Novel Evidence of Acute Kidney Injury in COVID-19


1
Department of Medical Laboratory, Cardinal-Tien Hospital, New Taipei City 231, Taiwan
2
Department of Education, Cardinal Tien Junior College of Healthcare and Management, New Taipei City 231, Taiwan
3
Research Center of Urology and Kidney, Taipei Medical University, Taipei 110, Taiwan
4
Division of Nephrology, Department of Internal Medicine, Taipei Medical University, Shuang Ho Hospital, Ministry of New Taipei City 235, Taiwan
5
Division of Nephrology, Department of Internal Medicine, School of Medicine, College of Medicine, Taipei Medical University, Taipei 110, Taiwan
6
Division of Nephrology, Department of Medicine, Cardinal-Tien Hospital, New Taipei City 231, Taiwan
7
School of Medicine, Fu Jen Catholic University, New Taipei City 242, Taiwan
8
Division of Nephrology, Department of Medicine, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei City 231, Taiwan
*
Author to whom correspondence should be addressed.
†
Contributed equally to this review article.
J. Clin. Med. 2020, 9(11), 3547; https://doi.org/10.3390/jcm9113547
Submission received: 2 October 2020
/
Revised: 29 October 2020
/
Accepted: 2 November 2020
/
Published: 3 November 2020
(This article belongs to the Special Issue Coronavirus Disease 2019 (COVID-19) Pandemic: Current Knowledge and Future Perspectives)
Abstract
:The coronavirus 2019 (COVID-19) pandemic has caused a huge impact on health and economic issues. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes cellular damage by entry mediated by the angiotensin-converting enzyme 2 of the host cells and its conjugation with spike proteins of SARS-CoV-2. Beyond airway infection and acute respiratory distress syndrome, acute kidney injury is common in SARS-CoV-2-associated infection, and acute kidney injury (AKI) is predictive to multiorgan dysfunction in SARS-CoV-2 infection. Beyond the cytokine storm and hemodynamic instability, SARS-CoV-2 might directly induce kidney injury and cause histopathologic characteristics, including acute tubular necrosis, podocytopathy and microangiopathy. The expression of apparatus mediating SARS-CoV-2 entry, including angiotensin-converting enzyme 2, transmembrane protease serine 2 (TMPRSS2) and a disintegrin and metalloprotease 17 (ADAM17), within the renal tubular cells is highly associated with acute kidney injury mediated by SARS-CoV-2. Both entry from the luminal and basolateral sides of the renal tubular cells are the possible routes for COVID-19, and the microthrombi associated with severe sepsis and the dysregulated renin–angiotensin–aldosterone system worsen further renal injury in SARS-CoV-2-associated AKI. In the podocytes of the glomerulus, injured podocyte expressed CD147, which mediated the entry of SARS-CoV-2 and worsen further foot process effacement, which would worsen proteinuria, and the chronic hazard induced by SARS-CoV-2-mediated kidney injury is still unknown. Therefore, the aim of the review is to summarize current evidence on SARS-CoV-2-associated AKI and the possible pathogenesis directly by SARS-CoV-2.
1. Introduction
The coronavirus disease 2019 (COVID-19) pandemic has had severe public health and economic impacts as a result of its rapid spread and association with severe acute respiratory distress syndrome. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) can lead to severe sepsis and systemic inflammation, which can induce multiple organ dysfunction. Acute kidney injury (AKI) is predictive of mortality and deteriorating organ dysfunction in patients with SARS-CoV-2 [1,2]. The most common risk factors related to SARS-Cov-2-mediated AKI include diabetes, obesity or hypertension and previous chronic kidney disease (CKD) [3]. Angiotensin-converting enzyme 2 (ACE2) and an assembly protein are known requirements for the entry of the virus into host cells. In patients with hypertension, diabetes mellitus and CKD, renin–angiotensin–aldosterone system activation is associated with ACE2 deficiency, which worsens renal tubulointerstitial fibrosis [4]. The modified phenotype of podocyte might facilitate SARS-CoV-2 entry into the reno-tubular system and worsen kidney damage. Moreover, SARS-CoV-2-associated sepsis might worsen AKI through the induction of a cytokine storm or hemodynamic dysregulation [5], but the pathogenesis and understanding of SARS-CoV-2-mediated kidney injury is still lacking. This review investigated the possible molecular mechanisms of acute kidney injury directly mediated by SARS-CoV-2.
2. High Levels of Angiotensin-Converting Enzyme 2 and Transmembrane Protease Serine 2 Expression in the Urinary System
According to data published by the Genotype-Tissue Expression project, ACE2 is highly expressed within the kidneys [6]. ACE2 is distributed throughout multiple cells within the kidneys and is most concentrated in proximal tubule cells [7]. Fan et al. suggested that specific ACE2 expression is high within proximal tubular cells but is not observed in immune cells or glomerular parietal epithelial cells [8]. Suryawanshi et al. analyzed the data of kidney tissues in single-cell RNA sequencing datasets and found that proximal tubular cells in the kidney co-express ACE2 and transmembrane protease serine 2 (TMPRSS2) [9]. Studies have reported high levels of ACE2 and TMPRSS2 expression in proximal renal tubular epithelial cells and kidney podocytes [10,11,12], and such characteristics facilitate the SARS-CoV replication within these cells [13].
3. TMPRSS2 and a Disintegrin and Metalloprotease 17 (ADAM17) Protect Against Viruses in Host Proteases
ACE2 within the cell membrane is important for SARS-CoV-2 entry into host cells [14]. However, in severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) or SARS-CoV-2 infection, poor prognosis is reportedly associated with ACE2 downregulation [15]. Theoretically, fewer viral entry routes should correspond to improved clinical outcomes. However, studies have demonstrated that decreased ACE2 expression levels lead to greater illness severity and more serious end-organ damage because ACE2 plays an anti-inflammatory role under renin–angiotensin–aldosterone system (RAAS) activation [16]. Classical RAAS involves the conversion of Angiotensin (Ang) I to Ang II and AngII binding with angiotensin-1 receptor (AT1R). The classic RAAS pathway involves vasoconstriction, oxidative stress, inflammation and fibrosis. The non-classical RAAS pathway, on the other hand, converts AngII to Ang(1–7) by ACE2, and the binding of Ang(1–7) with Mas receptor (MasR) provides vasodilatation and an anti-inflammatory effect [17]. Viral invasion into host cells occurs primarily through the ACE2 receptor; this receptor may mediate the entry of SARS-CoV-2 into host cells through two distinct routes. The first involves clathrin-dependent endocytosis and the second involves ACE2 receptor–mediated TMPRSS2-dependent membrane fusion [18].
ACE2 downregulation occurs as these proteins are shed from cell membranes and circulated throughout the body. The end product of ACE2 cleavage mediated by a disintegrin and metalloprotease 17 (ADAM17) and TMPRSS2 may play a protective role against SARS-CoV-2 entry. In SARS-CoV-2, this occurs as a result of the spike protein activating ACE2 expression by seizing two host proteases: TMPRSS2, which facilitates viral entry by cleaving the S antigen into S1 (the active binding site), and ADAM17, which downregulates ACE2 expression by shedding ACE2 proteins into soluble form. The soluble ACE2 directly attached virus within circulation and decreased SARS-CoV-2 entry [19,20]. Research has indicated that ACE2 within human embryonic kidney cells can be shed by ADAM17 and protect against SARS-S entry [21]. In the human embryonic kidney cell line, decreased ACE2 expression is responsible for SARS-CoV-2 complications and end-organ damage and may cause greater harm to the host through enhanced ACE2 toxic effects, such as the activation of proinflammatory cytokines [22]. Heurich et al. suggested that TMPRSS2 and ADAM17 participate in the differential internalization of SARS-S [23]. TMPRSS2 cleaves ACE2 in arginine and lysine residues within amino acids 697 to 716, and the cleaved ACE2 is essential for SARS-S entry. Entry through TMPRSS2 is unrelated to ADAM17 because TRPMSS2 competes with ADAM17. Therefore, ADAM17 was proposed to participate in ACE2 ectodomain shedding, and TMPRSS2 enhances the intracellular cleavage of ACE2; thus, ADAM17 and TMPRSS2 compete during ACE2 processing. Hoffmann et al. demonstrated that TMPRSS2 inhibitors can block SARS-S cell entry through S protein processing [24]. Research has indicated that recombinant human ACE2 (rhACE2) mimics soluble ACE2 and inhibits viral invasion into host cells through competitive binding to CoV with the ACE2 lying in cellular membrane. The circulatory rhACE2-CoV2 binding decreases the internalization of membranous ACE2. Therefore, the non-classical RAAS provides further protection by alleviating intracellular inflammation [25]. Monteil et al. also used rhACE2 to block SARS-CoV-2 entry into the kidney organoid, demonstrating the possible protective effect of cleaved ACE2 against COVID-19 entry into host cells [26]. Therefore, in addition to its absolute expression, the cleavage and shedding of ACE2 could influence COVID-19 entry into cells (Figure 1).
