
Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma
by
 , , and
, , and
Maria Bousali
1 ,
,
George Papatheodoridis
2,
Dimitrios Paraskevis
3 and
Timokratis Karamitros
1,4,* 1
Bioinformatics and Applied Genomics Unit, Department of Microbiology, Hellenic Pasteur Institute, 11521 Athens, Greece
2
Department of Gastroenterology, “Laiko” General Hospital of Athens, Medical School, National and Kapodistrian University of Athens, 11527 Athens, Greece
3
Department of Hygiene Epidemiology and Medical Statistics, School of Medicine, National and Kapodistrian University of Athens, 15772 Athens, Greece
4
Laboratory of Medical Microbiology, Department of Microbiology, Hellenic Pasteur Institute, 11521 Athens, Greece
*
Author to whom correspondence should be addressed.
Microorganisms 2021, 9(8), 1787; https://doi.org/10.3390/microorganisms9081787
Submission received: 29 July 2021
/
Revised: 13 August 2021
/
Accepted: 18 August 2021
/
Published: 23 August 2021
(This article belongs to the Special Issue Molecular Biology of Hepatitis B Virus—from Basic Concepts to Therapeutic Targets)
Abstract
:Hepatitis B Virus (HBV) is an Old World virus with a high mutation rate, which puts its origins in Africa alongside the origins of Homo sapiens, and is a member of the Hepadnaviridae family that is characterized by a unique viral replication cycle. It targets human hepatocytes and can lead to chronic HBV infection either after acute infection via horizontal transmission usually during infancy or childhood or via maternal–fetal transmission. HBV has been found in ~85% of HBV-related Hepatocellular Carcinomas (HCC), and it can integrate the whole or part of its genome into the host genomic DNA. The molecular mechanisms involved in the HBV DNA integration is not yet clear; thus, multiple models have been described with respect to either the relaxed-circular DNA (rcDNA) or the double-stranded linear DNA (dslDNA) of HBV. Various genes have been found to be affected by HBV DNA integration, including cell-proliferation-related genes, oncogenes and long non-coding RNA genes (lincRNAs). The present review summarizes the advances in the research of HBV DNA integration, focusing on the evolutionary and molecular side of the integration events along with the arising clinical aspects in the light of WHO’s commitment to eliminate HBV and viral hepatitis by 2030.
1. Introduction
In 1965, Dr. Baruch Blumberg and his colleagues, during studies completely unrelated to viral hepatitis, discovered the “Australia antigen” [1], which was afterwards identified as the hepatitis B virus surface antigen (HBsAg). This discovery was awarded with the Nobel Prize in Physiology or Medicine in 1976 and is often believed to mark the beginning of hepatitis research [2]. In fact, the research on the pathogenesis of viral hepatitis had started decades before Dr. Blumberg’s critical breakthrough. In 1865, Rudolf Carl Virchow, one of the most prominent physicians of the 19th century, provided an authoritative explanation of the transmission by human serum of the “catarrhal jaundice” [3], which had been described by Hippocrates [4].
In the early 1920s, the distinction between “infectious” and “serum” hepatitis was recognized [2] and in 1947, the terms “hepatitis A” and “hepatitis B” were introduced [5]. Chronic hepatitis was first reported in the 1940s after observations of patients with cirrhosis, who had no history of alcoholic liver disease, but who had recovered many years earlier from an episode of catarrhal jaundice [6], and the development of liver function tests led to the recognition of anicteric infections and the existence of chronic carriers in the 1950s.
After the discovery of the “Australia antigen”, the vast amount of accumulated epidemiological and clinical data, as well as the huge numbers of stored serum samples, enabled rapid progress in understanding hepatitis B, and revealed the existence of a vast population of chronically infected people in multiple regions of the world [2]. After all these critical breakthroughs, HBV-related studies grew in number. In 1972, the first vaccine against HBV was available [7], in 1986, the first clinical trial of interferon—a therapy of chronic hepatitis B—started [8], and, in 1998, the first direct-acting antiviral (lamivudine) for chronic hepatitis B was approved by the FDA [9].
The identification of integrated HBV DNA in human genomic DNA was first reported in the early 1980s from four separate research groups [10,11,12,13], while more research studies were subsequently carried out to further examine the HBV DNA integration process. Most of the HBV integrations were not found to be recurrent, as each integration site was identified in only one sample [14,15,16,17]. In the early 2000s, the first recurrent HBV DNA integration site was identified in humans TERT gene [18,19] and from them on more recurrent HBV DNA integration sites have been reported from different research groups, using multiple methodologies.
Despite the availability of a vaccine since the 1980s, HBV remains a global public health problem, with an estimated 257 million people with chronic HBV infection, 90% of whom are unaware of their infection [20,21] and, as a result, do not receive appropriate medical care. Moreover, in many low-income and remote regions worldwide, vaccine coverage is another barrier in the disease control, and Africa and the Western Pacific region are disproportionately affected by HBV (Figure 1), (www.who.int/publications/i/item/global-hepatitis-report-2017 accessed on 18 August 2021).
The total population living with chronic HBV infection is indicative of the historical prevalence of HBV, whereas the prevalence among children aged 5 years also reflects access to preventive strategies, particularly infant vaccination [20]. For example, most of the countries of the SEARO (South East Asia) regions have accepted vaccination against HBV in their EPI program (Expanded Program on Immunization), while in China—which has the greatest number of HBsAg-positive individuals—had a percentage of 99% HBV immunization coverage among one-year-olds in 2019 (Figure 1), (WHO Global Health Observatory data repository: apps.who.int/gho/data/view.main.80300 accessed on 18 August 2021).
