Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History

 ,
,
Abstract
:1. Introduction
2. ILD Overview
3. Pathophysiology
4. Comprehensive Medical History
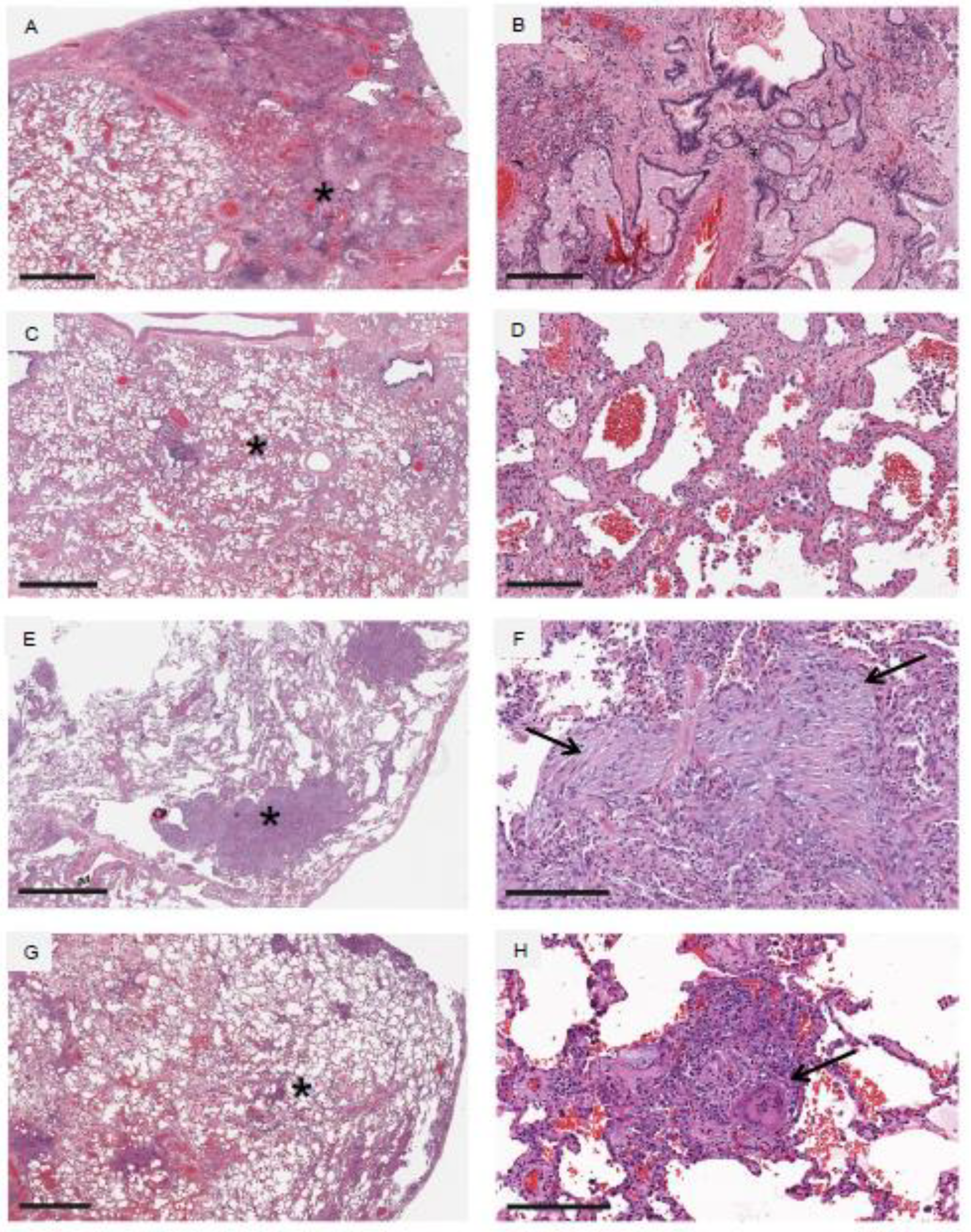
5. Histology
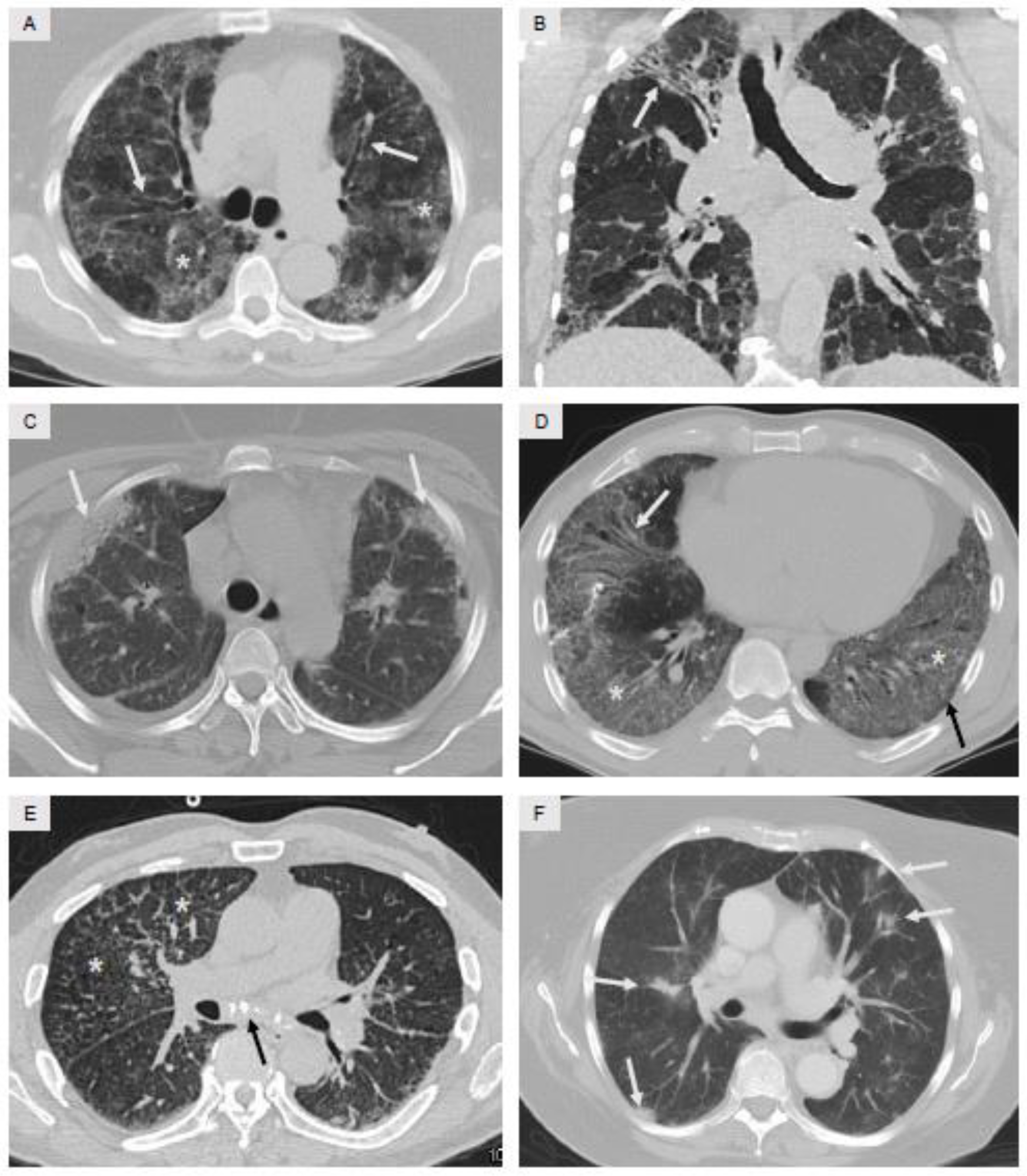
6. Radiology
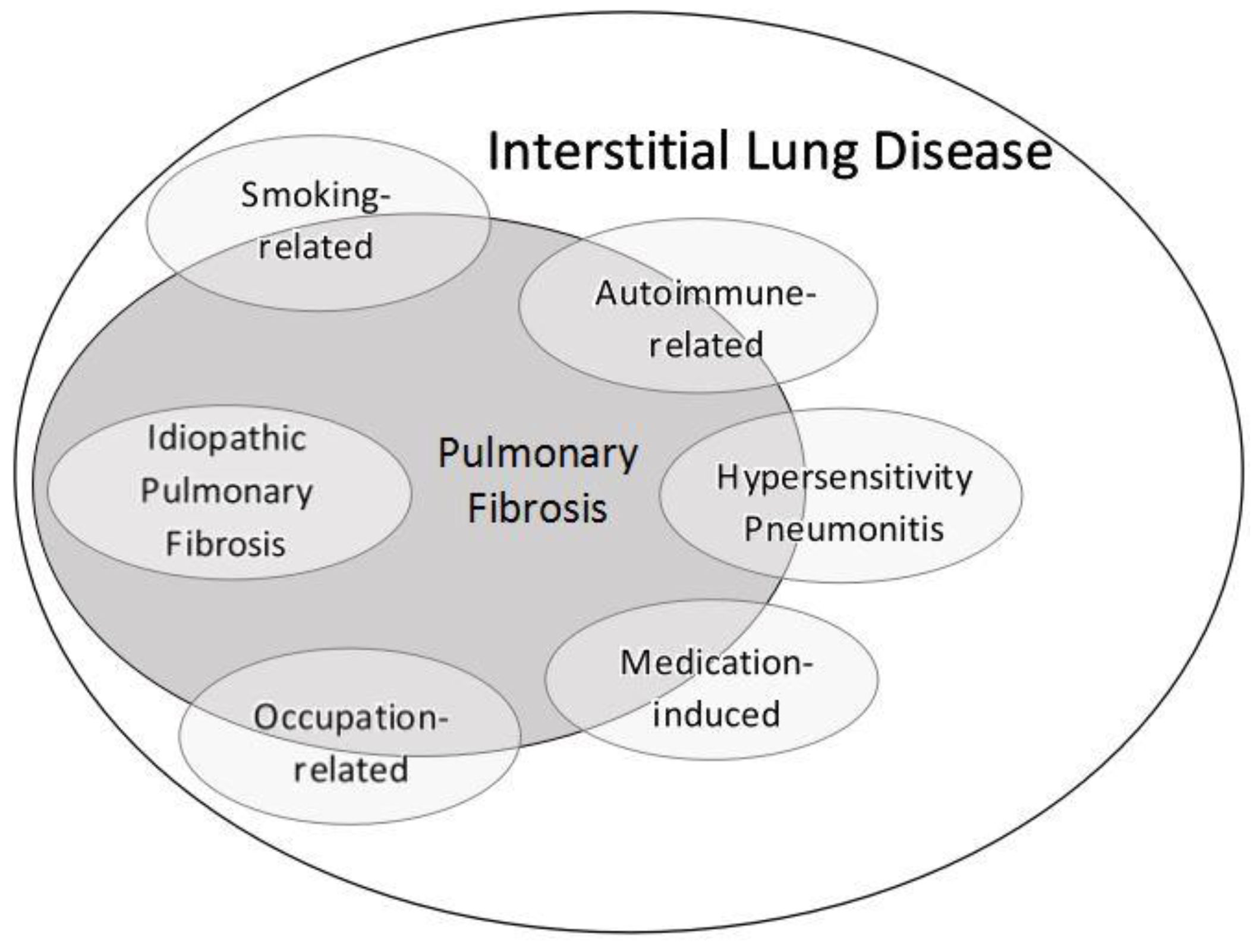
7. ILD Subgroups
7.1. Smoking Related-Interstitial Lung Disease
7.2. Hypersensitivity Pneumonitis
7.3. Connective Tissue Disease-Associated ILD
7.4. Occupation-Related ILD
7.5. Medication-Induced ILD
7.6. Idiopathic Pulmonary Fibrosis
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am. J. Respir. Crit. Care Med. 2002, 165, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.J.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Bradley, B.; Branley, H.M.; Egan, J.J.; Greaves, M.S.; Hansell, D.M.; Harrison, N.K.; Hirani, N.; Hubbard, R.; Lake, F.; Millar, A.B.; et al. Interstitial lung disease guideline: The British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax 2008, 63 (Suppl. 5), v1–v58. [Google Scholar] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.C.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Kinder, B.W.; Wells, A.U. The art and science of diagnosing interstitial lung diseases. Am. J. Respir. Crit. Care Med. 2009, 179, 974–975. [Google Scholar] [CrossRef] [PubMed]
- Leslie, K.O. My approach to interstitial lung disease using clinical, radiological and histopathological patterns. J. Clin. Pathol. 2009, 62, 387–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.J.; Flaherty, K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc. Am. Thorac. Soc. 2006, 3, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.L.F.; Wells, A.U.; Desai, S.R.; Poletti, V.; Piciucchi, S.; Dubini, A.; Nunes, H.; Valeyre, D.; Brillet, P.Y.; Kambouchner, M.; et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: A case-cohort study. Lancet Respir. Med. 2016, 4, 557–565. [Google Scholar] [CrossRef]