However, more studies are necessary to clarify the role of ADAM17 and other proteases on ACE2 shedding in the kidney and the importance of these proteases in SARS-CoV-2 infection [27].
4. SARS-CoV-2 Invades Host Cells Through a Novel Route: CD147-Spike Protein
Research on kidney epithelial cells (Vero E6 cells) has demonstrated that spike proteins bound to CD147 can mediate SARS-CoV-2 host invasion. The localization of CD147 and spike protein was observed in SARS-CoV-2–infected cells through an immunoelectron microscope, and the discovery of the new CD147-SP route for SARS-CoV-2 invasion of host cells has provided a critical target for the development of a specific antiviral medicine [28].
CD147 (Extracellular Matrix Metalloproteinase Inducer/Basigin) in Kidney Diseases
Basigin (Bsg)/CD147, also known as an extracellular matrix metalloproteinase (MMP) inducer (EMMPRIN), is the glycosylated transmembrane protein governing cell survival, cell migration and cancer invasion [29]. In SARS-CoV-2–mediated myocardial injury, EMMPRIN facilitates viral entry into the cardiomyocyte and induces the release of cytokines, such as interleukin-18 [30]. Moreover, the reactive oxygen species generated during inflammation dysregulate myocardial remodeling [31]. Atrial fibrillation is more inducible after SARS-CoV-2 entry and the downstream release of cytokines [32]. Wang et al. reported that the high affinity of CD147 and the spike protein to SARS-CoV-2 mediates viral entry into the kidney epithelium [28]. In normal kidneys, CD147 is highly expressed only on the basolateral side of tubular epithelial cells [29]. In the case of viremia, SARS-CoV-2 might invade renal proximal tubular epithelial cells through both the luminal surface and the basolateral side (Figure 2). Kato et al. demonstrated that Bsg/CD147 was associated with neutrophil recruitment in renal tubules with ischemic/reperfusion injury. In Bsg-deficient [Bsg(−/−)] mice, neutrophilic infiltration is suppressed after renal ischemic/reperfusion injury. Bsg/CD147 is a crucial physiologic ligand for E-selectin and therefore facilitates neutrophilic infiltration [33]. In an animal model of unilateral urinary obstruction, Bsg/CD147 promoted renal fibrosis by inducing MMP and hyaluronan expression. In a primary culture of the renal tubules of Bsg(−/−) mice, MMP expression was less responsive to transforming growth factor β [34].
The aforementioned research findings suggest that SARS-CoV-2 targets the renal tubules by entering from the basolateral side in adjunction with CD147. Yoshioka et al. reported that Bsg/CD147 is rarely expressed in healthy podocytes. In adriamycin-induced nephropathy, increases in Bsg expression and proteinuria are mediated by increases in focal adhesion kinase signaling [35]. In their clinical study, Musial et al. reported the serum concentrations of CD147 along with other fibrosis markers, such as transforming growth factor beta (TGF-β) levels, in children with CKD [36]. The findings indicated that patients with CKD may exhibit variations in CD147 expression that may contribute to pathological changes.
5. COVID-19 Prevents ACE2 from Converting ANG II into ANG I–VII and Increases Intracellular ANG II and Membrane ADAM17 Expression
Cytosolic ACE2 is a negative regulator for the activation of ACE and Ang I [37]. The carboxypeptidase ACE2 converts Ang II into Ang (1–7) and Ang I into Ang (1–9), thus preventing the conversion of Ang I into Ang II [38]. Under normal conditions, endothelial cells synthesize tissue factors and inhibitors of thrombosis to maintain immune and coagulation homeostasis [39]. COVID-19 enters cells through ACE2-mediated assembly. The inability of ACE2 to convert Ang I or Ang II when paired with COVID-19 results in endothelial senescence, caused by the upregulation of interleukin 6 and oxidative stress induced by disturbances in mitochondrial function [40]. In the hypoxia and sepsis, the un-countered AT1R activation by Ang II worsens the vasoconstriction and downstream fibrosis within lung tissue [37]. The excess Ang I and II induces immune and coagulation abnormalities and dysfunction. COVID-19/ACE2 complexes enter host cells through endocytosis, and those that are not endocytosed are shed by ADAM17 [40]. Increases in renal ADAM17 expression are known to mediate Ang II-induced growth of renal lesions in patients with CKD. The binding of Ang II to its AT1 receptor increases cytosolic ADAM17 expression and its translocation to the plasma membrane. In the vascular system, excessive ADAM17 expression induces the shedding of ACE2 and worsens further activation of the classical RAAS and inflammation [41]. ADAM17 induces the shedding of ACE2 and increases circulatory ACE2 (sACE2), thereby mitigating further entry of COVID-19 [42]. Additionally, ADAM17 modulates the shedding of transforming growth factor-α (TGF-α) from its transmembrane precursor and activates the epidermal growth factor receptor (EGFR), causing proteinuria, tubular hyperplasia, fibrosis and mononuclear cell infiltration [43]. TGF-α/EGFR-driven vitamin D receptor reduction impairs 1,25(OH)2D/VDR renoprotection [44]. ADAM17 is also expressed within the renal tubules, and its activation, mediated by classical RAAS activation, may prevent the COVID-19 shed by sACE2 from entering tubular cells [45]. However, this further activation of the classical RAAS may worsen the hypercoagulability of COVID-19 infection, thereby increasing the extent of tubular damage and the risk of damage to the interstitium.
6. Pathological Changes in AKI Mediated by COVID-19
As mentioned, COVID-19 can induce multiorgan dysfunction through direct transmission into cells via ACE2 or through the activation of a cytokine storm, which leads to the recruitment of inflammatory cells and activates apoptosis. AKI is common among patients with SARS-CoV-2, affecting up to 37%, and may occur concomitantly with the initiation of mechanical ventilation [46]. The mechanisms of AKI induced by SARS-CoV-2 also involve hemodynamic factors, direct cellular injury caused by SARS-CoV-2 invasion through ACE2 or the cytokine storm induced by SARS-CoV-2, because the SARS-CoV-2 could not be detected in the postmortem autopsy or in the graft of renal transplantation patients when using RNA in-situ hybridization or by immunochemistry for nucleocapsid [47,48,49]. Varga et al. suggested that SARS-CoV-2 is detectable within endothelial cells, reporting that in postmortem autopsy specimens, endotheliitis associated with inflammatory cell accumulation was common in different organs [50]. Pei et al. suggested that intrinsic AKI, rather than post- or pre-renal AKI, is the most common characteristic of renal involvement in patients with SARS-CoV-2 [51]. Therefore, understanding the role of AKI in COVID-19 is crucial for elucidating the pathology and clinical manifestations of COVID-19 (the summary is listed in the Table 1).
