchetti, P.; Lambiase, A.; De Vincentiis, M.; Greco, A. Immunotherapy in the Treatment of Metastatic Melanoma: Current Knowledge and Future Directions. J. Immunol. Res. 2020, 2020, 9235638. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, E.; Abadi, R.; Abbas, O. Imiquimod in dermatology: An overview. Int. J. Dermatol. 2016, 55, 831–844. [Google Scholar] [CrossRef]
- Winkler, J.; Schneiderbauer, R.; Bender, C.; Sedlaczek, O.; Fröhling, S.; Penzel, R.; Enk, A.; Hassel, J. Anti-programmed cell death-1 therapy in nonmelanoma skin cancer. Br. J. Dermatol. 2016, 176, 498–502. [Google Scholar] [CrossRef]
- Chang, A.L.S.; Tran, D.C.; Cannon, J.G.; Li, S.; Jeng, M.; Patel, R.; Van der Bokke, L.; Pague, A.; Brotherton, R.; Rieger, K.E.; et al. Pembrolizumab for advanced basal cell carcinoma: An investigator-initiated, proof-of-concept study. J. Am. Acad. Dermatol. 2019, 80, 564–566. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.V.; Kuo, K.Y.; Chang, A.L.S. Incidental regression of an advanced basal cell carcinoma after ipilimumab exposure for metastatic melanoma. JAAD Case Rep. 2016, 2, 13–15. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, S.; Goodman, A.M.; Cohen, P.R.; Jensen, T.J.; Ellison, C.K.; Frampton, G.; Miller, V.; Patel, S.P.; Kurzrock, R. Metastatic basal cell carcinoma with amplification of PD-L1: Exceptional response to anti-PD1 therapy. NPJ Genom. Med. 2016, 1, 16037. [Google Scholar] [CrossRef] [Green Version]
- Lipson, E.J.; Lilo, M.T.; Ogurtsova, A.; Esandrio, J.; Xu, H.; Brothers, P.; Schollenberger, M.; Sharfman, W.H.; Taube, J.M. Basal cell carcinoma: PD-L1/PD-1 checkpoint expression and tumor regression after PD-1 blockade. J. Immunother. Cancer 2017, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Cannon, J.G.; Russell, J.S.; Kim, J.; Chang, A.L.S. A case of metastatic basal cell carcinoma treated with continuous PD-1 inhibitor exposure even after subsequent initiation of radiotherapy and surgery. JAAD Case Rep. 2018, 4, 248–250. [Google Scholar] [CrossRef]
- Fischer, S.; Ali, O.H.; Jochum, W.; Kluckert, T.; Flatz, L.; Siano, M. Anti-PD-1 Therapy Leads to Near-Complete Remission in a Patient with Metastatic Basal Cell Carcinoma. Oncol. Res. Treat. 2018, 41, 391–394. [Google Scholar] [CrossRef]
- Burova, E.; Hermann, A.; Waite, J.; Potocky, T.; Lai, V.; Hong, S.; Liu, M.; Allbritton, O.; Woodruff, A.; Wu, Q.; et al. Characterization of the Anti–PD-1 Antibody REGN2810 and Its Antitumor Activity in Human PD-1 Knock-In Mice. Mol. Cancer Ther. 2017, 16, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Thudium, K.B.; Han, M.; Wang, X.-T.; Huang, H.; Feingersh, D.; Garcia, C.; Wu, Y.; Kuhne, M.; Srinivasan, M.; et al. In Vitro Characterization of the Anti-PD-1 Antibody Nivolumab, BMS-936558, and In Vivo Toxicology in Non-Human Primates. Cancer Immunol. Res. 2014, 2, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratigos, A.J.; Sekulic, A.; Peris, K.; Bechter, O.; Prey, S.; Kaatz, M.; Lewis, K.D.; Basset-Seguin, N.; Chang, A.L.S.; Dalle, S.; et al. Cemiplimab in locally advanced basal cell carcinoma after hedgehog inhibitor therapy: An open-label, multi-centre, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 848–857. [Google Scholar] [CrossRef]
- Davis, C.M.; Lewis, K.D. Brief overview: Cemiplimab for the treatment of advanced basal cell carcinoma: PD-1 strikes again. Ther. Adv. Med. Oncol. 2022, 14, 17588359211066147. [Google Scholar] [CrossRef] [PubMed]
- Shalhout, S.Z.; Kaufman, H.L.; Emerick, K.S.; Miller, D.M. Immunotherapy for Nonmelanoma Skin Cancer: Facts and Hopes. Clin. Cancer Res. 2022, 28, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Damsin, T.; Lebas, E.; Marchal, N.; Rorive, A.; Nikkels, A.F. Cemiplimab for locally advanced and metastatic basal cell carcinoma. Expert Rev. Anticancer Ther. 2022, 22, 243–248. [Google Scholar] [CrossRef]