- Flaherty, K.R.; King, T.E.J.; Raghu, G.; Lynch JP 3rd Colby, T.V.; Travis, W.D.; Gross, B.H.; Kazerooni, E.A.; Toews, G.B.; Long, Q.; Murray, S.; et al. Idiopathic interstitial pneumonia: What is the effect of a multidisciplinary approach to diagnosis? Am. J. Respir. Crit. Care Med. 2004, 170, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Visscher, D.W.; Myers, J.L. Histologic spectrum of idiopathic interstitial pneumonias. Proc. Am. Thorac. Soc. 2006, 3, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair. 2012, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnato, G.; Harari, S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur. Respir. Rev. 2015, 24, 102–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.L.; Katzenstein, A.L. Epithelial necrosis and alveolar collapse in the pathogenesis of usual interstitial pneumonia. Chest. 1988, 94, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Todd, N.W.; Atamas, S.P.; Luzina, I.G.; Galvin, J.R. Permanent alveolar collapse is the predominant mechanism in idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 2015, 9, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Lutz, D.; Gazdhar, A.; Lopez-Rodriguez, E.; Ruppert, C.; Mahavadi, P.; Gunther, A.; Klepetko, W.; Bates, J.H.; Smith, B.; Geiser, T.; et al. Alveolar derecruitment and collapse induration as crucial mechanisms in lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.Y.; Chen, J.J.-L.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A., 3rd; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Maitra, M.; Wang, Y.; Gerard, R.D.; Mendelson, C.R.; Garcia, C.K. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J. Biol. Chem. 2010, 285, 22103–22113. [Google Scholar] [CrossRef] [PubMed]
- Van Moorsel, C.H.M.; van Oosterhout, M.F.M.; Barlo, N.P.; de Jong, P.A.; van der Vis, J.J.; Ruven, H.J.T.; van Es, H.W.; van den Bosch, J.M.M.; Grutters, J.C. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- ACCP Interstitial and Diffuse Lung Disease Patient Questionnaire [Internet]. American College of Chest Physicians Website. p. 4. Available online: http://www.chestnet.org/sitecore modules/web/~/media/chesnetorg/Foundation/Documents/Lung Disease Questionaire.ashx (accessed on 12 September 2018).