6.1. Acute Tubular Necrosis
Acute tubular necrosis is the most common pathological change observed in patients with COVID-19 who have AKI [52,53,54]. Hemodynamic instability, such as in patients with cardiorenal syndrome preceded by myocardial injury [5] or a cytokine storm induced by SARS-CoV-2 [55], reduces renal perfusion, and up to 80% of patients with acute tubular necrosis (ATN) require dialysis [52]. In patients with COVID-19 and ATN, proximal tubular cells might be the most susceptible tubular cells because of their high expression of ACE2 [53,56]. Werion et al. reported that tubular lumen dilatation with cellular debris, accompanied by changes in the brush border membranes of proximal tubules and moderate proteinuria, is a hallmark of COVID-19 involvement in the kidneys. In addition to increases in urinary β2-microglobulin or albumin, up to 46% of patients exhibit aminoaciduria [53]. Singer et al. demonstrated the interaction between amino acid transporter B0AT1 and ACE2 [57]. Altogether, these research findings highlight the importance of monitoring renal function in patients with COVID-19 recovering from AKI. In sum, the renal tubules, especially proximal tubules, carry CD147 within the basolateral side. The direct entry of SARS-CoV-2 through CD147 activates the partner protein of CD147, such as cyclophilins and integrins [54]. Cyclophilin then arouses the inflammation and worsens renal tubular damage [58]. Besides, the membrane attacking complex (MAC) has been detected within the renal tubules, which might be triggered by virus-mediated innate immunity [59,60,61].
6.2. Acute Interstitial Nephritis
Acute interstitial nephritis (AIN) is associated with the infiltration of inflammatory cells within the interstitium. The newly exposed antigen or hapten provokes inflammatory processes within tissues by recruiting cytotoxic T cells and antibody-producing B cells [62]. In patients with COVID-19, the infiltration of lymphocytes and macrophages within the tubulointerstitium were common when acute tubular necrosis developed [59]. According to the consensus report from Acute Disease Quality Initiative, the most common etiology of acute interstitial nephritis might be related to antibiotics or other nephrotoxic agents [3]. From the study by Diao et al., the CD68+ macrophages were the major inflammatory cells, whereas the role of CD4+ T cells or natural killer cells was less dominant [59]. Westhoff et al. reported the case of a patient receiving pancreas–kidney transplantation who developed AKI after COVID-19 infection; renal biopsy revealed mononuclear cell infiltration within the interstitium and in situ hybridization of SARS-CoV-2 [63]. Zhu et al. revealed the specific spectrum of immunity in patients with COVID-19. In single-cell sequencing of peripheral mononuclear cells, patients with COVID-19 had highly specific activation of STAT1 and IRF3 compared with patients with influenza A infection [64]. STAT-1 overexpression has been reported to be associated with tubulointerstitial fibrosis and epithelial to mesenchymal transition [65,66]. Moreover, one study demonstrated that the expression of CXCL-10+/CCL2+ macrophages induced by interferon gamma and tumor necrosis factor alpha might be related to tissue inflammation [67]. CD147 expression is also predictive of specific autoimmune-mediated nephritis. In sum, the CD147-mediated immune response in patients with COVID-19 is a possible pathogenesis of AIN [68].
6.3. Podocytopathy/Collapsing Focal Segmental Glomerulosclerosis
Along with the acute tubular necrosis with proximal tubular vacuolization, the podocyte could be a target for SARS-CoV-2 entry (Figure 3). Kissling et al. reported the case of an African male patient with AKI after SARS-CoV-2 infection; light microscopy revealed collapsing focal segmental sclerosis, and electron microscopy (EM) revealed viral inclusion within the vacuoles of the podocyte [69], in contrast to the EM result reported by Magoon et al. [70]. The spectrum of COVID-19-mediated podocytopathy remained unclear until the case report by Gupta et al. of a patient with repeat renal biopsy within 2 weeks. According to the report, an Asian male patient with an initial clinical presentation of nephrotic syndrome transitioned from minimal change disease to collapsing focal segmental glomerulosclerosis (FSGS) [71]. The mechanisms of collapsing FSGS and podocytopathy are still under investigation, but possibilities include cytokine storms and microRNA regulation [72]. High-risk groups for FSGS include patients who are APOL1 carriers [48]. Whether SARS-CoV-2-associated FSGS and human immunodeficiency virus-associated nephropathy (HIVAN) share the same pathomechanism is unknown. In patients with HIVAN, the loss of differentiating ability of podocyte gives the glomerulus a pseudo-crescent pattern, which involves collapsing FSGS with parietal epithelial cells [73,74]. ACE2 within the podocyte is known to play a protective role against specific renal diseases, such as diabetic nephropathy [75,76]; however, it remains unknown whether the polymorphism of ACE2 is predictive of SARS-CoV-2–associated FSGS or nephrotic syndrome. As mentioned, the expression of Bsg/CD147 is only increased in injured podocytes (Figure 3). In patients with a high risk of podocytopathy, such as those with diabetic nephropathy or obesity-related FSGS, the increased expression of CD147 in the podocyte might facilitate the entry of SARS-CoV-2 and increase injury [77].
6.4. Microangiopathy
COVID-19-associated coagulopathy has been widely reported to induce hypoxia, and downstream disseminated intravascular coagulation resulting from severe inflammation is a potential mechanism [78,79]. Thrombotic microangiopathy has been documented in patients with AKI and COVID-19. Jhaveri et al. noted microthrombi within the glomeruli in a patient with acute respiratory distress syndrome (ARDS) and AKI that was refractory to eculizumab treatment [80]. Mostly, the coagulation cascade is aroused by the SARS-CoV-2-mediated innate immunity, where MAC forms systemic thrombi/fibrin [60]. In the initial phase of the disease outbreak, a correlation was noted between SARS-CoV-2 and antiphospholipid antibodies. Zhang et al. noted the presence of anticardiolipin immunoglobulin A (IgA) antibodies and anti–β2-glycoprotein I IgA and IgG antibodies in patients with COVID-19 who had hypoxemic respiratory failure [81]. By contrast, Siguret et al. reported the presence of antiphospholipid antibodies in 12% of patients with COVID-19 [82]. Researchers have suggested that IgG in patients of COVID-19 could recruit neutrophilic aggregation with a similar ability to that of antiphospholipid antibody; moreover, a high incidence of thrombotic events in the inferior vena cava was noted in mice treated with the IgG of patients with COVID-19 [83]. In sum, activation of the classical RAAS and insufficient ACE2 may promote hypercoagulability [84,85]. It has been noticed that MAC was strongly detected in the tubules, but the involvement of MAC within the glomerulus or endothelium was less obvious [59,61]. However, MAC activity might triggered by the virus-mediated innate immunity within the endothelium [60]. Therefore, thrombotic microangiopathy within the kidney requires further investigation to provide a potential therapeutic target in COVID-19 patients with AKI.
7. ACEi/ARB Use and SARS-CoV-2-Associated AKI: Current Evidence
As explained in the previous section, ACE/Ang I directly activates vasoconstriction and downstream inflammation within the tissue. The application of ACEi/ARB could increase cytosolic ACE2 generation and lessen vasoconstriction and downstream tissue fibrosis [86]. In the scenario of acute kidney injury, the glomerular efferent arteriolar constriction mediated by Ang II is crucial for maintaining glomerular filtration during hemodynamic instability or vasodilatory shock [87,88]. From the report of Hirsch et al., chronic use of ACEi/ARB was a risk of acute kidney injury during SARS-CoV-2 infection [46]. The possible protective role of ACEi/ARB in lessening SARS-CoV-2 entry into lung tissue has been notified. Milne et al. demonstrated that in ACEi users, ACE2 and TMPRSS2 gene expression were lower than in non-ACEi users in the lung tissue [89], which was contrary with the increased ACE2 expression in the selected animal model [90,91]. Recently, Wysocki et al. provided in vivo evidence of the altered ACE2 expression in renal tubular cells after been treated with ACEi/ARB. The expression of ACE2 within apical membrane decreased in the ACEi/ARB-treated mice due to the internalization into cytosol [92]. Therefore, ACEi/ARB might not potentiate the hazard or disease severity in the SARS-CoV-2 infection. From the meta-analysis by Liu et al., patients with ACEi/ARB had lower disease severity and lower all-cause mortality [93]. Therefore, ACEi/ARB should only be discontinued if contraindications occur in the patients with SARS-CoV-2, such as hyperkalemia or unstable hemodynamics [94].