- Nikolouzakis, T.K.; Falzone, L.; Lasithiotakis, K.; Krüger-Krasagakis, S.; Kalogeraki, A.; Sifaki, M.; Spandidos, D.A.; Chrysos, E.; Tsatsakis, A.; Tsiaoussis, J. Current and Future Trends in Molecular Biomarkers for Diagnostic, Prognostic, and Predictive Purposes in Non-Melanoma Skin Cancer. J. Clin. Med. 2020, 9, 2868. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.J.; Wadleigh, D.J.; Gangopadhyay, A.; Hama, S.; Grijalva, V.R.; Navab, M.; Fogelman, A.M.; Reddy, S.T. Paraoxonase-2 Is a Ubiquitously Expressed Protein with Antioxidant Properties and Is Capable of Preventing Cell-mediated Oxidative Modification of Low Density Lipoprotein. J. Biol. Chem. 2001, 276, 44444–44449. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, A.; Bourquard, N.; Hama, S.; Navab, M.; Grijalva, V.R.; Morvardi, S.; Clarke, C.F.; Vergnes, L.; Reue, K.; Teiber, J.F.; et al. Paraoxonase 2 Deficiency Alters Mitochondrial Function and Exacerbates the Development of Atherosclerosis. Antioxid. Redox. Sig. 2011, 14, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Bacchetti, T.; Salvolini, E.; Pompei, V.; Campagna, R.; Molinelli, E.; Brisigotti, V.; Togni, L.; Lucarini, G.; Sartini, D.; Campanati, A.; et al. Paraoxonase-2: A potential biomarker for skin cancer aggressiveness. Eur. J. Clin. Investig. 2020, 51, e13452. [Google Scholar] [CrossRef]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [CrossRef]
- Peng, Y.; Sartini, D.; Pozzi, V.; Wilk, D.; Emanuelli, M.; Yee, V.C. Structural Basis of Substrate Recognition in Human Nicotinamide N-Methyltransferase. Biochemistry 2011, 50, 7800–7808. [Google Scholar] [CrossRef] [Green Version]
- Campagna, R.; Pozzi, V.; Sartini, D.; Salvolini, E.; Brisigotti, V.; Molinelli, E.; Campanati, A.; Offidani, A.; Emanuelli, M. Beyond Nicotinamide Metabolism: Potential Role of Nicotinamide N-Methyltransferase as a Biomarker in Skin Cancers. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef]
- Pompei, V.; Salvolini, E.; Rubini, C.; Lucarini, G.; Molinelli, E.; Brisigotti, V.; Pozzi, V.; Sartini, D.; Campanati, A.; Offidani, A.; et al. Nicotinamide N-methyltransferase in nonmelanoma skin cancers. Eur. J. Clin. Investig. 2019, 49, e13175. [Google Scholar] [CrossRef]
- Smits, M.; van Rijn, S.; Hulleman, E.; Biesmans, D.; van Vuurden, D.G.; Kool, M.; Haberler, C.; Aronica, E.; Vandertop, W.P.; Noske, D.P.; et al. EZH2-Regulated DAB2IP Is a Medulloblastoma Tumor Suppressor and a Positive Marker for Survival. Clin. Cancer Res. 2012, 18, 4048–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R.; Chan, M.; Andrews, C.; Kahana, A. EZH2, Proliferation Rate, and Aggressive Tumor Subtypes in Cutaneous Basal Cell Carcinoma. JAMA Oncol. 2016, 2, 962–963. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Ma, L.; Wu, Z.; Zheng, G.; Li, J. Expression of miR-34a in basal cell carcinoma patients and its relationship with prognosis. Head Neck 2019, 24, 1283–1288. [Google Scholar]
- Zelin, E.; Maronese, C.A.; Dri, A.; Toffoli, L.; Di Meo, N.; Nazzaro, G.; Zalaudek, I. Identifying Candidates for Immunotherapy among Patients with Non-Melanoma Skin Cancer: A Review of the Potential Predictors of Response. J. Clin. Med. 2022, 11, 3364. [Google Scholar] [CrossRef]
- Maubec, E.; Boubaya, M.; Petrow, P.; Beylot-Barry, M.; Basset-Seguin, N.; Deschamps, L.; Grob, J.-J.; Dréno, B.; Scheer-Senyarich, I.; Bloch-Queyrat, C.; et al. Phase II Study of Pembrolizumab As First-Line, Single-Drug Therapy for Patients With Unresectable Cutaneous Squamous Cell Carcinomas. J. Clin. Oncol. 2020, 38, 3051–3061. [Google Scholar] [CrossRef]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 1555. [Google Scholar] [CrossRef]