Simultaneously with the continuation of mass vaccination against HBV, other challenges must be overcome, including the prevention of mother to child HBV transmission by the timely administration of the HBV birth dose vaccine (within 24 h of birth) [22], the improvement of injection safety, the routine screening of all blood donations for transfusion, as well as the provision of sterilized syringes to people who inject drugs (PWID) (WHO-Global Health Sector Strategy on Viral Hepatitis 2016–2021: apps.who.int/iris/bitstream/handle/10665/246177/WHO-HIV-2016.06-eng.pdf accessed on 18 August 2021). Moreover, the conduction of nation-wide and workplace-based [23] surveys on HBV prevalence, mortality, and genotype distribution will provide more insights and feedback on the HBV prevention and elimination process.
2. Evolution of HBV and Co-Existence with Human Populations
HBV is a small spherical virus with icosahedral symmetry that is classified under the Hepadnaviridae family. The virus combines a partial double-stranded DNA (ds-DNA) and virus-encoded RT, and consequently it is classified as Group VII in Baltimore’s classification system (also referred to as pararetroviruses). The Hepadnaviridae family is characterized by a unique viral replication cycle, as the virus-encoded polymerase has reverse transcriptase (RT), DNA-polymerase (pol), and protein priming activities, while the reverse transcription step takes place in the late stages of the genomic replication [24]. Moreover, the polymerase lacks proofreading activity and, as a result, creates genetic variability that is manifested as circulating viral quasi-species [25].
The HBV substitution rate is estimated as ~1.5–3.0 × substitutions per site per year, while the origin of the virus is estimated at 33,600–34,100 years ago [26,27]. Chronic infections, including extended asymptomatic periods and a slow progression to clinical disease—characteristics that are also observed in Mycobacterium tuberculosis [28] and Helicobacter pylori [29] infections—are features suggesting that HBV co-existed with human populations for many thousands of years [25].
HBV sequences have been isolated from 400-year-old mummies from Korea [30] and Italy [31], while the analysis of ancient DNA derived from 7000-year-old skeletal remains from across Eurasia gained more insights about the origin and age of HBV [32,33]. Recent descriptions of members of the Hepadnaviridae family isolated from fish, reptiles and amphibians [34] strengthened the HBV—host species co-evolution theory [35].
3. Geographical Distribution of Human HBV Genotypes and Their Role in the Natural History of the Infection
There are nine confirmed (HBV A-I) and one putative (HBV J) human HBV genotypes, while some of them are further subdivided into sub-genotypes (over 30 in number) with particular global distribution patterns [38], disease and clinical progression, response to antiviral treatment and prognosis. HBV genotypes are characterized by a >8% nucleotide difference, while sub-genotypes are characterized by 4–8% nucleotide differences [39]. Furthermore, the geographical distribution of HBV genotypes is possibly related to the route of transmission and whether the exposure to the virus is perinatal, vertical or horizontal.
As presented in Figure 1, genotype A is widespread in Africa, Southern Asia, Europe and North America, and it is related to early childhood transmission, while genotypes B, C and E are localized in high-endemic regions (SE Asia, Western and Central Africa, China, Japan and Australia) and have been related to maternal–fetal, perinatal exposure. Genotype D is distributed globally and has been associated with early childhood or adult horizontal transmission [39], while genotypes F–H are localized mainly in the USA. Finally, the recently identified genotypes I and J are localized in Asia and specifically in Laos, Vietnam, China (genotype I) and Japan (genotype J), respectively.
The HBV genotype distribution is heterogeneous, and there is a lack of accurate genotyping data and monitoring of the epidemic genotypes. This makes epidemiological studies reflect only snapshots of the epidemiology while different HBV genotypes have different dynamics and may drift and change dynamically over time [40]. For example, although genotypes B, C and E have been reported to be prevalent in Asian countries, recent studies have reported that Bangladeshi patients have three major HBV genotypes (A, C and D) in considerable proportions [41].
Specifically, Raihan et al. [41], found that there was an association between HBV genotype C and high levels of HBV DNA and ALT, liver cirrhosis and HCC, while genotypes D and A are prevalent in patients with persistently low HBV DNA and normal ALT levels. Absence of the HBV genotype B has been reported from India, as well [42]. Furthermore, a recent study conducted by Velkov et al. [36] regarding the global HBV genotype distribution underlined that, in Latin America the genotypes F, G and H—that are rare in other parts of the world—were found in significant proportions. Moreover, they revealed the domination of genotypes A and D in Brazil as well as the existence of genotypes G and H in Mexico that are not present in other populations (Figure 1).
4. Natural History of Chronic HBV Infection
The natural history of human HBV infection varies between the different genotypes [25], as the predominant mode of transmission, the timing of the Hepatitis B e antigen (HBeAg) seroconversion to the hepatitis B e antibody (anti-HBe), the progression of liver-associated diseases—including Hepatocellular Carcinoma (HCC), and the potential of Hepatitis B surface antigen (HBsAg) seroclearance, vary substantially Box 1 [38]. Specifically, patients infected with the genotype C remain HBeAg-positive for many years longer than patients infected with any of the genotypes A, B, D or F [43].
Box 1. Serological markers of HBV infection.
HBeAg (Hepatitis B e antigen): Protein molecule contained in the nucleocapsid core of HBV that is detected in the serum of persons with high virus titers indicating high infectivity.
HBcAg (Hepatitis B core antigen): Protein molecule on the surface of the nucleocapsid core that is not secreted and, as a result, cannot be detected in the serum of infected individuals (however, its antibody can). Its presence indicates ongoing HBV replication during active infection.
* HBeAg and HBcAg originate from the same ORF at the HBV genome.
HBsAg (Hepatitis B surface antigen): Protein molecule on the surface of HBV, which can be detected in high levels in serum during acute or chronic HBV infection and indicates that the person is infectious. HBsAg is the antigen used to make the vaccine against HBV.
anti-HBs (Hepatitis B surface antibody): The presence of anti-HBs indicates recovery and immunity from HBV infection as well as successful vaccination against HBV.
anti-HBc (Total hepatitis B core antibody): Appears at the onset of symptoms in acute hepatitis B and persists for life, indicating previous or ongoing HBV infection in an undefined time frame.