8. Conclusions
The COVID-19 pandemic has prompted the need for further understanding of how daily habits can influence multiple organ dysfunction by inducing severe inflammation or the direct invasion of SARS-CoV-2 into specific organs. Cell entry is mediated by ACE2 expression, the modulation of surface protein CD147 expression and the interplay between ADAM17 and TMPRSS2, which are commonly found in renal tubules. In SARS-CoV-2-associated AKI, virus-specific treatment might mitigate further cellular injury by influencing the protein associated with viral entry. Chronic follow-up for renal function in patients after SARS-CoV-2 infection should be important.
Author Contributions
Y.-C.H. drafted the manuscript. C.-M.Z. and T.-I.C. designed and corrected the figure. K.-C.L. confirmed the correction and the structure of the study. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACE2 | Angiotensin-Converting Enzyme 2 |
| ADAM17 | A Disintegrin and Metalloprotease 17 |
| AKI | Acute Kidney Injury |
| Ang | Angiotensin |
| APOL1 | Apolipoprotein L1 |
| ARDS | Acute Respiratory Distress Syndrome. |
| AT1R | Angiotensin Type 1 Receptor |
| COVID-19 | Coronavirus 2019 |
| C/EBPβ | CAAT-Enhancer Binding Protein-B |
| FSGS | Focal Segmental Glomerular Sclerosis |
| IRF3 | IFN Regulatory Factor 3 |
| MasR | Mas Receptor |
| MMP | Matrix Metalloproteinase |
| NEP | Neprilysin |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| SHPT | Secondary Hyperparathyroidism |
| STAT1 | Signal Transducer and Activator of Transcription 1 |
| TGF-α | Transforming Growth Factor-A |
| TMPRSS2 | Transmembrane Protease Serine 2 |
References
- Chen, Y.-T.; Shao, S.-C.; Lai, E.C.-C.; Hung, M.-J.; Chen, Y.-C. Mortality rate of acute kidney injury in SARS, MERS, and COVID-19 infection: A systematic review and meta-analysis. Crit. Care 2020, 24, 1–4. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, W.; Wu, L.; Cai, G. SARS-CoV-2-Related kidney injury: Current concern and challenges. SN Compr. Clin. Med. 2020, 1–10. [Google Scholar] [CrossRef]
- Nadim, M.K.; Forni, L.G.; Mehta, R.L.; Connor, M.J.; Liu, K.D.; Ostermann, M.; Rimmelé, T.; Zarbock, A.; Bell, S.; Bihorac, A.; et al. COVID-19-associated acute kidney injury: Consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup. Nat. Rev. Nephrol. 2020. [Google Scholar] [CrossRef]
- Nangaku, M.; Fujita, T. Activation of the renin-angiotensin system and chronic hypoxia of the kidney. Hypertens. Res. 2008, 31, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Li, K.; Ding, Y.; Lu, W.L.; Wang, J. ACE2 Expression in Kidney and Testis May Cause Kidney and Testis Damage After 2019-nCoV Infection. medRxiv 2020. [Google Scholar] [CrossRef]
- Suryawanshi, H.; Morozov, P.; Tuschl, T.; Muthukumar, T.; Tenoever, B.R.; Yamaji, M.; Williams, Z. Cell-Type-Specific Expression of Renin-Angiotensin-System Components in the Human Body and Its Relevance to SARS-CoV-2 Infection. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.-W.; Xu, D.; Zhang, H.; Zhou, W.; Wang, L.-H.; Cui, X.-G. Identification of a potential mechanism of acute kidney injury during the COVID-19 outbreak: A study based on single-cell transcriptome analysis. Intensiv. Care Med. 2020, 46, 1114–1116. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.; Zhang, J.; Ma, X.; Tan, J.; Chen, L.; Liu, S.; Xin, Y.; Zhuang, L. ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19. Biomed. Pharmacother. 2020, 131, 110678. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zhou, Y.; Hua, J.; Zhang, L.; Bian, J.; Liu, B.; Zhao, Z.; Jin, S. The scRNA-seq expression profiling of the receptor ACE2 and the cellular protease TMPRSS2 reveals human organs susceptible to COVID-19 infection. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Pacciarini, F.; Ghezzi, S.; Canducci, F.; Sims, A.; Sampaolo, M.; Ferioli, E.; Clementi, M.; Poli, G.; Conaldi, P.G.; Baric, R.; et al. Persistent replication of severe acute respiratory syndrome coronavirus in human tubular kidney cells selects for adaptive mutations in the membrane protein. J. Virol. 2008, 82, 5137–5144. [Google Scholar] [CrossRef] [Green Version]
- Kai, H.; Kai, M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens. Res. 2020, 43, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Liu, B.; Zhang, S. COVID-19: The role of excessive cytokine release and potential ACE2 down-regulation in promoting hypercoagulable state associated with severe illness. J. Thromb. Thrombolysis 2020, 1–17. [Google Scholar] [CrossRef]
- AlGhatrif, M.; Cingolani, O.; Lakatta, E.G. The Dilemma of Coronavirus Disease 2019, Aging, and Cardiovascular Disease. JAMA Cardiol. 2020, 5, 747. [Google Scholar] [CrossRef] [Green Version]
- Chappell, M.C. Nonclassical renin-angiotensin system and renal function. Compr. Physiol. 2012, 2, 2733–2752. [Google Scholar] [CrossRef] [Green Version]
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J.M. Trilogy of ACE2: A peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 2010, 128, 119–128. [Google Scholar] [CrossRef]
- Brill, A.; Chauhan, A.K.; Canault, M.; Walsh, M.T.; Bergmeier, W.; Wagner, D.D. Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc. Res. 2009, 84, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, D.W.; Yarski, M.A.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [Green Version]