- Kiefer, J.D.; Neri, D. Immunocytokines and bispecific antibodies: Two complementary strategies for the selective activation of immune cells at the tumor site. Immunol. Rev. 2016, 270, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Jahan, N.; Talat, H.; Curry, W.T. Agonist OX40 immunotherapy improves survival in glioma-bearing mice and is complementary with vaccination with irradiated GM-CSF–expressing tumor cells. Neuro-Oncology 2017, 20, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Li, X.; Zhang, L.; Zhu, Q.; Chen, C.; Bao, J.; Chen, Y. CD4+ T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol. 2019, 20, 27. [Google Scholar] [CrossRef] [Green Version]
- Workman, C.J.; Rice, D.S.; Dugger, K.J.; Kurschner, C.; Vignali, D.A. Phenotypic analysis of the murine CD4-related glyco-protein, CD223 (LAG-3). Eur. J. Immunol. 2002, 32, 2255–2263. [Google Scholar] [CrossRef]
- Grosso, J.F.; Goldberg, M.V.; Getnet, D.; Bruno, T.C.; Yen, H.-R.; Pyle, K.J.; Hipkiss, E.; Vignali, D.A.A.; Pardoll, D.M.; Drake, C.G. Functionally Distinct LAG-3 and PD-1 Subsets on Activated and Chronically Stimulated CD8 T Cells. J. Immunol. 2009, 182, 6659–6669. [Google Scholar] [CrossRef] [Green Version]
- Solinas, C.; De Silva, P.; Bron, D.; Willard-Gallo, K.; Sangiolo, D. Significance of TIM3 expression in cancer: From biology to the clinic. Semin. Oncol. 2019, 46, 372–379. [Google Scholar] [CrossRef]
- Malinga, N.Z.; Siwele, S.C.; Steel, H.C.; Kwofie, L.L.; Meyer, P.W.; Smit, T.; Anderson, R.; Rapoport, B.L.; Kgokolo, M.C. Systemic levels of the soluble co-inhibitory immune checkpoints, CTLA-4, LAG-3, PD-1/PD-L1 and TIM-3 are markedly increased in basal cell carcinoma. Transl. Oncol. 2022, 19, 101384. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Pricl, S.; Cortelazzi, B.; Col, V.D.; Marson, D.; Laurini, E.; Fermeglia, M.; Licitra, L.; Pilotti, S.; Bossi, P.; Perrone, F. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Mol. Oncol. 2014, 9, 389–397. [Google Scholar] [CrossRef]
- Myers, B.R.; Neahring, L.; Zhang, Y.; Roberts, K.J.; Beachy, P.A. Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proc. Natl. Acad. Sci. USA 2017, 114, E11141–E11150. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and Arsenic Trioxide Inhibit Hedgehog Pathway Activation and Tumor Growth Associated with Acquired Resistance to Smoothened Antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A.; et al. Open-Label, Exploratory Phase II Trial of Oral Itraconazole for the Treatment of Basal Cell Carcinoma. J. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef]
- Xin, M.; Ji, X.; De La Cruz, L.K.; Thareja, S.; Wang, B. Strategies to target the Hedgehog signaling pathway for cancer therapy. Med. Res. Rev. 2018, 38, 870–913. [Google Scholar] [CrossRef]
- Wang, C.; Wu, H.; Katritch, V.; Han, G.W.; Huang, X.-P.; Liu, W.; Siu, F.Y.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Structure of the human smoothened receptor bound to an antitumour agent. Nature 2013, 497, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L.S. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2016, 22, 1325–1329. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.; Andre, V.; Ho, A.; Kudchadkar, R.; Migden, M.; Infante, J.; Tiu, R.V.; Pitou, C.; Tucker, T.; Brail, L.; et al. Phase I Study of LY2940680, a Smo Antagonist, in Patients with Advanced Cancer Including Treatment-Naïve and Previously Treated Basal Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2082–2091. [Google Scholar] [CrossRef]
Figure 1.