IgM anti-HBc (IgM antibody to hepatitis B core antigen): Positively indicates recent (≤6 months) acute HBV infection.
The risk of developing chronic HBV infection depends on the age of exposure to HBV. Acute HBV infection within the first year of life has a 90% probability; in childhood, 20–30% probability; and in adulthood, a <5% probability of progression to chronic HBV infection [44]. Moreover, most of the neonatal and perinatal HBV infections become persistent [45]. The fact that, in early stages of the life cycle, HBV infections lead in such great percentages to chronic HBV infection is due to immune inhibitory mechanisms that either delete the HBV-specific T-cells in the thymus or render them dysfunctional in the periphery [46].
Chronic HBV infection is a complex and dynamic condition, as infected individuals can move from a phase of high viral load without liver disease to a phase of active liver disease, followed by a phase of inactivation and revert back to active liver disease [47]. Not all individuals with chronic HBV infection develop cirrhosis, as clinic-based longitudinal studies suggest that the overall incidence is 2–3% per year [48], while factors, such as the older age, the long-term presence of HBeAg and elevated levels of alanine aminotransferase (ALT) increase the risk [49].
There are five phases of chronic HBV infection: the immune tolerant phase, two immune active phases, the inactive chronic HBV infection phase and the recovery phase (Figure 2) [50]. The immune tolerant phase or HBeAg-positive chronic HBV phase (phase 1) lasts longer in patients infected via maternal–fetal transmission from HBeAg-positive mothers [43], which occurs more frequently in HBV genotype C infections. It can last for a few months or typically from years to even lifelong [51], the levels of ALT are normal, and there is either no or minimal liver inflammation or fibrosis.
During this phase, HBV integrates parts or whole of its genome into the host’s hepatocyte genomic DNA, as a result of the reverse transcription mediated by the HBV polymerase, and high levels of serum HBV DNA (>1 million DNA copies, >200,000 IU/mL) are present. During the HBeAg-positive immune active phase or HBeAg-positive chronic hepatitis B (CHB) (phase 2), inflammatory reaction occurs as a result of the host’s immune response, elevated ALT levels are observed and serum HBV DNA levels progressively decrease but remain high (>20,000 IU/mL). Active liver inflammation is usually present with or without significant liver fibrosis [47].
After the seroconversion from HBeAg to anti-HBe, most of the infected individuals (>80% of the cases) will transition into the HBeAg-negative inactive chronic infection phase, while the others will directly progress in the HBeAg-negative immune active phase or HBeAg-negative CHB [52]. The inactive chronic HBV infection or carrier phase (phase 3), is characterized by HBeAg seroclearance and anti-HBe positivity. The ALT levels are within the reference range, the HBV DNA levels are low (usually <2000 IU/mL), the liver inflammation recedes, and HBsAg is still present. This is also the phase when multiple integration events occur within the host’s genome.
The inactive HBV carrier phase may last the lifetime, while some patients may reverse to HBeAg seropositivity (usually within the first 12 months after HBeAg seroclearance) or a proportion of cases (20–30%) may develop HBeAg-negative CHB (phase 4). Except for the presence of HBeAg in serum, the characteristics of the immune active phase of HBeAg-negative CHB are similar to those of HBeAg-positive CHB, although the median serum HBV DNA levels are relatively lower. Finally, during the recovery phase (phase 5), neither HBV DNA nor HBsAg can be detected into the serum, while various antibodies have been produced against HBV (anti-HBs and anti-HBc) [52,53].
Gish et al. [54] proposed two more theoretical clearance phases; the clearance of cccDNA and the clearance of cells that have integrated HBV. These two phases have been extensively studied recently, as they are considered key points for the development of novel future therapies against HBV. The complete clearance of cccDNA in vivo is rare, while two immune mechanisms have been proposed. Based on the cytolytic mechanism, infected cells are killed by cytotoxic T lymphocytes and are being replaced by uninfected hepatocytes, while, based on the non-cytolytic cytokine-induced mechanism, the clearance is achieved through the activation of intracellular antiviral pathways [55].
In general, it is considered that clearance of HBV is performed via HBV-specific CD8+ T cells that escape central tolerance and attack the infected hepatocytes, while a small proportion of HBV-specific CD8+ T cells without the ability to produce antiviral cytokines possibly sustain long-term immunopathological responses and whether they manage to achieve HBV clearance is unknown [56]. The reverberation of HBV integrations in this process is an issue that needs to be explored as there is no reported evidence.
5. Genome Organization and Types of Genomic Material within the HBV Life Cycle
The HBV life cycle is a process with numerous steps and increased complexity that arises from the different types of HBV particles observed in the serum of infected patients, the different states in which HBV genomic DNA is found in the viral particles, as well as the HBV genome, itself, with the overlapping open reading frames (ORFs).
Four categories of HBV particles have been sufficiently described in the serum of infected patients; the Dane particles, the enveloped (nucleo)capsids containing immature DNA/RNA or as genome-free (empty virions), the subviral particles (SVPs) and the filament structures of variable length [58,59]. A fifth type is the envelope-less, naked nucleocapsids; however, this type has been described only in vitro [58]. From these, only the 42 nm—diameter spherical Dane particles are infectious, while the 22 nm—diameter enveloped (nucleo)capsids and SVPs are non-infectious but much more abundant in the patient serum.
The Dane particles [60], that were named by the scientist who first visualized them using an electron microscope in 1970, consist of a lipid membrane with three HBV surface antigens (HBs) with different sizes: large (L-HBs), middle (M-HBs), and small (S-HBs). The lipid membrane surrounds the nucleocapsid (composed of the HBc), the HBV polymerase (Pol) and the genomic DNA. Both the enveloped and the naked nucleocapsids may contain DNA, RNA or no genomic material [59]. Moreover, the genome-free enveloped capsids (empty virions) have been recently suggested as diagnostic markers for hepatic cccDNA [61] as well as candidates for a new generation of HBV vaccine [58].