- Luther, J.M.; Gainer, J.V.; Murphey, L.J.; Yu, C.; Vaughan, D.E.; Morrow, J.D.; Brown, N.J. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 2006, 48, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2013, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Deng, W.; Li, S.; Yang, X. Advances in research on ACE2 as a receptor for 2019-nCoV. Cell. Mol. Life Sci. 2020, 1–14. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Palau, V.; Riera, M.; Soler, M.J. ADAM17 inhibition may exert a protective effect on COVID-19. Nephrol. Dial. Transplant. 2020, 35, 1071–1072. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kosugi, T.; Maeda, K.; Sato, W.; Maruyama, S.; Kadomatsu, K. CD147 (EMMPRIN/Basigin) in kidney diseases: From an inflammation and immune system viewpoint. Nephrol. Dial. Transpl. 2014, 30, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, B.; Valente, A.J.; Prabhu, S.D.; Shanmugam, P.; Delafontaine, P.; Chandrasekar, B. EMMPRIN activates multiple transcription factors in cardiomyocytes, and induces interleukin-18 expression via Rac1-dependent PI3K/Akt/IKK/NF-kappaB andMKK7/JNK/AP-1 signaling. J. Mol. Cell Cardiol. 2010, 49, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Yuan, W.; Peng, M.; Mao, Z.; Zhao, Q.; Sun, X.; Yan, J. Role of CyPA in cardiac hypertrophy and remodeling. Biosci. Rep. 2019, 39, 12. [Google Scholar] [CrossRef] [Green Version]
- Gawałko, M.; Kapłon-Cieślicka, A.; Hohl, M.; Dobrev, D.; Linz, D. COVID-19 associated atrial fibrillation: Incidence, putative mechanisms and potential clinical implications. IJC Hear. Vasc. 2020, 30, 100631. [Google Scholar] [CrossRef]
- Kato, N.; Yuzawa, Y.; Kosugi, T.; Hobo, A.; Sato, W.; Miwa, Y.; Sakamoto, K.; Matsuo, S.; Kadomatsu, K. The E-Selectin Ligand Basigin/CD147 Is Responsible for neutrophil recruitment in renal ischemia/reperfusion. J. Am. Soc. Nephrol. 2009, 20, 1565–1576. [Google Scholar] [CrossRef] [Green Version]
- Kato, N.; Kosugi, T.; Sato, W.; Ishimoto, T.; Kojima, H.; Sato, Y.; Sakamoto, K.; Maruyama, S.; Yuzawa, Y.; Matsuo, S.; et al. Basigin/CD147 Promotes renal fibrosis after unilateral ureteral obstruction. Am. J. Pathol. 2011, 178, 572–579. [Google Scholar] [CrossRef]
- Yoshioka, T.; Kosugi, T.; Masuda, T.; Watanabe, T.; Ryuge, A.; Nagaya, H.; Maeda, K.; Sato, Y.; Katsuno, T.; Kato, N.; et al. CD147/basigin deficiency prevents the development of podocyte injury through FAK signaling. Am. J. Pathol. 2019, 189, 1338–1350. [Google Scholar] [CrossRef] [PubMed]
- Musiał, K.; Bargenda, A.; Zwolińska, D. Urine matrix metalloproteinases and their extracellular inducer EMMPRIN in children with chronic kidney disease. Ren. Fail. 2015, 37, 980–984. [Google Scholar] [CrossRef]
- Alfano, G.; Guaraldi, G.; Fontana, F.; Ferrari, A.; Magistroni, R.; Mussini, C.; Cappelli, G.; The Modena Covid-19 Working Group (MoCo19). The Role of the renin-angiotensin system in severe acute respiratory syndrome-CoV-2 infection. Blood Purif. 2020, 1–5. [Google Scholar] [CrossRef]
- Danser, A.J.; Epstein, M.; Batlle, D. Renin-angiotensin system blockers and the COVID-19 pandemic. Hypertension 2020, 75, 1382–1385. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Sfera, A.; Osorio, C.; Jafri, N.; Diaz, E.L.; Maldonado, J.E.C. Intoxication with endogenous angiotensin II: A COVID-19 hypothesis. Front. Immunol. 2020, 11, 1472. [Google Scholar] [CrossRef]
- De Queiroz, T.M.; Lakkappa, N.; Lazartigues, E. ADAM17-mediated shedding of inflammatory cytokines in hypertension. Front. Pharmacol. 2020, 11, 1154. [Google Scholar] [CrossRef]
- Xu, J.; Sriramula, S.; Xia, H.; Moreno-Walton, L.; Culicchia, F.; Domenig, O.; Poglitsch, M.; Lazartigues, E. Clinical relevance and role of neuronal AT1 receptors in ADAM17-mediated ACE2 shedding in neurogenic hypertension. Circ. Res. 2017, 121, 43–55. [Google Scholar] [CrossRef]
- Melenhorst, W.B.W.H.; Visser, L.; Timmer, A.; Heuvel, M.C.V.D.; Stegeman, C.A.; Van Goor, H. ADAM17 upregulation in human renal disease: A role in modulating TGF-α availability? Am. J. Physiol. Physiol. 2009, 297, F781–F790. [Google Scholar] [CrossRef]
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Valdivielso, J.M.; Ortiz, A.; Egido, J.; Ruiz-Ortega, M. Paricalcitol inhibits aldosterone-induced proinflammatory factors by modulating epidermal growth factor receptor pathway in cultured tubular epithelial cells. BioMed Res. Int. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Hagiyama, M.; Ito, A. Renal ADAM10 and 17: Their physiological and medical meanings. Front. Cell Dev. Biol. 2018, 6, 153. [Google Scholar] [CrossRef]
- Hirsch, J.S.; Ng, J.H.; Ross, D.W.; Sharma, P.; Shah, H.H.; Barnett, R.L.; Hazzan, A.D.; Fishbane, S.; Jhaveri, K.D.; Abate, M.; et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020, 98, 209–218. [Google Scholar] [CrossRef]
- Santoriello, D.; Khairallah, P.; Bomback, A.S.; Xu, K.; Kudose, S.; Batal, I.; Barasch, J.; Radhakrishnan, J.; D’Agati, V.; Markowitz, G. Postmortem kidney pathology findings in patients with COVID-19. J. Am. Soc. Nephrol. 2020, 31, 2158–2167. [Google Scholar] [CrossRef]
- Kudose, S.; Batal, I.; Santoriello, D.; Xu, K.; Barasch, J.; Peleg, Y.; Canetta, P.; Ratner, L.E.; Marasa, M.; Gharavi, A.G.; et al. Kidney biopsy findings in patients with COVID-19. J. Am. Soc. Nephrol. 2020, 31, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Akilesh, S.; Nast, C.C.; Yamashita, M.; Henriksen, K.; Charu, V.; Troxell, M.L.; Kambham, N.; Bracamonte, E.; Houghton, D.; Ahmed, N.I.; et al. Multicenter clinicopathologic correlation of kidney biopsies performed in COVID-19 patients presenting with acute kidney injury or proteinuria. Am. J. Kidney Dis. 2020. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Pei, G.; Zhang, Z.; Peng, J.; Liu, L.; Zhang, C.; Yu, C.; Ma, Z.; Huang, Y.; Liu, W.; Yao, Y.; et al. Renal involvement and early prognosis in patients with COVID-19 pneumonia. J. Am. Soc. Nephrol. 2020, 31, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Uppal, N.N.; Wanchoo, R.; Shah, H.H.; Yang, Y.; Parikh, R.; Khanin, Y.; Madireddy, V.; Larsen, C.P.; Jhaveri, K.D.; et al. COVID-19-Associated kidney injury: A case series of kidney biopsy findings. J. Am. Soc. Nephrol. 2020, 31, 1948–1958. [Google Scholar] [CrossRef]