- Hong, S.J.; Kim, T.J.; Lee, J.-H.; Park, J.-S. Nontuberculous mycobacterial pulmonary disease mimicking lung cancer: Clinicoradiologic features and diagnostic implications. Medicine 2016, 95, e3978. [Google Scholar] [CrossRef] [PubMed]
- Franquet, T.; Lee, K.S.; Muller, N.L. Thin-section CT findings in 32 immunocompromised patients with cytomegalovirus pneumonia who do not have AIDS. AJR Am. J. Roentgenol. 2003, 181, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Kanne, J.P.; Yandow, D.R.; Meyer, C.A. Pneumocystis jiroveci pneumonia: High-resolution CT findings in patients with and without HIV infection. AJR Am. J. Roentgenol. 2012, 198, W555–W561. [Google Scholar] [CrossRef] [PubMed]
- Ryerson, C.J.; Corte, T.J.; Lee, J.S.; Richeldi, L.; Walsh, S.L.F.; Myers, J.L.; Behr, J.; Cottin, V.; Danoff, S.K.; Flaherty, K.R.; et al. A Standardized Diagnostic Ontology for Fibrotic Interstitial Lung Disease. An International Working Group Perspective. Am. J. Respir. Crit. Care Med. 2017, 196, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Katzenstein, A.L.; Fiorelli, R.F. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am. J. Surg. Pathol. 1994, 18, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Hunninghake, G.; King, T.E.J.; Lynch, D.A.; Colby, T.V.; Galvin, J.R.; Brown, K.K.; Chung, M.P.; Cordier, J.-F.; du Bois, R.M.; et al. Idiopathic nonspecific interstitial pneumonia: Report of an American Thoracic Society project. Am. J. Respir. Crit. Care Med. 2008, 177, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Todd, N.W.; Marciniak, E.T.; Sachdeva, A.; Kligerman, S.J.; Galvin, J.R.; Luzina, I.G.; Atamas, S.P.; Burke, A.P. Organizing pneumonia/non-specific interstitial pneumonia overlap is associated with unfavorable lung disease progression. Respir. Med. 2015, 109, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Cordier, J.F. Organising pneumonia. Thorax 2000, 55, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.R.; Travis, W.D.; Colby, T.V.; Toews, G.B.; Kazerooni, E.A.; Gross, B.H.; Jain, A.; Strawderman, R.L.; Flint, A.; Lynch, J.P.; et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2001, 164, 1722–1727. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, D.S.; Park, I.-N.; Jang, S.J.; Kitaichi, M.; Nicholson, A.G.; Colby TV Pr1 Park, J.H.; Kim, D.S.; Park, I.-N.; Jang, S.J.; et al. Prognosis of fibrotic interstitial pneumonia: Idiopathic versus collagen vascular disease-related subtypes. Am. J. Respir. Crit. Care Med. 2007, 175, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Leslie, K.O.; Gruden, J.F.; Parish, J.M.; Scholand, M.B. Transbronchial biopsy interpretation in the patient with diffuse parenchymal lung disease. Arch. Pathol. Lab. Med. 2007, 131, 407–423. [Google Scholar] [PubMed]
- Utz, J.P.; Ryu, J.H.; Douglas, W.W.; Hartman, T.E.; Tazelaar, H.D.; Myers, J.L.; Allen, M.S.; Schroeder, D.R. High short-term mortality following lung biopsy for usual interstitial pneumonia. Eur. Respir. J. 2001, 17, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collard, H.R.; Moore, B.B.; Flaherty, K.R.; Brown, K.K.; Kaner, R.J.; King, T.E.J.; Lasky, J.A.; Loyd, J.E.; Noth, I.; Olman, M.A.; et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V. Lung biopsy in interstitial lung disease: Balancing the risk of surgery and diagnostic uncertainty. Eur. Respir. J. 2016, 48, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
- Strollo, D.C.; Franks, T.J.; Galvin, J.R. The idiopathic interstitial pneumonias: Histology and imaging. Semin. Roentgenol. 2015, 50, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.I.S.; Churg, A.; Muller, N.L. Hypersensitivity pneumonitis: Spectrum of high-resolution CT and pathologic findings. AJR Am. J. Roentgenol. 2007, 188, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Staats, P.; Kligerman, S.; Todd, N.; Tavora, F.; Xu, L.; Burke, A. A comparative study of honeycombing on high resolution computed tomography with histologic lung remodeling in explants with usual interstitial pneumonia. Pathol. Res. Pract. 2015, 211, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Sumikawa, H.; Johkoh, T.; Colby, T.V.; Ichikado, K.; Suga, M.; Taniguchi, H.; Kondoh, Y.; Ogura, T.; Arakawa, H.; Fujimoto, K.; et al. Computed tomography findings in pathological usual interstitial pneumonia: Relationship to survival. Am. J. Respir. Crit. Care Med. 2008, 177, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.L.F.; Sverzellati, N.; Devaraj, A.; Wells, A.U.; Hansell, D.M. Chronic hypersensitivity pneumonitis: High resolution computed tomography patterns and pulmonary function indices as prognostic determinants. Eur. Radiol. 2012, 22, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.L.F.; Sverzellati, N.; Devaraj, A.; Keir, G.J.; Wells, A.U.; Hansell, D.M. Connective tissue disease related fibrotic lung disease: High resolution computed tomographic and pulmonary function indices as prognostic determinants. Thorax 2014, 69, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.R.; Fell, C.; Aubry, M.-C.; Brown, K.; Colby, T.; Costabel, U.; Franks, T.J.; Gross, B.H.; Hansell, D.M.; Kazerooni, E.; et al. Smoking-related idiopathic interstitial pneumonia. Eur. Respir. J. 2014, 44, 594–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attili, A.K.; Kazerooni, E.A.; Gross, B.H.; Flaherty, K.R.; Myers, J.L.; Martinez, F.J. Smoking-related interstitial lung disease: Radiologic-clinical-pathologic correlation. Radiographics 2008, 28, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Kligerman, S.; Franks, T.J.; Galvin, J.R. Clinical-Radiologic-Pathologic Correlation of Smoking-Related Diffuse Parenchymal Lung Disease. Radiol. Clin. N. Am. 2016, 54, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Martinez, F.J.; Pardo, A. Why an Aging Smoker Lung Develops IPF and Not COPD? Am. J. Respir. Crit. Care Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Schwartz, D.A. Epigenetics of idiopathic pulmonary fibrosis. Transl Res. 2015, 165, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margaritopoulos, G.A.; Vasarmidi, E.; Jacob, J.; Wells, A.U.; Antoniou, K.M. Smoking and interstitial lung diseases. Eur. Respir. Rev. 2015, 24, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.H.; Myers, J.L.; Capizzi, S.A.; Douglas, W.W.; Vassallo, R.; Decker, P.A. Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest 2005, 127, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Niewoehner, D.E.; Kleinerman, J.; Rice, D.B. Pathologic changes in the peripheral airways of young cigarette smokers. N. Engl. J. Med. 1974, 291, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Godbert, B.; Wissler, M.-P.; Vignaud, J.-M. Desquamative interstitial pneumonia: An analytic review with an emphasis on aetiology. Eur. Respir. Rev. 2013, 22, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Liebow, A.A.; Steer, A.; Billingsley, J.G. Desquamative interstitial pneumonia. Am. J. Med. 1965, 39, 369–404. [Google Scholar] [CrossRef]