The HBV genome is mainly composed of overlapping ORFs (Figure 3G) from which multiple functional proteins are produced. HBc, HBeAg and 22-kDa precore protein (p22cr) are produced from ORF-C; Pol is produced from ORF-P; L-HBs, M-HBs and S-HBs are produced from ORF-S; while the HBV X protein is produced from ORF-X.
During the life cycle of the virus, the HBV genomic DNA, can be found in four different states, two of which are candidates for HBV integration into the host genome. After the glycosaminoglycan-mediated viral entry—through the sodium taurocholate cotransporting polypeptide (NTCP) receptor [62], which is located in the basolateral membrane, into the hepatocytes, nucleocapsids enter the cytoplasm and are directed to the nucleus along with the microtubules [63,64]. At this stage, the nucleocapsids, contain relaxed circular DNA (rcDNA) or, more rarely, double-stranded linear DNA (dslDNA).
In the nucleus, the rcDNA is modified by cellular factors, including Tyrosyl DNA Phosphodiesterase-2 (TDP2) [65,66], DNA polymerase kappa (Pol kappa) [67], Pol alpha [68], DNA ligase 1 (LIG1) and LIG3 [69] and flap endonuclase 1 (FEN1) [70]. Specifically, the Pol-linked terminal redundant sequence in the 5 end of the minus strand DNA and the RNA oligonucleotide attached at the 5 end of the plus strand DNA are removed from the rcDNA [59,71,72].
The gaps in both strands are filled and ligated to generate covalently closed circular DNA (cccDNA), which is a stable episomal transcriptional template for the HBV mRNAs [73]. The cccDNA is transcribed into five HBV RNAs of different length, under the action of the host’s RNA polymerase II [74]. One of the five transcripts is the 3.5 kb length pre-genomic RNA (pgRNA). The pgRNA can be encapsidated into viral capsids, in a process mediated by the HBV polymerase [64]. In the nucleocapsids, reverse transcription of the pgRNA results in the formation of rcDNA or dslDNA.
The intranuclear HBV dslDNA is the main candidate of DNA molecule that can be integrated into the host genome. It has been shown that the integration occurs at a frequency of 1 in 10 to 10 cells in the woodchuck and duck models of HBV infection [75,76,77] and in chronically infected HBV patients [78,79,80]. Based on cell-culture experiments, the integration of HBV DNA into the host genome occurs within a week after the infection [81] via double-stranded DNA breaks by nonhomologous end joining (NHEJ) or micro-homology mediated end joining (MMEJ), as discussed below.
However, a series of NGS-based studies [82,83,84] reported that some of the identified HBV integration sites are uncorrelated to the dslDNA termini; thus, other HBV DNA forms may be important contributors in the integration process as well. The cohesive-end linear DNA, that occurs via denaturation of the 5′ cohesive overlap region of HBV rcDNA and subsequent extension of the recessed 3′ ends and formation of terminal redundancies between the direct repeat 1 (DR1) and DR2 as well as spliced variants of HBV RNA, may be the missing key points in the deciphering of the HBV integration process into the host genome [85].
6. Proposed Molecular Mechanisms of HBV DNA Integration
A series of molecular mechanisms have been described for the integration of HBV DNA into the human genome (Figure 3). All of the above-mentioned molecular mechanisms were proposed after thorough examination of the virus-host junction sites and the effect of the integration in the cellular DNA, which can be from minimal to disruptive.
The “single-stranded gap” model (Figure 3A) is based on the restriction maps and nucleotide sequences of the human-HBV DNA junctions, and it indicates that, during cellular DNA replication, the DNA polymerase switches from the human genomic DNA strand to the single-stranded region of nearby HBV genome [11]. This switch is restricted to the gap region close to the 5 end as procession of another DNA polymerase complex starting from the 3 end of the short strand of HBV limits the available single-stranded region. A recombination event then occurs and joins the long strand of the HBV dsDNA with the original human genomic DNA and after filling in and ligation, a full-length HBV integration site is present in the human genome.
Two more models were described after the observation that a 11 bp length sequence of the host–viral junctions matches the Direct Repeat (DR) sequences that are located close to the cohesive-end region of the HBV sequence [86]. Both of the models are based on the observation that integration of the DR sequence into the host genome occurs after deletion of the first two nucleotides of the DR sequence, the conservation of the DR1/DR2 polarity and the presence of viral-specific and non-specific sites of integration. The first of the two models assumes that one of the DRs is used as the target site for the formation of an initial host–viral junction via site-specific recombination, which is followed by a second recombination event, which forms the integration site (Figure 3B).
The second model that includes the DR sequences is based on the assumption that HBV DNA is re-organized prior to integration, forming ‘head-to-head’ structures that give rise to inverted direct repeats [86]. HBV integration is mediated by two inverted DR sequences [75,77,78,82,83,84,87,88,89]. Inverted DRs are known to be unstable, and thus deletions within the HBV DNA are expected to occur, leading to further re-organization of the HBV DNA that possibly results in the loss of at least one DR sequence.
Another proposed mechanism is based on the partial sequence homology between the HBV DNA and the host genomic DNA at the site of the integration (Figure 3C) [90], as it has been found that integrations result in the termination of transcription at a cellular termination signal. Based on this molecular mechanism, the integrations result in the expression of viral–human chimeric fusion proteins that potentially contribute to cancer progression as will be discussed below.
The next model is based on the idea of a site-specific mechanism of recombination [91,92], in which the 3′ free end of the minus HBV strand facilitates strand invasion into a double-stranded break or gap that is formed by the HBV infection or endogenous metabolism at the 5′ end of the host DNA in a process that leads to a temporary base pairing between the two DNA molecules because of a short homology region (Figure 3D). This process has been described as a “roll-in” method [14], and it leads to full or partial HBV DNA integration.