- Werion, A.; Belkhir, L.; Perrot, M.; Schmit, G.; Aydin, S.; Chen, Z.; Penaloza, A.; De Greef, J.; Yildiz, H.; Pothen, L.; et al. SARS-CoV-2 causes a specific dysfunction of the kidney proximal tubule. Kidney Int. 2020, 98, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yang, M.; Wan, C.; Yi, L.-X.; Tang, F.; Zhu, H.-Y.; Yi, F.; Yang, H.-C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-M.; Zhang, H. Genetic roadmap for kidney involvement of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Clin. J. Am. Soc. Nephrol. 2020, 15, 1044–1046. [Google Scholar] [CrossRef] [Green Version]
- Singer, D.; Camargo, S.M.R. Collectrin and ACE2 in renal and intestinal amino acid transport. Channels 2011, 5, 410–423. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Sato, Y.; Sasaki, T. Suppression of coronavirus replication by cyclophilin inhibitors. Viruses 2013, 5, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; Liu, Y.; Liu, Y.; et al. Human Kidney is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). medRxiv 2020, 2. [Google Scholar] [CrossRef] [Green Version]
- Merrill, J.T.; Erkan, D.; Winakur, J.; James, J.A. Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat. Rev. Rheumatol. 2020, 16, 581–589. [Google Scholar] [CrossRef]
- Rodríguez, E.; Gimeno, J.; Arias-Cabrales, C.; Barrios, C.; Redondo-Pachón, D.; Soler, M.J.; Crespo, M.; Sierra-Ochoa, A.; Riera, M.; Pascual, J. Membrane attack complex and factor H in humans with acute kidney injury. Kidney Blood Press. Res. 2018, 43, 1655–1665. [Google Scholar] [CrossRef]
- Clive, D.M.; Vanguri, V.K. The Syndrome of Tubulointerstitial Nephritis with Uveitis (TINU). Am. J. Kidney Dis. 2018, 72, 118–128. [Google Scholar] [CrossRef]
- Westhoff, T.H.; Seibert, F.S.; Bauer, F.; Stervbo, U.; Anft, M.; Doevelaar, A.A.N.; Rohn, B.J.; Winnekendonk, G.; Dittmer, U.; Schenker, P.; et al. Allograft infiltration and meningoencephalitis by SARS-CoV-2 in a pancreas-kidney transplant recipient. Arab. Archaeol. Epigr. 2020. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, P.; Zhao, Y.; Zhuang, Z.; Wang, Z.; Song, R.; Zhang, J.; Liu, C.; Gao, Q.; Xu, Q.; et al. Single-cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of COVID-19 and influenza patients. Immunity 2020, 53, 685–696.e3. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Wang, Q.; Guo, F.; Zhao, Y.; Ji, L.; An, T.; Song, Y.; Liu, Y.; He, Y.; Qin, G. FoxO1-mediated inhibition of STAT1 alleviates tubulointerstitial fibrosis and tubule apoptosis in diabetic kidney disease. eBioMedicine 2019, 48, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Nightingale, J. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via Jak/Stat pathway activation. J. Am. Soc. Nephrol. 2004, 15, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Mears, J.R.; Shakib, L.; Beynor, J.I.; Shanaj, S.; Korsunsky, I.; Nathan, A.; Donlin, L.T.; Raychaudhuri, S. IFN-γ and TNF-α drive a CXCL10+ CCL2+ macrophage phenotype expanded in severe COVID-19 and other diseases with tissue inflammation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Maeda-Hori, M.; Kosugi, T.; Kojima, H.; Sato, W.; Inaba, S.; Maeda, K.; Nagaya, H.; Sato, Y.; Ishimoto, T.; Ozaki, T.; et al. Plasma CD147 reflects histological features in patients with lupus nephritis. Lupus 2014, 23, 342–352. [Google Scholar] [CrossRef]
- Kissling, S.; Rotman, S.; Gerber, C.; Halfon, M.; Lamoth, F.; Comte, D.; Lhopitallier, L.; Sadallah, S.; Fakhouri, F. Collapsing glomerulopathy in a COVID-19 patient. Kidney Int. 2020, 98, 228–231. [Google Scholar] [CrossRef]
- Magoon, S.; Bichu, P.; Malhotra, V.; Alhashimi, F.; Hu, Y.; Khanna, S.; Berhanu, K. COVID-19-related glomerulopathy: A report of 2 cases of collapsing focal segmental glomerulosclerosis. Kidney Med. 2020, 2, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Bhargava, R.; Shaukat, A.-A.; Albert, E.; Leggat, J. Spectrum of podocytopathies in new-onset nephrotic syndrome following COVID-19 disease: A report of 2 cases. BMC Nephrol. 2020, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Widiasta, A.; Sribudiani, Y.; Nugrahapraja, H.; Hilmanto, D.; Sekarwana, N.; Rachmadi, D. Potential role of ACE2-related microRNAs in COVID-19-associated nephropathy. Non-Coding RNA Res. 2020, 5, 153–166. [Google Scholar] [CrossRef]
- Rednor, S.J.; Ross, M.J. Molecular mechanisms of injury in HIV-associated nephropathy. Front. Med. 2018, 5, 177. [Google Scholar] [CrossRef] [Green Version]
- Nasr, S.H.; Kopp, J.B. COVID-19-associated collapsing glomerulopathy: An emerging entity. Kidney Int. Rep. 2020, 5, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C. Podocyte ACE2 protects against diabetic nephropathy. Kidney Int. 2012, 82, 255–256. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Wysocki, J.; William, J.; Soler, M.J.; Cokic, I.; Batlle, D. Glomerular localization and expression of angiotensin-converting enzyme 2 and angiotensin-converting enzyme: Implications for Albuminuria in diabetes. J. Am. Soc. Nephrol. 2006, 17, 3067–3075. [Google Scholar] [CrossRef] [Green Version]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of ACE2, CD147, CD26, and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy 2020. [Google Scholar] [CrossRef]