- Vassallo, R.; Ryu, J.H.; Colby, T.V.; Hartman, T.; Limper, A.H. Pulmonary Langerhans’-cell histiocytosis. N. Engl. J. Med. 2000, 342, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Katzenstein, A.-L.A.; Mukhopadhyay, S.; Zanardi, C.; Dexter, E. Clinically occult interstitial fibrosis in smokers: Classification and significance of a surprisingly common finding in lobectomy specimens. Hum. Pathol. 2010, 41, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, Y.; Hoshi, E.; Murai, K.; Ikeya, T.; Takahashi, N.; Saitou, Y.; Kurashima, K.; Ubukata, M.; Takayanagi, N.; Sugita, H.; et al. Smoking-related changes in the background lung of specimens resected for lung cancer: A semiquantitative study with correlation to postoperative course. Histopathology 2008, 53, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Cherian, S.V.; Vassallo, R.; Yi, E.S.; Ryu, J.H. Current Concepts in Pathogenesis, Diagnosis, and Management of Smoking-Related Interstitial Lung Diseases. Chest 2018, 154, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Jankowich, M.D.; Rounds, S.I.S. Combined pulmonary fibrosis and emphysema syndrome: A review. Chest 2012, 141, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Todd, N.W.; Jeudy, J.; Lavania, S.; Franks, T.J.; Galvin, J.R.; Deepak, J.; Britt, E.J.; Atamas, S.P. Centrilobular emphysema combined with pulmonary fibrosis results in improved survival. Fibrogenesis Tissue Repair. 2011, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checa, M.; Hagood, J.S.; Velazquez-Cruz, R.; Ruiz, V.; Garcia-De-Alba, C.; Rangel-Escareno, C.; Urrea, F.; Becerril, C.; Montano, M.; Garcia-Trejo, S.; et al. Cigarette Smoke Enhances the Expression of Profibrotic Molecules in Alveolar Epithelial Cells. PLoS ONE 2016, 11, e0150383. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R. Epidemiology of idiopathic pulmonary fibrosis. Clin. Epidemiol. 2013, 5, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Portnoy, J.; Veraldi, K.L.; Schwarz, M.I.; Cool, C.D.; Curran-Everett, D.; Cherniack, R.M.; King, T.E.J.; Brown, K.K. Respiratory bronchiolitis-interstitial lung disease: Long-term outcome. Chest 2007, 131, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Johannson, K.; Ryerson, C.J. Making an accurate diagnosis of chronic hypersensitivity pneumonitis. Can. Respir. J. 2014, 21, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A.; King, T.E.J. Hypersensitivity pneumonitis: Insights in diagnosis and pathobiology. Am. J. Respir. Crit. Care Med. 2012, 186, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, T.; Miyazaki, Y.; Kuramochi, J.; Uchida, K.; Eishi, Y.; Inase, N. The amount of avian antigen in household dust predicts the prognosis of chronic bird-related hypersensitivity pneumonitis. Ann. Am. Thorac. Soc. 2015, 12, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Lacasse, Y.; Pardo, A.; Cormier, Y. Hypersensitivity pneumonitis caused by fungi. Proc. Am. Thorac. Soc. 2010, 7, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Ancillol, A.; Dominguez-Noche, C.; Gil-Adrados, A.C.; Cosmes, P.M. Hypersensitivity pneumonitis due to occupational inhalation of fungi-contaminated corn dust. J. Investig. Allergol Clin. Immunol. 2004, 14, 165–167. [Google Scholar] [PubMed]
- May, S.; Romberger, D.J.; Poole, J.A. Respiratory health effects of large animal farming environments. J. Toxicol. Environ. Health B Crit. Rev. 2012, 15, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Suga, M.; Nishiura, Y.; Arima, K.; Yoneda, R.; Tamura, M.; Ando, M. Occupational hypersensitivity pneumonitis in Japan: Data on a nationwide epidemiological study. Occup. Environ. Med. 1995, 52, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Seed, M.J.; Enoch, S.J.; Agius, R.M. Chemical determinants of occupational hypersensitivity pneumonitis. Occup. Med. 2015, 65, 673–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riario Sforza, G.G.; Marinou, A. Hypersensitivity pneumonitis: A complex lung disease. Clin. Mol. Allergy. 2017, 15, 6. [Google Scholar] [CrossRef] [PubMed]
- Apostolakos, M.J.; Rossmoore, H.; Beckett, W.S. Hypersensitivity pneumonitis from ordinary residential exposures. Environ. Health Perspect. 2001, 109, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Grandstaff, K. American Industrial Hygiene Association: Position Statement on Mold and Dampness in the Built Environment. [Internet]. American Industrial Hygiene Association. 2013, p. 12. Available online: https://www.aiha.org/government-affairs/PositionStatements/P-Mold-03-26-13.pdf (accessed on 27 August 2018).
- Cramer, C.; Schlunssen, V.; Bendstrup, E.; Stokholm, Z.A.; Vestergaard, J.M.; Frydenberg, M.; Kolstad, H.A. Risk of hypersensitivity pneumonitis and interstitial lung diseases among pigeon breeders. Eur. Respir. J. 2016, 48, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, J.; Inase, N.; Takayama, K.; Miyazaki, Y.; Yoshizawa, Y. Detection of indoor and outdoor avian antigen in management of bird-related hypersensitivity pneumonitis. Allergol. Int. 2010, 59, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Vourlekis, J.S.; Schwarz, M.I.; Cherniack, R.M.; Curran-Everett, D.; Cool, C.D.; Tuder, R.M.; King, T.E.J.; Brown, K.K. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am. J. Med. 2004, 116, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Hanak, V.; Golbin, J.M.; Ryu, J.H. Causes and presenting features in 85 consecutive patients with hypersensitivity pneumonitis. Mayo Clin. Proc. 2007, 82, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Hirschmann, J.V.; Pipavath, S.N.J.; Godwin, J.D. Hypersensitivity pneumonitis: A historical, clinical, and radiologic review. Radiographics 2009, 29, 1921–1938. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Perez, E.R.; Swigris, J.J.; Forssen, A.V.; Tourin, O.; Solomon, J.J.; Huie, T.J.; Olson, A.L.; Brown, K.K. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest 2013, 144, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.H.; Williams, G.V.; Woolcock, A.J. Bird breeder’s hypersensitivity pneumonitis: Progress studies of lung function after cessation of exposure to the provoking antigen. Am. Rev. Respir. Dis. 1976, 114, 555–566. [Google Scholar] [PubMed]
- Salisbury, M.L.; Myers, J.L.; Belloli, E.A.; Kazerooni, E.A.; Martinez, F.J.; Flaherty, K.R. Diagnosis and Treatment of Fibrotic Hypersensitivity Pneumonia. Where We Stand and Where We Need to Go. Am. J. Respir. Crit. Care Med. 2017, 196, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Adegunsoye, A.; Oldham, J.M.; Fernandez Perez, E.R.; Hamblin, M.; Patel, N.; Tener, M.; Bhanot, D.; Robinson, L.; Bullick, S.; Chen, L.; et al. Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ Open Res. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Monkare, S. Influence of corticosteroid treatment on the course of farmer’s lung. Eur. J. Respir. Dis. 1983, 64, 283–293. [Google Scholar] [PubMed]
- Kokkarinen, J.I.; Tukiainen, H.O.; Terho, E.O. Recovery of pulmonary function in farmer’s lung. A five-year follow-up study. Am. Rev. Respir. Dis. 1993, 147, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Morisset, J.; Johannson, K.A.; Vittinghoff, E.; Aravena, C.; Elicker, B.M.; Jones, K.D.; Fell, C.D.; Manganas, H.; Dube, B.-P.; Wolters, P.J.; et al. Use of Mycophenolate Mofetil or Azathioprine for the Management of Chronic Hypersensitivity Pneumonitis. Chest 2017, 151, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, M.L.; Gu, T.; Murray, S.; Gross, B.H.; Chughtai, A.; Sayyouh, M.; Kazerooni, E.A.; Myers, J.L.; Lagstein, A.; Konopka, K.E.; et al. Hypersensitivity Pneumonitis: Radiologic Phenotypes Are Associated With Distinct Survival Time and Pulmonary Function Trajectory. Chest 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Vij, R.; Strek, M.E. Diagnosis and treatment of connective tissue disease-associated interstitial lung disease. Chest 2013, 143, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Corte, T.J.; Copley, S.J.; Desai, S.R.; Zappala, C.J.; Hansell, D.M.; Nicholson, A.G.; Colby, T.V.; Renzoni, E.; Maher, T.M.; Wells, A.U. Significance of connective tissue disease features in idiopathic interstitial pneumonia. Eur. Respir. J. 2012, 39, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Vij, R.; Noth, I.; Strek, M.E. Autoimmune-featured interstitial lung disease: A distinct entity. Chest 2011, 140, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Kinder, B.W.; Collard, H.R.; Koth, L.; Daikh, D.I.; Wolters, P.J.; Elicker, B.; Jones, K.D.; King, T.E.J. Idiopathic nonspecific interstitial pneumonia: Lung manifestation of undifferentiated connective tissue disease? Am. J. Respir. Crit. Care Med. 2007, 176, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Arulkumaran, N.; Periselneris, N.; Gaskin, G.; Strickland, N.; Ind, P.W.; Pusey, C.D.; Salama, A.D. Interstitial lung disease and ANCA-associated vasculitis: A retrospective observational cohort study. Rheumatology 2011, 50, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Antoniou, K.M.; Brown, K.K.; Cadranel, J.; Corte, T.J.; du Bois, R.M.; Lee, J.S.; Leslie, K.O.; Lynch, D.A.; Matteson, E.L.; et al. An official European Respiratory Society/American Thoracic Society research statement: Interstitial pneumonia with autoimmune features. Eur. Respir. J. 2015, 46, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Jindal, S.K.; Agarwal, R. Autoimmunity and interstitial lung disease. Curr. Opin. Pulm Med. 2005, 11, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Noonan, C.W.; Pfau, J.C.; Larson, T.C.; Spence, M.R. Nested case-control study of autoimmune disease in an asbestos-exposed population. Environ. Health Perspect. 2006, 114, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, T.; Maeda, M.; Murakami, S.; Hayashi, H.; Miura, Y.; Kusaka, M.; Nakano, T.; Fukuoka, K.; Kishimoto, T.; Hyodoh, F.; et al. Immunological effects of silica and asbestos. Cell. Mol. Immunol. 2007, 4, 261–268. [Google Scholar] [PubMed]