Another proposed mechanism is based on the observation that integrated HBV DNA into the human genome is flanked on both sides by a 12 bp repeated sequence that is neither viral nor cellular but has been generated due to the integration itself [93]. Termination of the reverse transcription of the pgRNA before the completion of the transcription of the minus strand results in the formation of an incomplete minus strand with the absence of the HBcAg.
The 3′ end of the minus strand is joined to the 5′ overhang at the site of a staggered cut, the covalently bound viral DNA polymerase at the 5′ end of the minus strand is removed and the staggered ends are filled in and ligated creating two equal-sized direct repeats of the human flanking sequence (Figure 3E). In addition to other proposed models, this particular model assumes that there is no deletion or loss of human genomic sequences at the site of the integration.
The last method is based on the non-homologous end joining (NHEJ) molecular mechanism mediated by cellular enzymes and, in particular the topoisomerase I, leading to a 11 bp deletion of the host genome [94,95]. This mechanism takes into account the absence of sequence homology between the HBV and host DNA at the majority of the virus-host junctions [81,84]. Due to the microhomology that has been described in some of the junctions, the microhomology-mediated end joining (MMEJ) has also been described as a proposed mechanism of HBV DNA integration (Figure 3F) [81,83,96,97,98].
Finally, a major key point that should be taken into account in order to better understand the molecular mechanisms that describe the HBV integration process and, in general. the process of viral integrations into genomes, is the fact that the immediate integration events that occur after HBV infection are followed by additional post-integration changes due to genomic instability at the sites of integration. Moreover, integrations that occur within transposable and retrotransposable elements are very important since the transposition of these elements increase the “pool” of integrations, while the capacity of the integrated HBV to be excised from one genomic loci and transported to another genomic loci through reintegration [99] should be further investigated.
7. Hepatocellular Carcinoma (HCC)
HCC is the most common subtype of liver cancer and the forth leading cause of cancer death worldwide. Specifically, it has been estimated that HCC is the fifth most common cancer in men and the ninth in women, with approximately 500,000 and 200,000 new cases per year worldwide, respectively. As for the global distribution of HCC, 76% of the annual reported cases originate from Asia, 8.1% from Europe, 7.5% from Africa, 4.2% from North America, 3.8% from Latin America and the Caribbean and 0.4% from Oceania (Figure 1)(WHO data: www.gco.iarc.fr—Accessed on 8 June 2021).
The combination of the host genetic background and the access to health care and prevention facilities has an influence on the outcome of the disease, as belonging to a specific population group increases the susceptibility of HBV carrier to develop HCC [100]. Specifically, mortality due to HCC is increased in African and Asian populations (WHO data: www.gco.iarc.fr—Accessed on 8 June 2021), and, at the same time in these two populations, more than 70% of HCC develop in cases with HBV infection [101].
HBV infection causes chronic hepatitis and, subsequently, liver cirrhosis, while it has been identified as one of the most important contributors of HCC, as ~50% of the cases are due to HBV infection [102]. Specifically, it has been estimated that the development of HCC is 25–37-fold higher in carriers of HBsAg compared with non-infected individuals [103,104].
Research suggested that approximately 80% of tumor derived tissues are carrying integrated HBV DNA [15,105,106]. Adjacent tissues also harbor HBV integration events [107], and it has been proposed that viral integration occurs during chronic HBV infection, before HCC initiation [15,105]. Specifically, it has been suggested that hepatocytes with HBV integration undergo certain rounds of expansion during chronic hepatitis and some of them participate in the formation of focal proliferative lesions. As a result, they gain growth advantages, and they clonally expand during tumorigenesis [107].
In general, HBV integration is more frequent in tumor samples than in adjacent liver tissues, at percentages of 86.4% and 30.7%, respectively [82], while there is no reported difference in the chromosome distribution between tumor- and adjacent non-tumor-derived samples [105]. Alu-PCR, NGS and in silico approaches revealed that there is a preference for HBV integration to chromosomes 3 [108], 11 [107,109] and 17 [109].
8. HBV Integration Sites into the Human Genome and Target Genes
HBV integration into human host chromosomes occurs in the infected hepatocytes since early stages of natural acute infections [15,100,105]. Specifically, it has been shown that integration starts <3 days after the infection in cell cultures [81] and in the early stages of infection in vivo [75,110]. Deletions, duplications, inversions, copy number variations (CNVs) and other types of rearrangements of the HBV sequence have been observed in the sites of HBV integration, while sequencing analysis of the viral-host junctions revealed only a few micro-deletions, micro-insertions, point mutations and translocations with no significant difference being found between tumor-derived and non-tumor-derived samples [82,105].
HBV integrations induce chromosome changes, genome instability and changes in the expression of host genes, while they have been associated with chromosome fragile sites or repetitive sequences, followed by local rearrangements, which further increase the genomic instability [111]. It should also be taken into account that HBV integrations have the potential to endlessly continue during persistent and chronic infections [56], and thus the number of HBV integration sites per sample may be associated with the time of sample collection that reflects the phase of the HBV infection.
More than 10,000 unique HBV integration sites into the Human Genome have been reported in 25 different research studies, and ~51% (6864/13,572) of which have been found to target genomic regions that refer to 4183 different genes. It has been suggested that genomic areas that refer to cellular genes (Figure 4) are favored target sites for HBV integration [105,112], and specifically there is a preference of HBV integration into transcriptional and chromosomal regulatory regions [113]. DR1 and the topoisomerase I motif are the preferred break-points of the inserted viral fragments in both orientations [105]. Moreover, 2602 integration sites are recurrent (~19.2%), or, in other words, they have been reported more than once in different cases from independent research groups.
The main target genes of HBV integrations are the TERT, FN1, KMT2B, ALB, CCNA2, LINC00486, CPS1, SH3RF3, CCNE1, GLI2, KCNT2, LINGO2, PRKN, FAM157A, SOX5, GTF2I, PDE3A and CHRM3 genes (Supplementary Figure S1), that have been reported in more than twenty studied cases as presented in Table 1 and Figure 4.