Clinical features of basal cell carcinoma. (A) Nodular BCC. Translucent small papulonodule accompanied by telangiectasias and ulceration in the center of the tumor. (B) Morpheic/sclerosing BCC. Scar-like shiny area is observed (arrow). Boundary is very inconspicuous.
Figure 1.
Clinical features of basal cell carcinoma. (A) Nodular BCC. Translucent small papulonodule accompanied by telangiectasias and ulceration in the center of the tumor. (B) Morpheic/sclerosing BCC. Scar-like shiny area is observed (arrow). Boundary is very inconspicuous.

Figure 2.
Histopathological features of basal cell carcinoma. (A) Nodular BCC. Basaloid tumor cells arranged in a palisading manner. Tumor stroma cleftings are also found (arrow). Boundary is relatively clear (dotted line, original magnification ×200). (B) Morpheic/sclerosing BCC. Chords or thin nests of basaloid tumor cells infiltrate into the dermis. Tumor stroma clefting is not clear. Boundary is also unclear (x200).
Figure 2.
Histopathological features of basal cell carcinoma. (A) Nodular BCC. Basaloid tumor cells arranged in a palisading manner. Tumor stroma cleftings are also found (arrow). Boundary is relatively clear (dotted line, original magnification ×200). (B) Morpheic/sclerosing BCC. Chords or thin nests of basaloid tumor cells infiltrate into the dermis. Tumor stroma clefting is not clear. Boundary is also unclear (x200).

Figure 3.
Hedgehog pathway basal cell carcinoma. (A) Inactive state. PTCH1/2 inhibits SMO in the absence of Hh. (B) Active state. On ligand (Hh) binding, PTCH1/2-mediated repression of SMO is relieved (left). Alternatively, SMO is constitutively relieved if PTCH1/2 is mutated (right). SMO inhibitors (vismodegib, sonidegib, and taladegib) bind to drug-binding pockets of SMO then inhibit SMO. However, D473H SMO mutation causes resistance to vismodegib and sonidegib. PTCH1/2, patched 1/2; Hh, hedgehog ligand; SMO, smoothened; pocket, drug-binding pocket; SUFU, suppressor of fused; GLI, glioma-associated oncogene.
Figure 3.
Hedgehog pathway basal cell carcinoma. (A) Inactive state. PTCH1/2 inhibits SMO in the absence of Hh. (B) Active state. On ligand (Hh) binding, PTCH1/2-mediated repression of SMO is relieved (left). Alternatively, SMO is constitutively relieved if PTCH1/2 is mutated (right). SMO inhibitors (vismodegib, sonidegib, and taladegib) bind to drug-binding pockets of SMO then inhibit SMO. However, D473H SMO mutation causes resistance to vismodegib and sonidegib. PTCH1/2, patched 1/2; Hh, hedgehog ligand; SMO, smoothened; pocket, drug-binding pocket; SUFU, suppressor of fused; GLI, glioma-associated oncogene.