- Papiris, S.A.; Kagouridis, K.; Bouros, D. Serologic evaluation in idiopathic interstitial pneumonias. Curr. Opin. Pulm. Med. 2012, 18, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Connors, G.R.; Oaks, J.; Han, S.; Truong, A.; Richardson, B.; Lechtzin, N.; Mammen, A.L.; Casciola-Rosen, L.; Christopher-Stine, L.; et al. Clinical and pathologic differences in interstitial lung disease based on antisynthetase antibody type. Respir. Med. 2014, 108, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, L.-S.; Wei, Y.-R.; Du, S.-S.; Du, Y.-K.; He, X.; Li, N.; Zhou, Y.; Li, Q.-H.; Su, Y.-L.; et al. Clinical Characteristics of Connective Tissue Disease-Associated Interstitial Lung Disease in 1,044 Chinese Patients. Chest 2016, 149, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Jee, A.S.; Adelstein, S.; Bleasel, J.; Keir, G.J.; Nguyen, M.; Sahhar, J.; Youssef, P.; Corte, T.J. Role of Autoantibodies in the Diagnosis of Connective-Tissue Disease ILD (CTD-ILD) and Interstitial Pneumonia with Autoimmune Features (IPAF). J. Clin. Med. 2017, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Henry, T.S.; Little, B.P.; Veeraraghavan, S.; Bhalla, S.; Elicker, B.M. The Spectrum of Interstitial Lung Disease in Connective Tissue Disease. J. Thorac. Imaging. 2016, 31, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Moua, T.; Zamora Martinez, A.C.; Baqir, M.; Vassallo, R.; Limper, A.H.; Ryu, J.H. Predictors of diagnosis and survival in idiopathic pulmonary fibrosis and connective tissue disease-related usual interstitial pneumonia. Respir. Res. 2014, 15, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyldgaard, C. A cohort study of Danish patients with interstitial lung diseases: Burden, severity, treatment and survival. Dan Med. J. 2015, 62, B5069. [Google Scholar] [PubMed]
- Aggarwal, R.; McBurney, C.; Schneider, F.; Yousem, S.A.; Gibson, K.F.; Lindell, K.; Fuhrman, C.R.; Oddis, C.V. Myositis-associated usual interstitial pneumonia has a better survival than idiopathic pulmonary fibrosis. Rheumatology 2017, 56, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Alhamad, E.H.; Al-Kassimi, F.A.; Alboukai, A.A.; Raddaoui, E.; Al-Hajjaj, M.S.; Hajjar, W.; Shaik, S.A. Comparison of three groups of patients with usual interstitial pneumonia. Respir. Med. 2012, 106, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.C.; Raghu, G.; Baughman, R.P.; Brown, K.K.; Costabel, U.; du Bois, R.M.; Drent, M.; Haslam, P.L.; Kim, D.S.; Nagai, S.; et al. An official American Thoracic Society clinical practice guideline: The clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, S.; Latsi, P.I.; Wells, A.U.; Pantelidis, P.; Nicholson, A.G.; Colby, T.V.; Haslam, P.L.; Renzoni, E.A.; du Bois, R.M. BAL findings in idiopathic nonspecific interstitial pneumonia and usual interstitial pneumonia. Eur. Respir. J. 2003, 22, 239–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omote, N.; Taniguchi, H.; Kondoh, Y.; Watanabe, N.; Sakamoto, K.; Kimura, T.; Kataoka, K.; Johkoh, T.; Fujimoto, K.; Fukuoka, J.; et al. Lung-Dominant Connective Tissue Disease: Clinical, Radiologic, and Histologic Features. Chest 2015, 148, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Tansey, D.; Wells, A.U.; Colby, T.V.; Ip, S.; Nikolakoupolou, A.; du Bois, R.M.; Hansell, D.M.; Nicholson, A.G. Variations in histological patterns of interstitial pneumonia between connective tissue disorders and their relationship to prognosis. Histopathology 2004, 44, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Kocheril, S.V.; Appleton, B.E.; Somers, E.C.; Kazerooni, E.A.; Flaherty, K.R.; Martinez, F.J.; Gross, B.H.; Crofford, L.J. Comparison of disease progression and mortality of connective tissue disease-related interstitial lung disease and idiopathic interstitial pneumonia. Arthritis Rheum. 2005, 53, 549–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Chida, K.; Suda, T.; Hayakawa, H.; Iwata, M.; Imokawa, S.; Tsuchiya, T.; Ida, M.; Gemma, H.; Yasuda, K.; et al. Nonspecific interstitial pneumonia in collagen vascular diseases: Comparison of the clinical characteristics and prognostic significance with usual interstitial pneumonia. Sarcoidosis Vasc. Diffuse Lung Dis. 2003, 20, 235–241. [Google Scholar] [PubMed]
- Wells, A.U.; Denton, C.P. Interstitial lung disease in connective tissue disease--mechanisms and management. Nat. Rev. Rheumatol. 2014, 10, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.; Vummidi, D.; Khanna, D. Management of connective tissue diseases associated interstitial lung disease: A review of the published literature. Curr. Opin. Rheumatol. 2016, 28, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Dheda, K.; Lalloo, U.G.; Cassim, B.; Mody, G.M. Experience with azathioprine in systemic sclerosis associated with interstitial lung disease. Clin. Rheumatol. 2004, 23, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.; Ngian, G.-S.; Elford, K.; Moore, O.; Stevens, W.; Nikpour, M.; Rabusa, C.; Proudman, S.; Roddy, J.; Zochling, J.; et al. Mycophenolate mofetil is an effective and safe option for the management of systemic sclerosis-associated interstitial lung disease: Results from the Australian Scleroderma Cohort Study. Clin. Exp. Rheumatol. 2016, 34 (Suppl. 1), 170–176. [Google Scholar] [PubMed]
- Oldham, J.M.; Lee, C.; Valenzi, E.; Witt, L.J.; Adegunsoye, A.; Hsu, S.; Chen, L.; Montner, S.; Chung, J.H.; Noth, I.; et al. Azathioprine response in patients with fibrotic connective tissue disease-associated interstitial lung disease. Respir. Med. 2016, 121, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas-Serrano, J.; Herrera-Bringas, D.; Perez-Roman, D.I.; Perez-Dorame, R.; Mateos-Toledo, H.; Mejia, M. Rheumatoid arthritis-related interstitial lung disease (RA-ILD): Methotrexate and the severity of lung disease are associated to prognosis. Clin. Rheumatol. 2017, 36, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.D.; Kremer, J.M. Successful treatment of interstitial lung disease in systemic lupus erythematosus with methotrexate. J. Rheumatol. 1995, 22, 967–969. [Google Scholar] [PubMed]
- Yamasaki, Y.; Yamada, H.; Yamasaki, M.; Ohkubo, M.; Azuma, K.; Matsuoka, S.; Kurihara, Y.; Osada, H.; Satoh, M.; Ozaki, S. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology 2007, 46, 124–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, P.D.; Bavaliya, M.; Sashidharan, S.; Nalianda, K.; Sreenath, S. Cyclophosphamide versus mycophenolate mofetil in scleroderma interstitial lung disease (SSc-ILD) as induction therapy: A single-centre, retrospective analysis. Arthritis Res. Ther. 2016, 18, 123. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, E.R.; Tashkin, D.P.; Li, N.; Roth, M.D.; Khanna, D.; Hoffmann-Vold, A.-M.; Kim, G.; Goldin, J.; Clements, P.J.; Furst, D.E.; et al. Mycophenolate Mofetil Versus Placebo for Systemic Sclerosis-Related Interstitial Lung Disease: An Analysis of Scleroderma Lung Studies I and II. Arthritis Rheumatol. 2017, 69, 1451–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashkin, D.P.; Roth, M.D.; Clements, P.J.; Furst, D.E.; Khanna, D.; Kleerup, E.C.; Goldin, J.; Arriola, E.; Volkmann, E.R.; Kafaja, S.; et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): A randomised controlled, double-blind, parallel group trial. Lancet Respir. Med. 2016, 4, 708–719. [Google Scholar] [CrossRef]