- Chng, W.J.; Lai, H.C.; Earnest, A.; Kuperan, P. Haematological parameters in severe acute respiratory syndrome. Int. J. Lab. Hematol. 2005, 27, 15–20. [Google Scholar] [CrossRef]
- Fan, B.E.; Chong, V.C.L.; Chan, S.S.W.; Lim, G.H.; Lim, K.G.E.; Tan, G.B.; Mucheli, S.S.; Kuperan, P.; Ong, K.H. Hematologic parameters in patients with COVID-19 infection. Am. J. Hematol. 2020, 95, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhaveri, K.D.; Meir, L.R.; Flores Chang, B.S.; Parikh, R.; Wanchoo, R.; Barilla-LaBarca, M.L.; Bijol, V.; Hajizadeh, N. Thrombotic microangiopathy in a patient with COVID-19. Kidney Int. 2020, 98, 509–512. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. New Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef]
- Siguret, V.; Voicu, S.; Neuwirth, M.; Delrue, M.; Gayat, E.; Stépanian, A.; Mégarbane, B. Are antiphospholipid antibodies associated with thrombotic complications in critically ill COVID-19 patients? Thromb. Res. 2020, 195, 74–76. [Google Scholar] [CrossRef]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic antiphospholipid antibodies in COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Fraga-Silva, R.A.; Da Silva, D.G.; Montecucco, F.; Mach, F.; Stergiopulos, N.; da Silva, R.F.; Santos, R.A. The angiotensin-converting enzyme 2/angiotensin-(1–7)/Mas receptor axis: A potential target for treating thrombotic diseases. Thromb. Haemost. 2012, 108, 1089–1096. [Google Scholar] [CrossRef]
- Shi, W.; Lv, J.; Lin, L. Coagulopathy in COVID-19: Focus on vascular thrombotic events. J. Mol. Cell. Cardiol. 2020, 146, 32–40. [Google Scholar] [CrossRef]
- South, A.M.; Brady, T.M.; Flynn, J.T. ACE2 (Angiotensin-Converting Enzyme 2), COVID-19, and ACE inhibitor and Ang II (Angiotensin II) receptor blocker use during the pandemic. Hypertension 2020, 76, 16–22. [Google Scholar] [CrossRef]
- Abuelo, J.G. Normotensive ischemic acute renal failure. New Engl. J. Med. 2007, 357, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Tumlin, J.A.; Murugan, R.; Deane, A.M.; Ostermann, M.; Busse, L.W.; Ham, K.R.; Kashani, K.; Szerlip, H.M.; Prowle, J.R.; Bihorac, A.; et al. Outcomes in patients with vasodilatory shock and renal replacement therapy treated with intravenous Angiotensin II. Crit. Care Med. 2018, 46, 949–957. [Google Scholar] [CrossRef]
- Milne, S.; Yang, C.X.; Timens, W.; Bossé, Y.; Sin, D.D. SARS-CoV-2 receptor ACE2 gene expression and RAAS inhibitors. Lancet Resp. Med. 2020, 8, e50–e51. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of Angiotensin-converting enzyme inhibition and Angiotensin II receptor blockers on cardiac Angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of Angiotensin-converting enzyme 2 after myocardial infarction by blockade of Angiotensin II receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef] [Green Version]
- Wysocki, J.; Lores, E.; Ye, M.; Soler, M.J.; Batlle, D. Kidney and Lung ACE2 Expression after an ACE Inhibitor or an Ang II receptor blocker: Implications for COVID-19. J. Am. Soc. Nephrol. 2020, 31, 1941–1943. [Google Scholar] [CrossRef]
- Liu, X.; Long, C.; Xiong, Q.; Chen, C.; Ma, J.; Su, Y.; Hong, K. Association of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers with risk of COVID-19, inflammation level, severity, and death in patients with COVID-19: A rapid systematic review and meta-analysis. Clin. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Kovacs, R.; Harrington, B. Joint HFSA/ACC/AHA Statement addresses concerns re: Using RAAS antagonists in COVID-19. J. Card. Fail. 2020, 26, 370. [Google Scholar] [CrossRef]
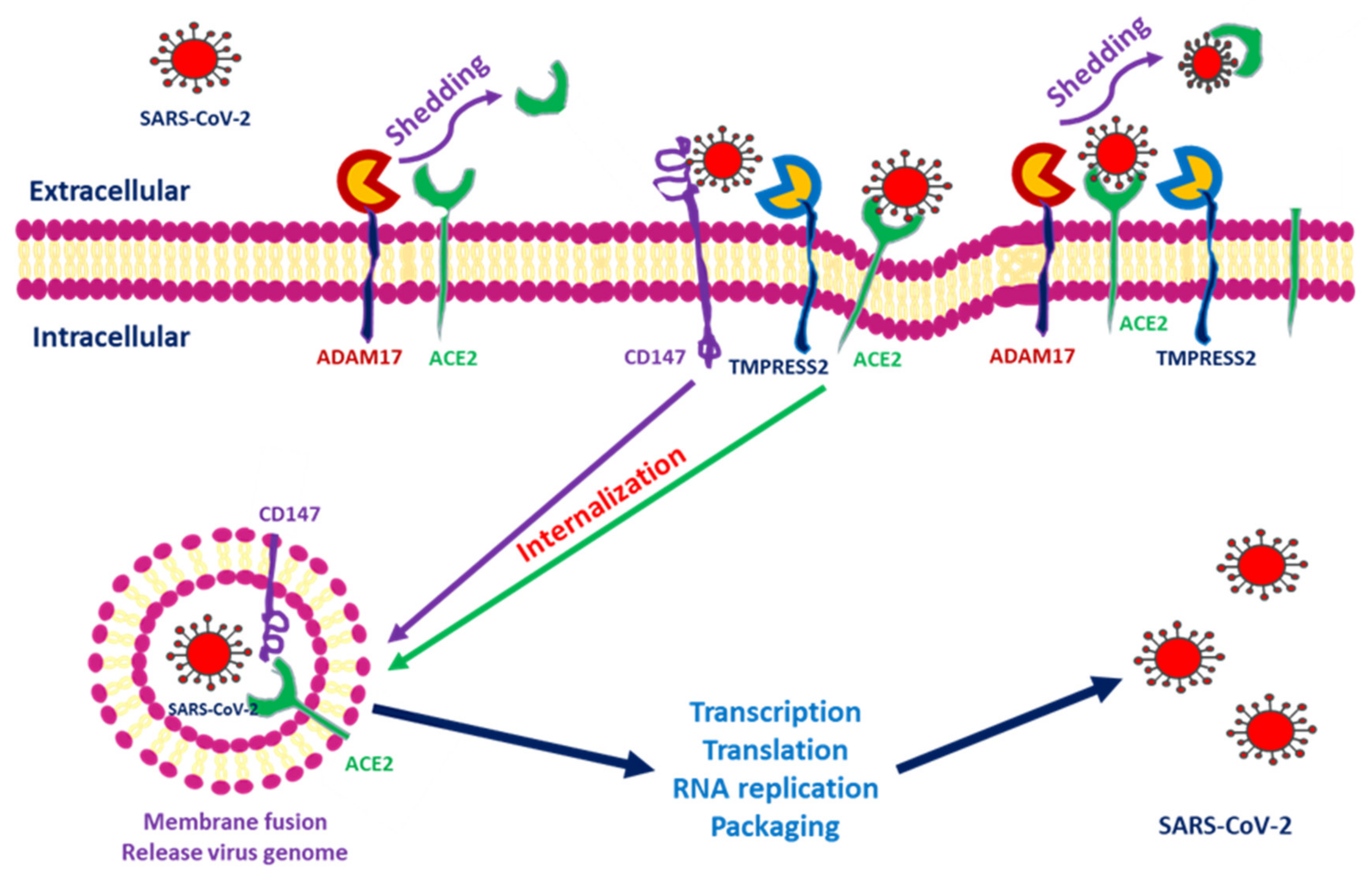
Figure 1.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entry into cells through interactions among angiotensin-converting enzyme 2 (ACE2), transmembrane protease serine 2 (TMPRSS2), a disintegrin and metalloprotease 17 (ADAM17) and CD147. After SARS-CoV-2 interacts with the cells, ACE2 conjugates with the spike protein of SARS-CoV-2. TMPRSS2 interacts with the spike protein and facilitates internalization. ADAM17 serves to shed SARS-CoV-2 by transforming ACE2 into a soluble form. CD147 expressed within the transmembrane also facilitates entry and induces downstream inflammatory cytokine expression.
Figure 1.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entry into cells through interactions among angiotensin-converting enzyme 2 (ACE2), transmembrane protease serine 2 (TMPRSS2), a disintegrin and metalloprotease 17 (ADAM17) and CD147. After SARS-CoV-2 interacts with the cells, ACE2 conjugates with the spike protein of SARS-CoV-2. TMPRSS2 interacts with the spike protein and facilitates internalization. ADAM17 serves to shed SARS-CoV-2 by transforming ACE2 into a soluble form. CD147 expressed within the transmembrane also facilitates entry and induces downstream inflammatory cytokine expression.

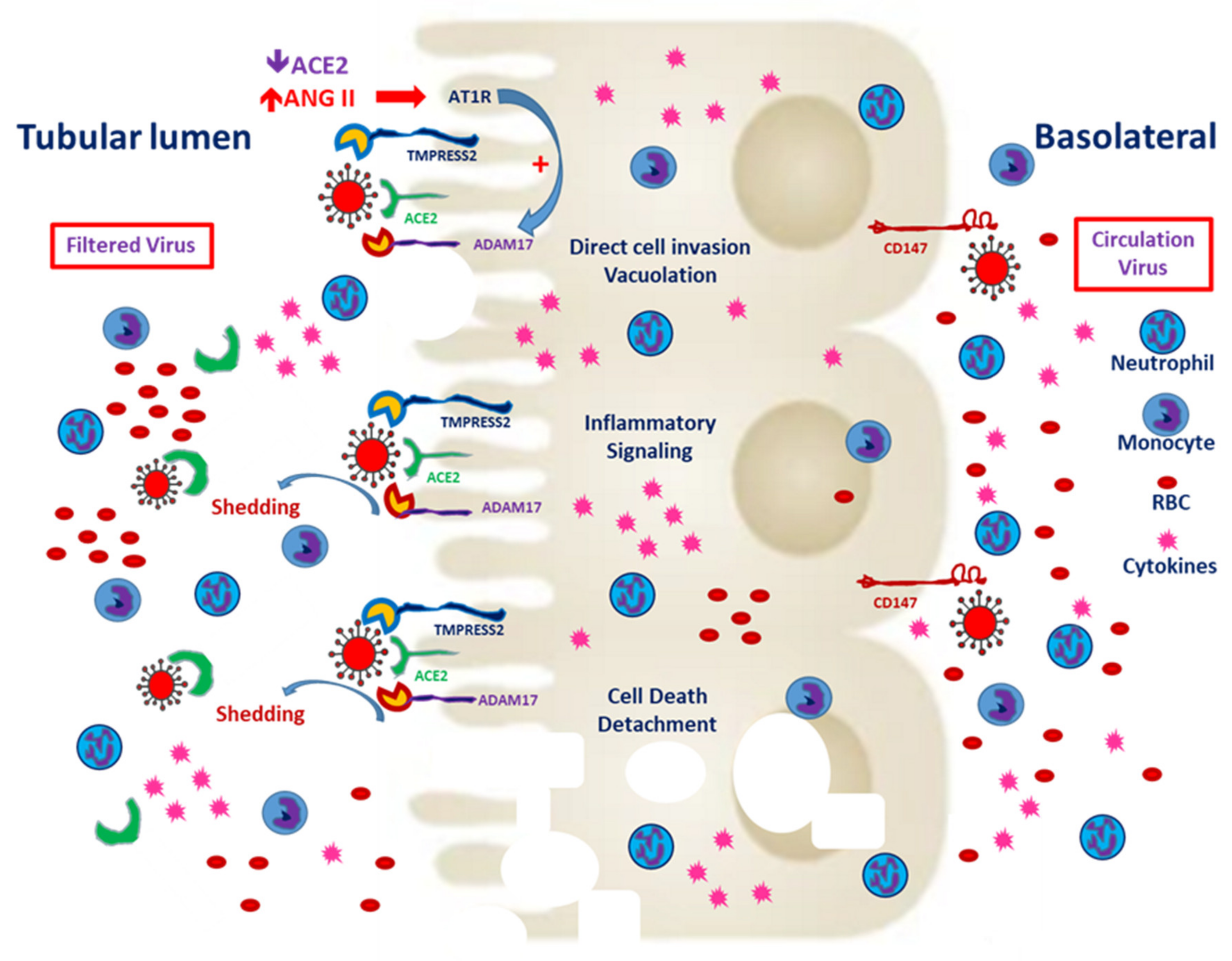
Figure 2.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entry from the apical and basolateral sides of proximal tubular cells. CD147 expression is mainly distributed on the basolateral side of the proximal tubules. In the tubules predisposed to injury, enhanced CD147 might facilitate the entry of SARS-CoV-2. Classical RAAS activation is common in CKD patients, and local expression of angiotensin-converting enzyme 2 (ACE2) enhances hypercoagulability and induces microthrombi. Besides, filtered SARS-CoV-2 might enter the proximal tubules through ACE2 and TMPRESS2. Entry through either route could worsen tubular inflammation and increase cell death.