8.1. Genes Involved in the Cell Cycle G1/S Transition (Gene Ontology (GO) ID:0044843)
The accurate transition from G1 to S phase of the cell cycle is critical for the control of the eukaryotic cell proliferation, while it has been found that misregulation promotes oncogenesis [131]. The CCNA2, CCNE1 and TERT genes take part in the G1/S transition process, and they have been found to be major genomic locations of recurrent HBV DNA integrations.
The proteins encoded by the CCNA2 and CCNE1 genes, cyclin A2 and cyclin E1, respectively, belong to the highly conserved cyclin family. All the members of the cyclin family are characterized by a dramatic periodicity in protein abundance through the cell cycle. Both proteins regulate the cell cycle by promoting the S phase entry and progression. Specifically, the cyclin A2 promotes the transition through G1/S and G2/M via activation of the cyclin-dependent kinase 2 while cyclin E1 accumulates at the G1/S phase boundary and is degraded as cells progress through S phase. Both genes are disrupted by HBV DNA integrations more frequently in tumors than in non-tumor samples. Moreover, cyclin A2 and E1 alterations define a homogenous entity of aggressive HCC, which is mostly developed in non-cirrhotic patients [130].
Cyclin-driven HCC displays a unique signature of structural rearrangements with hundreds of tandem duplications and templated insertions frequently activating the TERT promoter [130]. The HBx/HBsAg genes are the parts of the HBV genome that are mainly integrated into the CCNE1 [127,132,133]. It has been proposed that the disregulation of cyclins and especially interruption of the CCNA2 gene plays a direct role in cellular transformation and oncogenesis through the loss of cell cycle control [118,134]. As for the gene expression values, researchers suggested that up-regulation of cyclin-encoded genes results in chromosome instability and contributes to cancer progression, while it has been found that HBV integrations—particularly in the promoter region—elevate the gene expression levels [124].
The TERT gene is a protein-coding gene that contributes in the formation of the telomerase enzyme whose role is the maintenance of the telomeres. Telomeres role is the protection of chromosomes from abnormal sticking together and degradation. In most cells, including the hepatocytes, telomeres are progressively shortened because of cell division, and, when they reach a critical length, they trigger the cell to stop division and undergo apoptosis. The role of the telomerase is crucial in this process as it adds small repeated segments of DNA to the ends of the chromosomes each time the cell divides.
In general, the expression levels of the TERT gene are either undetectable or very low. Abnormal activation of telomerase has been reported in various types of cancers (including mostly melanoma and acute myeloid leukemia) and leads to cell growth and division without order. TERT gene disruptions have been associated with HCC, and particularly HBV integration in the TERT locus possibly confers a clonal advantage in the early phase of HBV-related liver carcinogenesis [114].
Over-expression of the TERT gene has been reported due to HBV DNA integration [82,107,121,129], while the possible functional impact of the HBs-TERT chimera that has been reported to occur after integration, should be further investigated [118]. Moreover, the gene disruption mediated by HBV integration can cause uncontrolled cell growth and can be a contributing factor in the formation of HCC in patients with chronic HBV infections.
Studies have found that the integration is more frequent in the promoter region than in the intronic region of the TERT gene [107,116,121,125,135] and that this possibly results in a “cis” effect on the transcription of the gene [17,82,125]. It has been also suggested that the HBV-TERT integration frequency may differ in different populations and HBV subtypes [127].
8.2. Genes Involved in the DNA Replication (GO:0006260)
In addition to the CCNA2, CCNE1 genes, the GLI2 gene is also involved in DNA replication, and it has been found to be affected by HBV integrations. The GLI2 gene encodes a protein that belongs to the Gii family and specifically to the C2H2-type zinc finger protein subclass, which has been characterized as a transcription factior (TF) with the ability to bind DNA through zinc finger motifs. The genes of this subclass have been described as potential oncogenes in the embryonal carcinoma cell while the GLI2 gene possibly plays a role during embryogenesis. Moreover, GLI2 is considered a marker of activation of the sonic hedgehog signaling pathway, which has been associated with aggressive types of renal cell carcinoma [136,137,138] as well as HCC [139,140].
HBV DNA integrations within the GLI2 gene are more frequent in tumor samples, while there is no reported evidence regarding the gene expression values in hepatocytes with HBV integrations. Studies regarding the expression of GLI2 in HCC tissues have revealed that it is up-regulated [139,141], and it has been associated with a poor prognosis in patients with HCC after hepatectomy [141].
8.3. Genes Involved in the Histone Modification (GO:0016570)
The KMT2B gene takes part in the histone modification process along with the CCNA2 gene. The KMT2B is an oncogene [142] that encodes the lysine-specific methyltransferase 2D enzyme that is found in many organs and tissues, including the liver, while its main role is the regulation of developmental-related genes. Moreover, it has been described as a tumor suppressor gene as it helps in the prevention of uncontrolled cell growing and division.
HBV integration sites in the KMT2B gene are more frequent in tumor samples, while it has been reported that the gene is upregulated in tumor samples relative to the adjacent normal tissue samples [82]. Another observation that should be further examined is that tumor samples with HBV integrations in the KMT2B gene do not have HBV integrations in the TERT gene and vice versa [115].
8.4. Genes Involved in the Stem Cell Differentiation (GO:0048863)
The genes SOX5 and the FN1 are involved in stem cell differentiation while being recurrently affected by HBV DNA integrations. The SOX5 gene encodes a member of the SRY-related HMG-box (SOX) family of transcription factors, which play important roles in the maintenance of embryonic stem (ES) cell self-renewal and pluripotency. This gene is involved in the regulation of embryonic development and determination of the cell fate, and it has also been suggested that it possibly acts as a transcriptional regulator after the formation of a protein complex.