- Mira-Avendano, I.C.; Parambil, J.G.; Yadav, R.; Arrossi, V.; Xu, M.; Chapman, J.T.; Culver, D.A. A retrospective review of clinical features and treatment outcomes in steroid-resistant interstitial lung disease from polymyositis/dermatomyositis. Respir Med. 2013, 107, 890–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Brown, K.K.; Du Bois, R.M.; Frankel, S.K.; Cosgrove, G.P.; Fernandez-Perez, E.R.; Huie, T.J.; Krishnamoorthy, M.; Meehan, R.T.; Olson, A.L.; et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J. Rheumatol. 2013, 40, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Mahler, M.; Miller, F.W.; Fritzler, M.J. Idiopathic inflammatory myopathies and the anti-synthetase syndrome: A comprehensive review. Autoimmun. Rev. 2014, 13, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lega, J.-C.; Fabien, N.; Reynaud, Q.; Durieu, I.; Durupt, S.; Dutertre, M.; Cordier, J.-F.; Cottin, V. The clinical phenotype associated with myositis-specific and associated autoantibodies: A meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun. Rev. 2014, 13, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.; Thapamagar, S. Amyotrophic dermatomyositis. CMAJ 2017, 189, E164. [Google Scholar] [CrossRef] [PubMed]
- Labrador-Horrillo, M.; Martinez, M.A.; Selva-O’Callaghan, A.; Trallero-Araguas, E.; Balada, E.; Vilardell-Tarres, M.; Juarez, C. Anti-MDA5 antibodies in a large Mediterranean population of adults with dermatomyositis. J. Immunol. Res. 2014, 2014, 290797. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Kuwana, M. Clinically amyopathic dermatomyositis. Curr. Opin. Rheumatol. 2010, 22, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Lega, J.-C.; Reynaud, Q.; Belot, A.; Fabien, N.; Durieu, I.; Cottin, V. Idiopathic inflammatory myopathies and the lung. Eur. Respir. Rev. 2015, 24, 216–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, L.J.; Curran, J.J.; Strek, M.E. The Diagnosis and Treatment of Antisynthetase Syndrome. Clin. Pulm. Med. 2016, 23, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.C.; Casciola-Rosen, L.; Samedy, L.-A.; Werner, J.; Owoyemi, K.; Danoff, S.K.; Christopher-Stine, L. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: Expanding the clinical spectrum. Arthritis Care Res. 2013, 65, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, K.; Hagino, N. Dermatomyositis with Rapidly Progressive Interstitial Lung Disease Treated with Rituximab: A Report of 3 Cases in Japan. Intern. Med. 2017, 56, 1399–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hozumi, H.; Fujisawa, T.; Nakashima, R.; Johkoh, T.; Sumikawa, H.; Murakami, A.; Enomoto, N.; Inui, N.; Nakamura, Y.; Hosono, Y.; et al. Comprehensive assessment of myositis-specific autoantibodies in polymyositis/dermatomyositis-associated interstitial lung disease. Respir. Med. 2016, 121, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Harington, J.S. Investigative techniques in the laboratory study of coal workers’ pneumoconiosis: Recent advances at the cellular level. Ann. N. Y. Acad. Sci. 1972, 200, 816–834. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, T.; Mackler, A.D.; Langer, A.M.; Selikoff, I.J. Indetification and characterization of pulmonary dust burden in pneumoconiosis. Ann. Clin. Lab. Sci. 1973, 3, 118–131. [Google Scholar] [PubMed]
- Parkes, W.R. Asbestos-related disorders. Br. J. Dis. Chest 1973, 67, 261–300. [Google Scholar] [CrossRef]
- Moitra, S.; Puri, R.; Paul, D.; Huang, Y.-C.T. Global perspectives of emerging occupational and environmental lung diseases. Curr. Opin. Pulm. Med. 2015, 21, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Wyman, A.E.; Hines, S.E. Update on metal-induced occupational lung disease. Curr. Opin. Allergy Clin. Immunol. 2018, 18, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Chan-Yeung, M.; Wong, R.; MacLean, L.; Tan, F.; Dorken, E.; Schulzer, M.; Dennis, R.; Grzybowski, S. Respiratory survey of workers in a pulp and paper mill in Powell River, British Columbia. Am. Rev. Respir. Dis. 1980, 122, 249–257. [Google Scholar] [PubMed]
- Boehlecke, B.A.; Merchant, J.A. The use of pulmonary function testing and questionnaires as epidemiologic tools in the study of occupational lung disease. Chest 1981, 79 (Suppl. 4), 114S–122S. [Google Scholar] [CrossRef]
- Papali, A.; Hines, S.E. Evaluation of the patient with an exposure-related disease: The occupational and environmental history. Curr. Opin. Pulm. Med. 2015, 21, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Lee, K.S.; Chung, M.J.; Han, J.; Kwon, O.J.; Kim, T.S. Pneumoconiosis: Comparison of imaging and pathologic findings. Radiographics 2006, 26, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Stark, P.; Jacobson, F.; Shaffer, K. Standard imaging in silicosis and coal worker’s pneumoconiosis. Radiol. Clin. N. Am. 1992, 30, 1147–1154. [Google Scholar] [PubMed]
- Friedman, A.C.; Fiel, S.B.; Fisher, M.S.; Radecki, P.D.; Lev-Toaff, A.S.; Caroline, D.F. Asbestos-related pleural disease and asbestosis: A comparison of CT and chest radiography. AJR Am. J. Roentgenol. 1988, 150, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Fireman, E.; Haimsky, E.; Noiderfer, M.; Priel, I.; Lerman, Y. Misdiagnosis of sarcoidosis in patients with chronic beryllium disease. Sarcoidosis Vasc. Diffuse Lung Dis. 2003, 20, 144–148. [Google Scholar] [PubMed]
- Ribeiro, M.; Fritscher, L.G.; Al-Musaed, A.M.; Balter, M.S.; Hoffstein, V.; Mazer, B.D.; Maier, L.A.; Liss, G.M.; Tarlo, S.M. Search for chronic beryllium disease among sarcoidosis patients in Ontario, Canada. Lung 2011, 189, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.V.; Gotsch, A.R.; Brooks, S.; Landrigan, P.J.; Hankinson, J.L.; Merchant, J.A. Prevention of occupational lung disease. Task Force on Research and Education for the Prevention and Control of Respiratory Diseases. Chest 1992, 102 (Suppl. 3), 257S–276S. [Google Scholar] [CrossRef] [PubMed]
- Cummings, K.J.; Deubner, D.C.; Day, G.A.; Henneberger, P.K.; Kitt, M.M.; Kent, M.S.; Kreiss, K.; Schuler, C.R. Enhanced preventive programme at a beryllium oxide ceramics facility reduces beryllium sensitisation among new workers. Occup. Environ. Med. 2007, 64, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Litow, F.K.; Petsonk, E.L.; Bohnker, B.K.; Brodkin, C.A.; Cowl, C.T.; Guidotti, T.L.; Harber, P.; Biggs, J.J.; Hegmann, K.T. Occupational Interstitial Lung Diseases. J. Occup. Environ. Med. 2015, 57, 1250–1254. [Google Scholar] [PubMed]
- Redding, R.A.; Hardy, H.L.; Gaensler, E.A. Beryllium disease: A 16-year follow-up case study. Respiration 1968, 25, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.; Beckett, W.S.; Cullen, M.R. Variable response to long-term corticosteroid therapy in chronic beryllium disease. Chest 2004, 126, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Geddes, D.M.; Brostoff, J. Pulmonary fibrosis associated with hypersensitivity to gold salts. Br. Med. J. 1976, 1, 1444. [Google Scholar] [CrossRef] [PubMed]
- Hadjinicolaou, A.V.; Nisar, M.K.; Bhagat, S.; Parfrey, H.; Chilvers, E.R.; Ostor, A.J.K. Non-infectious pulmonary complications of newer biological agents for rheumatic diseases—A systematic literature review. Rheumatology 2011, 50, 2297–2305. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, O. Drug-induced interstitial lung disease: Mechanisms and best diagnostic approaches. Respir. Res. 2012, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Camus, P. Pneumotox On Line [Internet]. Available online: https://www.pneumotox.com/drug/index/ (accessed on 28 September 2018).