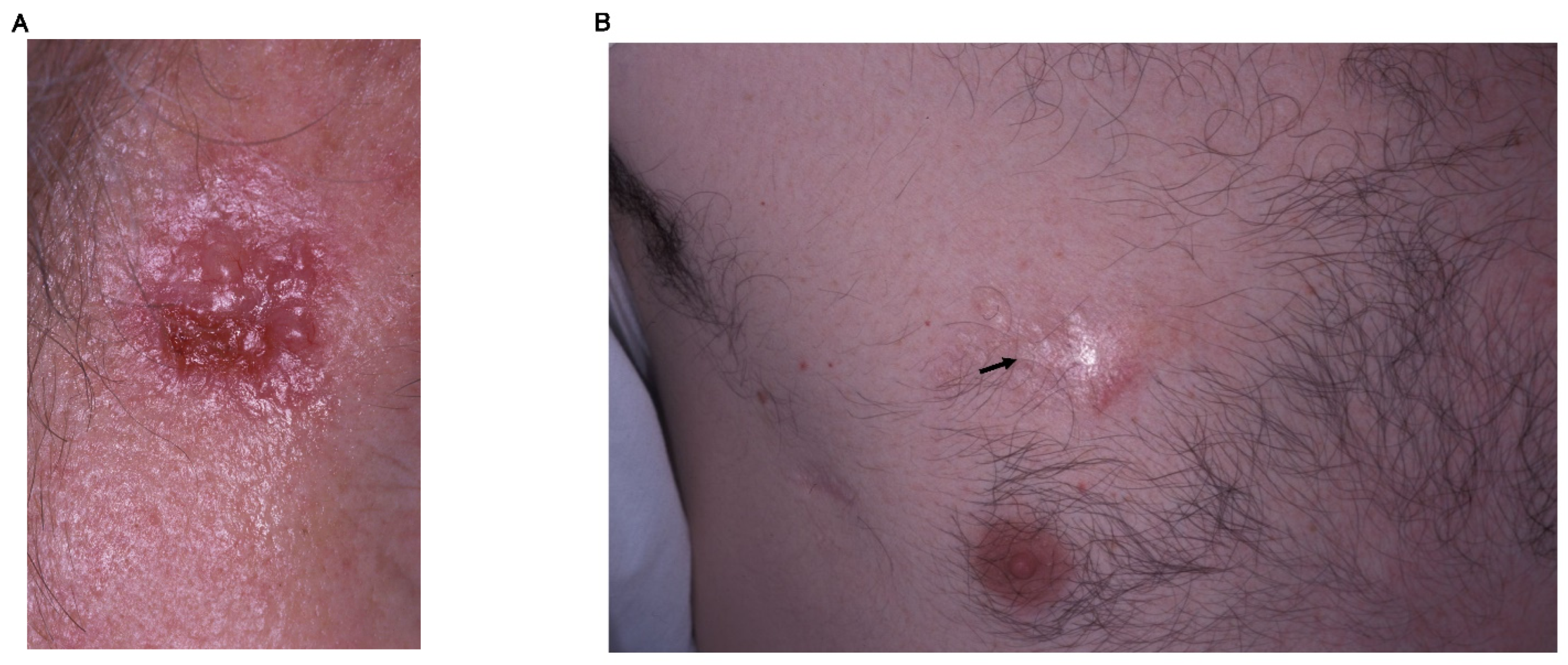{kind=link}
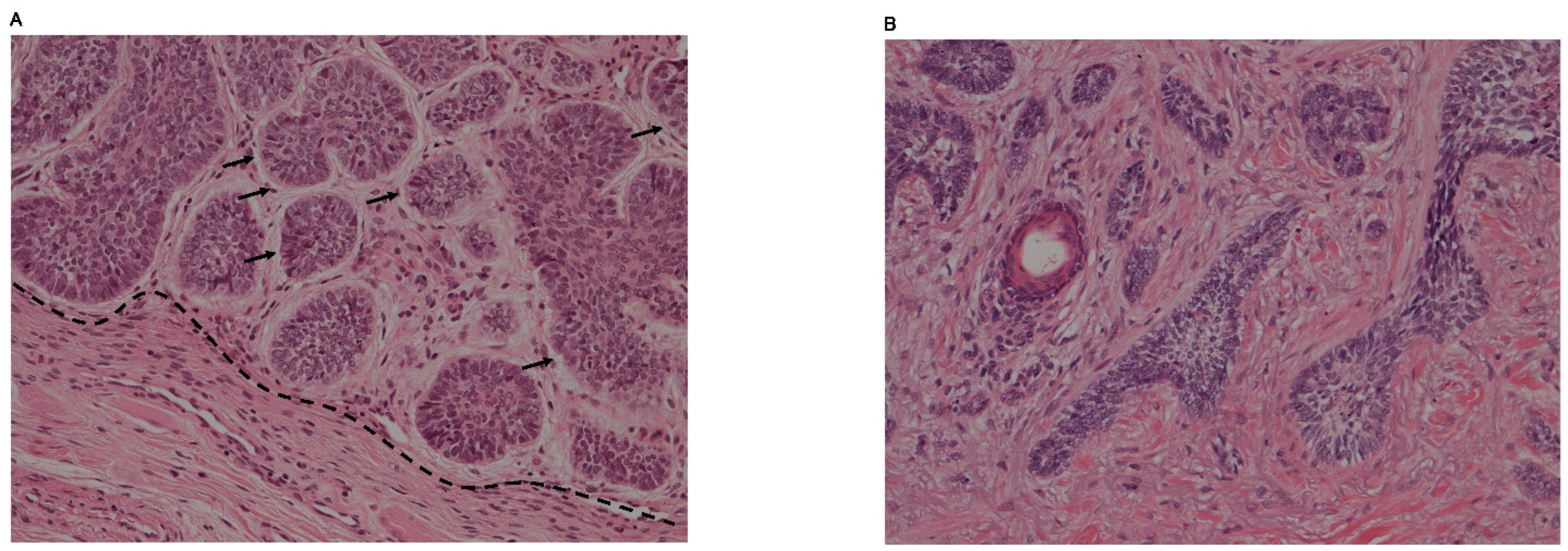