It has been associated with various types of cancer, including HCC, gastric, breast and prostate cancer. HBV integrations within the SOX5 have been reported mostly in non-tumor samples. In general, the expression of the SOX gene family is frequently down-regulated in gastric cancer and HCC [143], while studies focusing particularly on the SOX5 gene reported up-regulation in HCC tissues and cell lines as well as in gastric cancer [144,145]. Up-regulation and over-expression of the SOX5 gene has been associated with metastasis and lower progression-free survival [145].
The FN1 gene encodes the soluble plasma fibronectin-1 and the insoluble cellular fibronectin-1 proteins. The soluble plasma fibronectin-1 protein is produced by the liver cells, and it is released into the bloodstream where it is involved in blood clotting and wound healing while it also assists in the formation of the extracellular matrix. Moreover, both fibronectin-1 proteins take part in the expansion, migration and differentiation of the cells. FN1 has been described as a cancer-related gene.
Mutations at the FN1 gene have been associated with fibronectin glumerulopathy—a kidney disease that results in irreversible kidney failure, while there is an association with various types of cancers, including liver cancer but mostly thyroid cancer. HBV integrations within the FN1 gene have been reported mostly in non-tumor samples, and it has been found that the gene is downregulated in tumor samples [82].
These observations imply that HBV integrations occur in normal liver tissue but are not directly associated with HCC [82]. Detected HBV-FN1 fusion transcripts have been associated with the 5′ side of the FN1 exons [123] and are predicted to cause alternative splicing [146]. Given that FN1 is a key modulator of fibrosis [147], the HBV-FN1 fusion protein is possibly involved in the pathogenesis of liver fibrosis [123]. Moreover, HBV integrations in the FN1 gene occur preferentially in cases with a high liver fibrosis stage rather than in cases with a low fibrosis stage [122].
8.5. Genes Involved in the Wnt Signaling Pathway (GO:0016055, GO:0198738)
Three genes recurrently affected by HBV DNA integrations are involved in the Wnt signaling pathway and cell–cell signaling. These genes are CCNE1, TERT and PRKN. Specifically, the PRKN gene is involved in negative regulation of the Wnt signaling pathway (GO:0090090), while the TERT gene is involved in positive regulation of the Wnt signaling pathway (GO:0030177).
The PRKN gene encodes the parkin protein, a member of the E3 ubiquitin ligases group, that plays a role in the cell machinery by degrading damaged and excess proteins after tagging them with ubiquitin molecules. Ubiquitin-tagged unneeded proteins are moved to the proteasome for degradation. Moreover, the ubiquitin–proteasome system regulates the availability of proteins that are involved in several critical cell activities, including the timing of cell division and growth, and thus it was proposed that parkin acts as a tumor suppressor protein [148].
The PRKN gene is an extremely large gene (1.5 Mb) that is located within the FRA6E—the third most active common fragile site [149]. It is frequently altered in ovarian, breast and hepatocellular carcinomas, while it has also been associated with Parkinson and Hansen (also known as leprosy) diseases. Moreover, it has been found that the gene is down-regulated in HCC samples compared to non-tumor samples [148].
8.6. Genes Involved in Angiogenesis (GO:0001525)
The FN1, TERT and GTF2I genes are involved in angiogenesis or, in other words, the formation of new blood cells, and they have been found to be affected by HBV DNA integrations. The GTF2I gene belongs to the general transcription factors family and encodes the TFII-I transcription factor, which has been associated with the coordination of cell growth and division as well as the control of the calcium flow into the cells. TFII-I is ubiquitously expressed in human cells. It is localized at the cytoplasm, and it translocates to the nucleus at the time of transcription [150].
Genomic alterations within the GTF2I gene have been recurrently associated with thymic epithelial tumors and sporadically with other types of cancer [151,152,153,154,155]. HBV integrations within the GTF2I gene have been reported mostly in non-tumor samples, while there is no evidence regarding the effect of the integrations in the gene expression values.
8.7. Genes Involved in Blood Circulation (GO:0008015)
The CHRM3, PDE3A and CPS1 genes are involved in blood circulation and are affected by HBV DNA integrations, while the ALB and FN1 genes are directly related to the blood as blood microparticles (GO:0072562).
The PDE3A gene encodes a member of the cGMP-inhibited cyclic nucleotide phosphodiesterase (cGI-PDE) family, whose role is the hydrolyzation of cAMP and cGMP and, thus, is a key point in the regulation of intracellular cyclic nucleotide signals [156]. PDE3A has been associated with breast cancer [157], lung adenocarcinoma, cervical carcinoma, melanoma [158] and HCC [159], and it has been found to be overexpressed when compared to non-tumor samples [158,159]. HBV integrations are mainly reported in tumor samples.
The CHRM3 gene encodes a member of the G protein-coupled receptor family, with the ability of acetylcholine binding and, consequently, the inhibition of adenylate cyclase and degeneration of phosphoinositide mediation of potassium channels and, generally, synaptic transmission. CHRM3 overexpression has been associated with endometrial carcinoma [160], colorectal cancer [161,162], breast cancer [163], prostate cancer [164], lung cancer [165] and gastric cancer [166]. HBV integrations are mostly reported in non-tumor samples, while the role of this gene in HCC as well as the effect of the integrations in gene expression values remains unknown.
The CPS1 gene encodes the liver-specific rate-limiting carbamoyl phosphate synthetase I enzyme, which participates in the urea cycle and facilitates the efficient removal of ammonia from the body. Specifically, carbamoyl phosphate synthetase I is the enzyme that catalyzes the first step of the urea cycle in a reaction in which excess nitrogen compounds are incorporated into the cycle to be processed.
Furthermore, research found that CPS1 knockdown reduced the cell growth [119,167] and decreased the metabolite levels associated with nucleic acid biosynthesis pathway [167]. HBV integrations in the CPS1 gene have been found mostly in non-tumor samples. The expression levels of CPS1 in HBV-integrated genomes has not been studied, although studies regarding the role of CPS1 in HCC suggest that, in HCC samples, the CPS1 is hypermethylated, and the gene is down-regulated [121].