Figure 2.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entry from the apical and basolateral sides of proximal tubular cells. CD147 expression is mainly distributed on the basolateral side of the proximal tubules. In the tubules predisposed to injury, enhanced CD147 might facilitate the entry of SARS-CoV-2. Classical RAAS activation is common in CKD patients, and local expression of angiotensin-converting enzyme 2 (ACE2) enhances hypercoagulability and induces microthrombi. Besides, filtered SARS-CoV-2 might enter the proximal tubules through ACE2 and TMPRESS2. Entry through either route could worsen tubular inflammation and increase cell death.

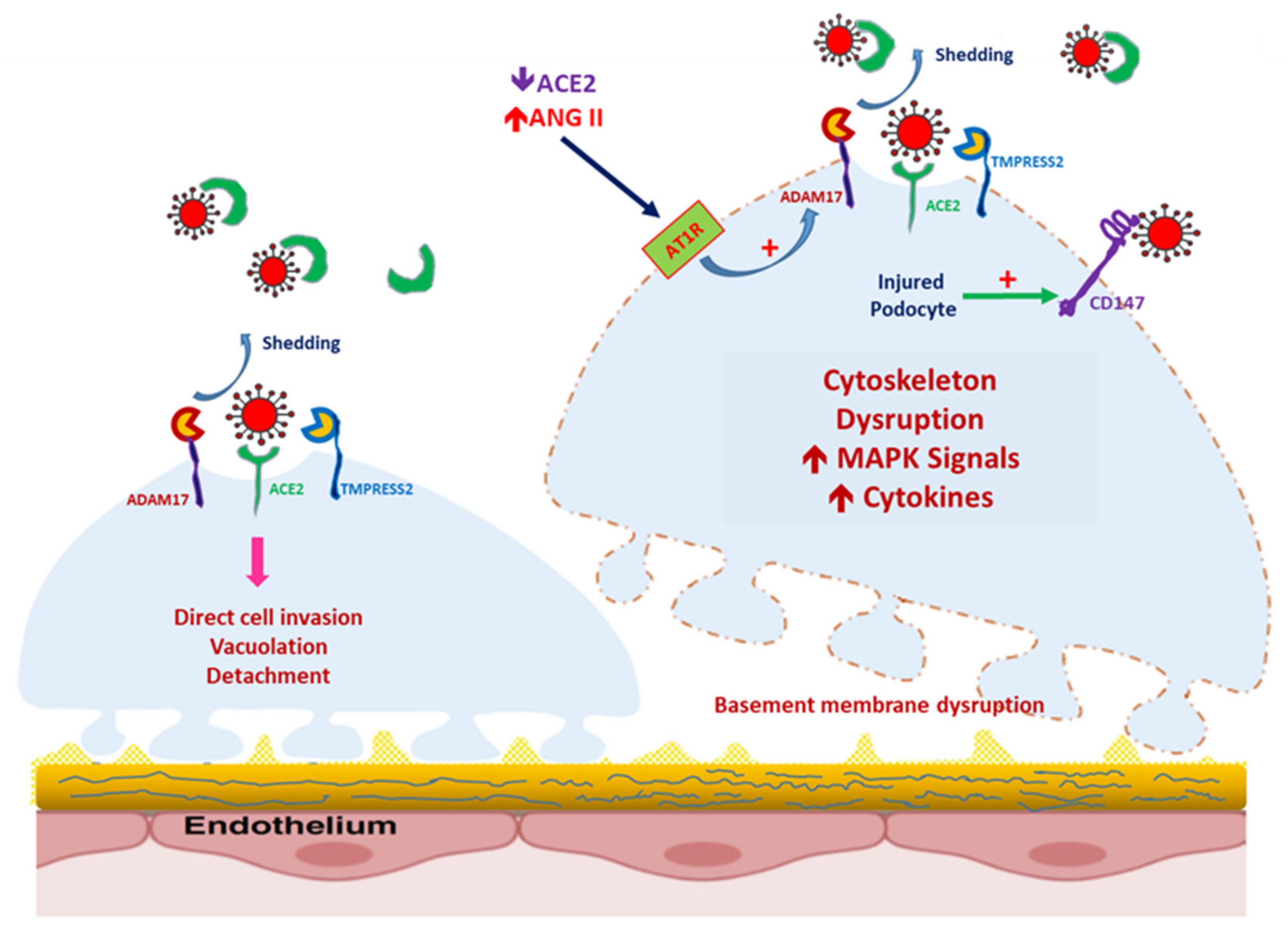
Figure 3.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-induced podocyte injury, mediated by the increased expression of CD147. In healthy podocytes, CD147 is seldom expressed. In podocytes damaged by the overexpression of the renin–angiotensin–aldosterone system, CD147 expression is increased, even though a disintegrin and metalloprotease 17 (ADAM17) expression is also modulated by Ang II-mediated AT1R activation. SARS-CoV-2 can enter the podocyte and induce podocytopathy.
Figure 3.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-induced podocyte injury, mediated by the increased expression of CD147. In healthy podocytes, CD147 is seldom expressed. In podocytes damaged by the overexpression of the renin–angiotensin–aldosterone system, CD147 expression is increased, even though a disintegrin and metalloprotease 17 (ADAM17) expression is also modulated by Ang II-mediated AT1R activation. SARS-CoV-2 can enter the podocyte and induce podocytopathy.


{kind=link}
{kind=link}
{kind=link}
Table 1.
The histopathologic and clinical characteristics of acute kidney injury in COVID-19 patients and the possible pathogenesis based on current evidence.
Table 1.
The histopathologic and clinical characteristics of acute kidney injury in COVID-19 patients and the possible pathogenesis based on current evidence.
| Injured Site | Histologic Change | Postulated Pathogenesis | Clinical Manifestations | References |
|---|---|---|---|---|
| Acute tubular Necrosis |
|
|
| [5,28,55,59] |
| Acute Interstitial Nephritis | Mononuclear Cell Infiltration Within Interstitium |
| Decrease In GFR | [64,65,66,67] |
| Podocytopathy |
|
| Proteinuria | [73,74,77] |
| Thrombotic Microangiopathy | Microthrombi Within the Glomeruli |
| Decrease In GFR Hematuria | [60,80,81,83] |
* Collapsing glomerulopathy is characterized by segmental or global glomerular tuft collapse with hypertrophy and hyperplasia of the overlying podocytes and dedifferentiated podocytes or parietal epithelial cells. Along with acute tubular injury, tubular dilation with microcyst formation and interstitial inflammation are common.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chueh, T.-I.; Zheng, C.-M.; Hou, Y.-C.; Lu, K.-C. Novel Evidence of Acute Kidney Injury in COVID-19. J. Clin. Med. 2020, 9, 3547. https://doi.org/10.3390/jcm9113547
AMA Style
Chueh T-I, Zheng C-M, Hou Y-C, Lu K-C. Novel Evidence of Acute Kidney Injury in COVID-19. Journal of Clinical Medicine. 2020; 9(11):3547. https://doi.org/10.3390/jcm9113547
Chicago/Turabian StyleChueh, Ti-I, Cai-Mei Zheng, Yi-Chou Hou, and Kuo-Cheng Lu. 2020. "Novel Evidence of Acute Kidney Injury in COVID-19" Journal of Clinical Medicine 9, no. 11: 3547. https://doi.org/10.3390/jcm9113547
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.