- Bledsoe, T.J.; Nath, S.K.; Decker, R.H. Radiation Pneumonitis. Clin. Chest Med. 2017, 38, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Bargagli, E.; Bonti, V.; Bindi, A.; Scotti, V.; Pistolesi, M.; Voltolini, L.; Ferrari, K. Fibrotic lung toxicity induced by cytotoxic drugs, radiation and immunotherapy in patients treated for lung cancer. Monaldi Arch. Chest Dis. 2018, 88, 917. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.E.; Erasmus, J.J.; McAdams, H.P.; Sporn, T.A.; Goodman, P.C. Pulmonary drug toxicity: Radiologic and pathologic manifestations. Radiographics 2000, 20, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Huggins, J.T.; Sahn, S.A. Drug-induced pleural disease. Clin. Chest Med. 2004, 25, 141–153. [Google Scholar] [CrossRef]
- Flieder, D.B.; Travis, W.D. Pathologic characteristics of drug-induced lung disease. Clin. Chest Med. 2004, 25, 37–45. [Google Scholar] [CrossRef]
- Ludwig, H.; Khayat, D.; Giaccone, G.; Facon, T. Proteasome inhibition and its clinical prospects in the treatment of hematologic and solid malignancies. Cancer 2005, 104, 1794–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizawa, K.; Mukai, H.Y.; Miyazawa, M.; Miyao, M.; Ogawa, Y.; Ohyashiki, K.; Katoh, T.; Kusumoto, M.; Gemma, A.; Sakai, F.; et al. Bortezomib therapy-related lung disease in Japanese patients with multiple myeloma: Incidence, mortality and clinical characterization. Cancer Sci. 2014, 105, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappasodi, P.; Dore, R.; Castagnola, C.; Astori, C.; Varettoni, M.; Mangiacavalli, S.; Lazzarino, M.; Corso, A. Rapid response to high-dose steroids of severe bortezomib-related pulmonary complication in multiple myeloma. J. Clin. Oncol. 2007, 25, 3380–3381. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Khunger, M.; Rakshit, S.; Pasupuleti, V.; Hernandez, A.V.; Mazzone, P.; Stevenson, J.; Pennell, N.A.; Velcheti, V. Incidence of Pneumonitis With Use of Programmed Death 1 and Programmed Death-Ligand 1 Inhibitors in Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis of Trials. Chest 2017, 152, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, T.F.; Shachar, S.S.; Nyrop, K.A.; Muss, H.B. Safety and Tolerability of PD-1/PD-L1 Inhibitors Compared with Chemotherapy in Patients with Advanced Cancer: A Meta-Analysis. Oncologist 2017, 22, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Widmann, G.; Nguyen, V.A.; Plaickner, J.; Jaschke, W. Imaging Features of Toxicities by Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Radiol. Rep. 2016, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Postow, M.; Lao, C.D.; Schadendorf, D. Management of Adverse Events Following Treatment With Anti-Programmed Death-1 Agents. Oncologist 2016, 21, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Diab, A.; Abdallah, K.; Bingham, B.C., 3rd; Dadu, R.; Hamad, L.; Kim, S.; Lacouture, M.E.; LeBoeuf, N.R.; Lenihan, D.; et al. Managing toxicities associated with immune checkpoint inhibitors: Consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J. Immunother Cancer. 2017, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M. Pathogenesis and natural history of usual interstitial pneumonia: The whole story or the last chapter of a long novel. Chest. 2005, 128 (Suppl. 1), 526S–532S. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Amatto, V.C.; Behr, J.; Stowasser, S. Comorbidities in idiopathic pulmonary fibrosis patients: A systematic literature review. Eur. Respir J. 2015, 46, 1113–1130. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Brown, K.K.; Bradford, W.Z.; Starko, K.; Noble, P.W.; Schwartz, D.A.; King, T.E.J. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2004, 350, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Demedts, M.; Behr, J.; Buhl, R.; Costabel, U.; Dekhuijzen, R.; Jansen, H.M.; MacNee, W.; Thomeer, M.; Wallaert, B.; Laurent, F.; et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2005, 353, 2229–2242. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.J.; Albera, C.; Bradford, W.Z.; Costabel, U.; Hormel, P.; Lancaster, L.; Noble, P.W.; Sahn, S.A.; Szwarcberg, J.; Thomeer, M.; et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): A multicentre, randomised, placebo-controlled trial. Lancet 2009, 374, 222–228. [Google Scholar] [CrossRef]
- Daniels, C.E.; Lasky, J.A.; Limper, A.H.; Mieras, K.; Gabor, E.; Schroeder, D.R. Imatinib treatment for idiopathic pulmonary fibrosis: Randomized placebo-controlled trial results. Am. J. Respir Crit. Care Med. 2010, 181, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Anstrom, K.J.; King, T.E.J.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [PubMed]