{kind=link}
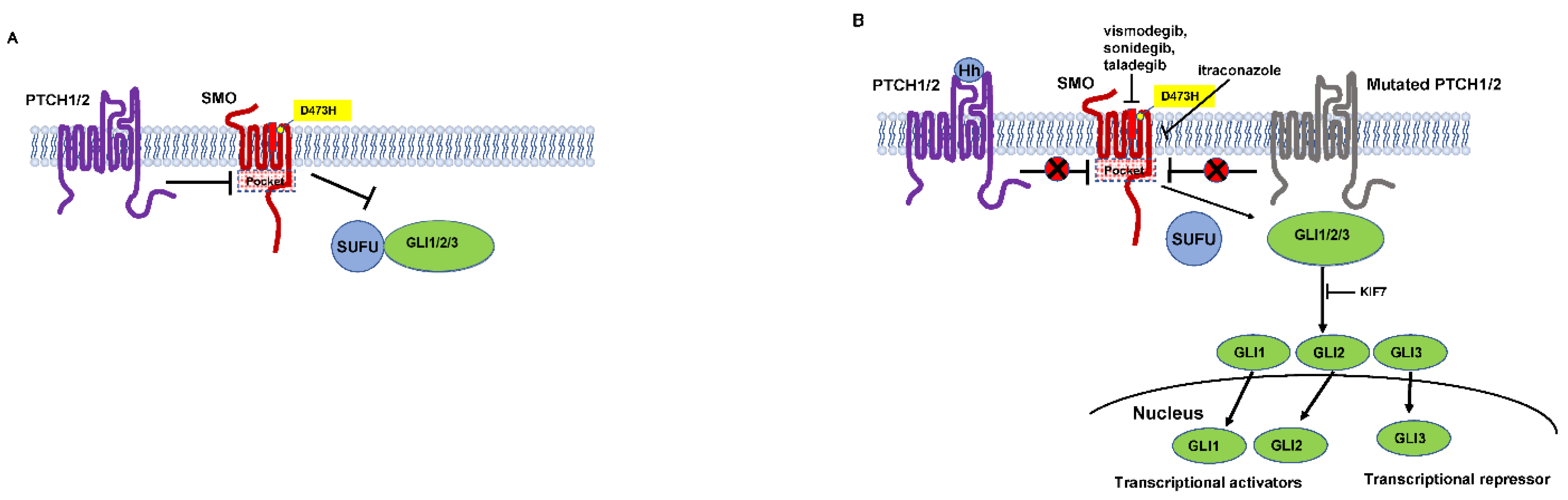{kind=link}
Table 1.