- Raghu, G.; Behr, J.; Brown, K.K.; Egan, J.J.; Kawut, S.M.; Flaherty, K.R.; Martinez, F.J.; Nathan, S.D.; Wells, A.U.; Collard, H.R.; et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann. Intern. Med. 2013, 158, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Mukae, H.; Sakamoto, N.; Kakugawa, T.; Yoshioka, S.; Soda, H.; Oku, H.; Urata, Y.; Kondo, T.; Kubota, H.; et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008, 82, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Hostettler, K.E.; Zhong, J.; Papakonstantinou, E.; Karakiulakis, G.; Tamm, M.; Seidel, P.; Sun, Q.; Mandal, J.; Lardinois, D.; Lambers, C.; et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir. Res. 2014, 15, 157. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E.J.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- King, T.E.J.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; du Bois, R.M.; Fagan, E.A.; Fishman, R.S.; Glaspole, I.; Glassberg, M.K.; Glasscock, K.F.; et al. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: Analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax 2016, 71, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.K.; Raghu, G. Inherited interstitial lung disease. Clin. Chest Med. 2004, 25, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Harari, S.; Torre, O.; Cassandro, R.; Moss, J. The changing face of a rare disease: Lymphangioleiomyomatosis. Eur. Respir. J. 2015, 46, 1471–1485. [Google Scholar] [CrossRef] [PubMed]
- Vourlekis, J.S. Acute interstitial pneumonia. Clin. Chest Med. 2004, 25, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Akira, M. Computed tomography and pathologic findings in fulminant forms of idiopathic interstitial pneumonia. J. Thorac. Imaging 1999, 14, 76–84. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ILD Subgroup | Medical History Clues | Radiographic Findings | Histologic Patterns | Autoimmune Serology Evaluation | Therapeutic Considerations |
|---|---|---|---|---|---|
| Smoking-Related | Prior or current tobacco smoking, biomass combustion exposure | GGO, emphysema with fibrosis, mosaic attenuation | RB, DIP, PLCH, emphysema, fibrosis | Normal or non-reactive | Smoking cessation, removal from exposure, anti-inflammatory therapy in advanced disease |
| Hypersensitivity Pneumonitis | Exposure to environmental organic antigens (avian proteins, mold, farming-related) | GGO, mosaic attenuation, upper-lobe fibrosis, centrilobular nodules | Poorly-formed granulomas with multinucleated giant cells | Normal or non-reactive; consider serum precipitating antibody panel | Removal from exposure, remediation of environment, anti-inflammatory and/or immunosuppressive therapy in advanced disease |
| Connective Tissue Disease-Related | Constitutional symptoms, arthralgia/arthritis, rash, Raynaud’s phenomenon | GGO, reticular opacities, consolidation, fibrosis, peripheral sparing | Chronic inflammation, UIP, NSIP, or OP | Autoimmune evaluation recommended | Anti-inflammatory and/or immunosuppressive therapy |
| Occupation-Related | Current or prior at-risk occupation | GGO, reticular opacities, fibrosis, nodules, pleural plaques (asbestos) | Chronic inflammation, UIP, NSIP, OP, or granulomatous inflammation | Normal or non-reactive; lymphocyte proliferation test if beryllium exposure suspected | Removal from exposure, use of respiratory protective equipment |
| Medication-Induced | Current or prior use of possible offending medications or therapies | GGO, reticular opacities, consolidation, nodules, fibrosis | Chronic inflammation, UIP, NSIP, OP, or granulomatous inflammation | Usually normal or non-reactive | Medication discontinuation if possible, anti-inflammatory and/or immunosuppressive therapy in advanced disease |
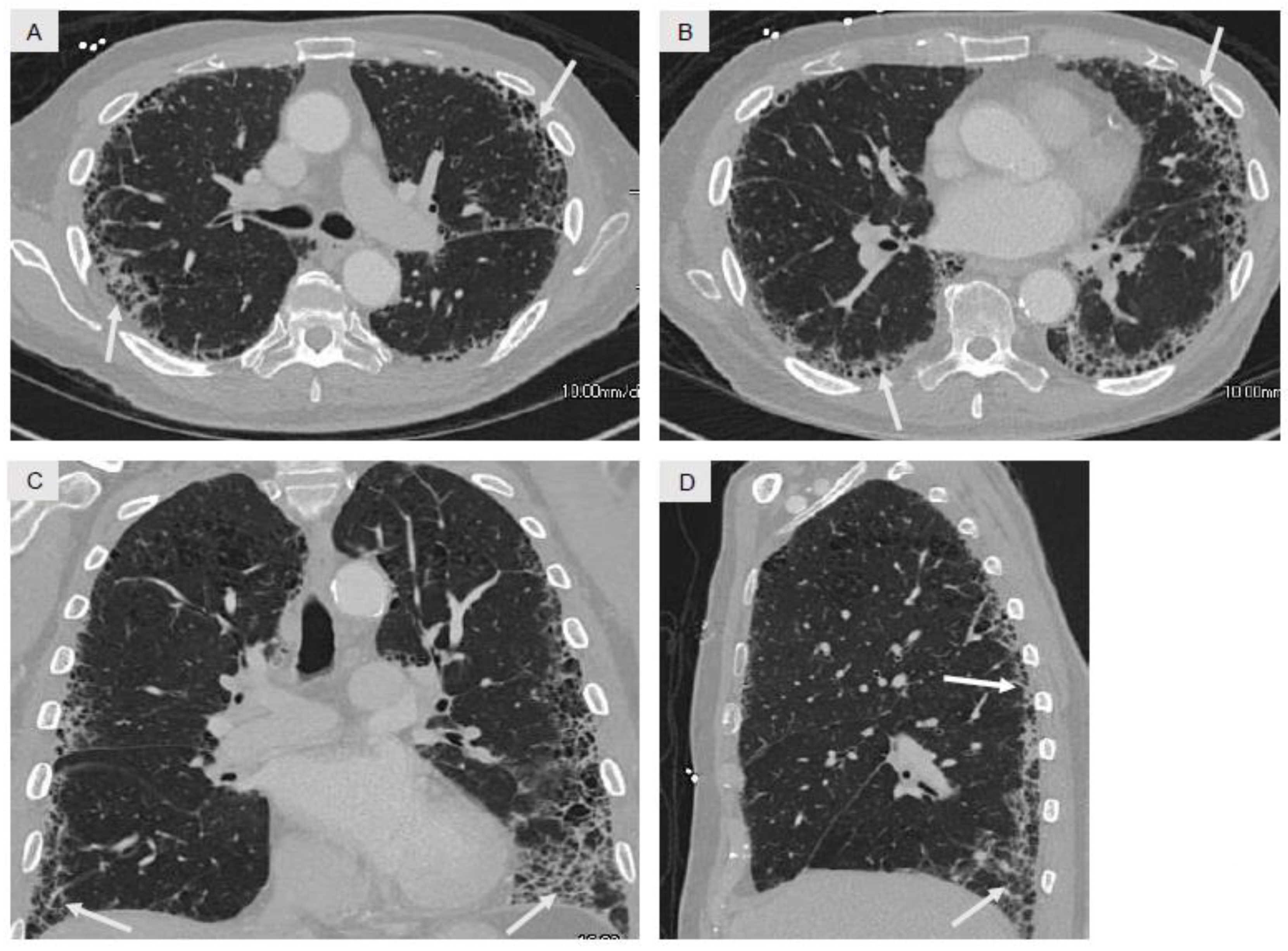
| Idiopathic Pulmonary Fibrosis | Male predominance, 7th–8th decade, past history of tobacco smoking or dust exposure | Peripheral and basilar fibrosis with honeycombing (Figure 4) | UIP | Normal or non-reactive | Consideration of antifibrotic therapies, lung transplantation evaluation in selected patients |
| Serum Markers of Systemic Inflammation | |||
| ● erythrocyte sedimentation rate (ESR) | |||
| ● c-reactive protein (CRP) | |||
| Autoantibodies | |||
| Anti-neutrophil cytoplasmic antibodies (ANCAs) | |||
| ● c-ANCA, p-ANCA, atypical p-ANCA | |||
| ● anti-MPO | |||
| ● anti-PR3 | |||
| Rheumatoid arthritis | |||
| ● anti-CCP | |||
| ● rheumatoid factor (RF) | |||
| SLE and related autoimmune diseases (MCTD, Sjögren’s syndrome) | |||
| ● ANA | |||
| ● anti-dsDNA | |||
| ● anti-SS-A (Ro) | |||
| ● anti-SS-B (La) | |||
| ● anti-Smith | |||
| ● anti-RNP | |||
| Systemic sclerosis | |||
| ● anti-Scl-70 | |||
| ● anti-Centromere | |||
| Myositis-specific and myositis-associated antibodies | |||
| ● anti-Jo1 | ● anti-PL7 | ● anti-PL12 | ● anti-EJ |
| ● anti-OJ | ● anti-SRP | ● anti-Mi-2α | ● anti-Mi-2β |
| ● anti-MDA5 | ● anti-TIF-1γ | ● anti-NXP-2 | ● anti-KS |
| ● anti-Zo | ● anti-Ku | ||
| Muscle Enzymes | |||
| ● CPK | |||
| ● aldolase | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalchiem-Dekel, O.; Galvin, J.R.; Burke, A.P.; Atamas, S.P.; Todd, N.W. Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History. J. Clin. Med. 2018, 7, 476. https://doi.org/10.3390/jcm7120476
Kalchiem-Dekel O, Galvin JR, Burke AP, Atamas SP, Todd NW. Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History. Journal of Clinical Medicine. 2018; 7(12):476. https://doi.org/10.3390/jcm7120476
Chicago/Turabian StyleKalchiem-Dekel, Or, Jeffrey R. Galvin, Allen P. Burke, Sergei P. Atamas, and Nevins W. Todd. 2018. "Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History" Journal of Clinical Medicine 7, no. 12: 476. https://doi.org/10.3390/jcm7120476