FDA approved target therapies.
| Drug | Indication | Mechanism | ORR | Median Follow-Up Time | Identifier | AE |
|---|---|---|---|---|---|---|
| Vismodegib | laBCC | SMO inhibitor | 60.3% | 39 months | ERIVANCE; NCT00833417 | muscle spasms, alopecia, taste loss, weight loss, decreased appetite, fatigue, nausea, diarrhea |
| mBCC | 48.5% | 39 months | ||||
| Sonidegib | laBCC | SMO inhibitor | 56.1% | 30 months | BOLT trial; NCT01327053 | muscle spasms, alopecia, dysgeusia |
| mBCC | 7.7% | 30 months | ||||
| Cemiplimab | laBCC (HH failure) | anti-PD-1 | 31% | 15.1 months | Study 1620; NCT03132636 | fatigue, diarrhea, pruritus |
| mBCC (HH failure) | 21% | 38.9 weeks |
Abbreviations: ORR, overall response rate; AE, adverse effect; BCC, basal cell carcinoma; laBCC, locally advanced BCC; mBCC, metastatic BCC; SMO, smoothened; PD-1, program death-1; HH, hedgehog. Note: Reported ORR in this table are from FDA-approved labels for each agent (Drugs@FDA).
Table 2.
Clinical trials of target therapies.
| Drug | Indication | Mechanism | Other Drugs | Mechanism | Phase | ORR | Identifier |
|---|---|---|---|---|---|---|---|
| XmAb20717 | BCC | bispecific (anti-PD-1 and anti-CTLA-4) | Phase I | NCT03517488 | |||
| Pemrolizumab | advanced BCC | anti-PD-1 | vismodegib | SMO inhibitor | Phase I/II | 44% | NCT02690948 |
| Pembrolizumab | |||||||
| 29% | |||||||
| Pembrolizumab + vismodegib | |||||||
| Cemiplimab | advanced BCC | anti-PD-1 | sonidegib | SMO inhibitor | Phase II | NCT04679480 | |
| Nivolumab | laBCC/mBCC | anti-PD-1 | relatlimab | anti-LAG-3 | Phase II | NCT03521830 | |
| Ipilimumab | anti-CTLA-4 | ||||||
| Itraconazole | BCC | SMO inhibitor | Phase II | NCT01108094 | |||
Abbreviations: ORR, overall response rate; BCC, basal cell carcinoma; laBCC, locally advanced BCC; mBCC, metastatic BCC; SMO, smoothened; PD-1, program death-1; CTLA-4, cytotoxic T-lymphocyte antigen-4; LAG-3, lymphocyte activation gene 3. Note: Reported ORR in this table are from FDA-approved labels for each agent (Drugs@FDA).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hoashi, T.; Kanda, N.; Saeki, H. Molecular Mechanisms and Targeted Therapies of Advanced Basal Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 11968. https://doi.org/10.3390/ijms231911968
AMA Style
Hoashi T, Kanda N, Saeki H. Molecular Mechanisms and Targeted Therapies of Advanced Basal Cell Carcinoma. International Journal of Molecular Sciences. 2022; 23(19):11968. https://doi.org/10.3390/ijms231911968
Chicago/Turabian StyleHoashi, Toshihiko, Naoko Kanda, and Hidehisa Saeki. 2022. "Molecular Mechanisms and Targeted Therapies of Advanced Basal Cell Carcinoma" International Journal of Molecular Sciences 23, no. 19: 11968. https://doi.org/10.3390/ijms231911968
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.