ALB encodes the most abundant protein in the blood plasma, the albumin, and, as a consequence, it has an enhanced role in the liver. The encoded protein functions in the regulation of the blood plasma colloid osmotic pressure and acts as a carrier protein for a wide range of endogenous molecules. ALB expression has been reported to be restricted in the liver, and mutations at this gene have been associated with liver cancer. HBV integrations that disrupt the ALB gene have been reported in multiple studies, and it has been shown that they more commonly occur in non-tumor samples than in tumor samples. As for the expression levels, it has been reported that the gene is up-regulated and highly expressed in tumor samples with HBV integrations [130].
8.8. Other Protein-Coding Affected Genes
The SH3RF3 gene encodes the SH3 domain containing ring finger 3, also known as the POSH2 protein (plenty of SH3 domains protein 2). This particular protein has been recently identified, and it has four Src homology 3 (SH3) domains and a Ring finger domain [168]. SH3RF3 has been associated with acute lymphoblastic leukemia [169], HIV-associated neurocognitive disorder [170], late-onset familial Alzheimer’s disease [171] as well as breast cancer [172]. Although, the functional role and mechanism of SH3RF3 in pathological processes, especially in cancer, are largely unexplored [172]. HBV integrations have been reported to affect the SH3RF3 gene, and HBV integrations are more frequent in tumor-derived samples.
The KCNT2 gene belongs to a large gene family whose members encode proteins for the generation of potassium channels. The KCNT2 gene encodes a subunit of a voltage-gated potassium channel named SLICK (sequence like an intermediate calcium channel) that is activated by an increase of cytoplasmic Na+ concentration and contributes to slow after-hyperpolarization in certain neuronal populations in the brain [173]. This gene has been associated with various types of cancer, including skin melanoma. HBV integrations have been reported to interrupt the KCNT2 gene mostly in non-tumor samples. Regarding HCC, it has been found that the KCNT2 gene is up-regulated when compared with normal samples [174,175], but there is no other evidence regarding the role of HBV integrations in the observed overexpression of the gene in HCC samples.
The LINGO2 gene belongs to the LINGO gene family and encodes a novel protein that has a role in the central nervous system during early developmental stages as well as in the limbic system and cerebral cortex in adult tissues. The role of the gene in health and disease remains unclear as it has been reported to play a role in multiple diseases including asthma [176], chronic obstructive pulmonary disease [177], Parkinson’s and inflammatory bowel disease [178], as well as various types of cancer (esophageal squamous cell carcinoma, gastric cancer, pancreatic cancer, renal cell carcinoma, lung cancer, breast cancer, bladder cancer and prostate cancer) [179,180,181]. Specifically, in gastric cancer, LINGO2 up-regulation has been associated with advanced clinical stage and decreased Overall Survival (OS) [180].
8.9. Long Non-Coding RNA Genes (lincRNAs)
The LINC00486 and FAM157A genes are long non-coding RNA genes (lincRNAs) that are recurrently affected by HBV integrations. The LINC00486 gene is a member of the Long Intergenic Non-Protein Coding RNAs (LINC) group, while the FAM157A gene is a member of the Long non-coding RNAs with the FAM root symbol. HBV integrations within the LINC00486 gene are more frequent in non-tumor samples, while HBV integrations within the FAM157A have been found almost only in tumor samples.
There is no extended bibliography regarding the role of those two genes in tumor progression and association with HCC. Although, in the last couple of years, there has been a growing body of evidence that lincRNAs are induced by cellular stress, thus, promoting cell proliferation and the invasion of various types of cancer [119,182,183]. Moreover, the functional impact of mutations, genomic alterations and chromosomal deletions, insertions and translocations in these genes is unknown. Although, it has been proposed that genetic alterations at the genomic sites referring to lincRNAs may influence the binding affinity for proteins and possibly result in higher-order structure of the lincRNAs [184].
It should be also mentioned that the interplay between HBV and generally viral infections and the miRNA machinery of the host is a subject of intensive investigations, as there is evidence of existence of viral mechanisms that lead to reprogramming of the miRNA machinery [185]. Specifically, the HBV X protein triggers selective miRNA downregulation [186] while at the same time, HBV integration sites within miRNA genes (MIR4457 and MIR4424) have been reported [115].
Moreover, associations of the number of HBV integration sites with the patient survival have been reported. Specifically, a positive association between the large numbers of integration sites and the increased serum HBsAg and alpha-fetoprotein (AFP) levels has been reported, while, at the same time, cases with HCC and large numbers of HBV integrations have been reported to survive a significantly shorter time than those with no or low numbers of HBV integrations [82]. Finally, individuals with HBV integrations seem to develop HCC at younger ages [82].
Analysis of the correlation between HBV genotypes and their integration frequencies would be necessary to understand whether they exhibit different capacity of integration. At the same time, the role of HBV integration events in different subclasses of HCC should also be investigated. Moreover, different technologies might have different false positive and false negative HBV integration identification rates.
False Positive (FP) results can be obtained from different methodologies used during library preparation and ligation procedures, while the False Negative (FN) rate may be related to the sequencing depth, quality as well as the mapping parameters. Another major issue is the bioinformatics methodology used to declare an integration event and, in brief, the number of supporting mapped reads and whether these reads are concordantly mapped paired-end reads or chimeric reads consisting of both viral and host genomic parts.
9. Molecular Mechanisms of Hepatocarcinogenesis Caused by HBV Integration
The oncogenic contribution of HBV DNA integration in HCC is not yet clear. It has been proposed that the insertion of viral DNA into cellular genomic regulatory regions and coding regions results in the modification of gene expression (cis-activation) and production of structurally and functionally aberrant cellular or hybrid proteins. Specifically, HBV can promote HCC in a variety of ways, including the accumulation of genetic damage due to immune-mediated hepatic inflammation [188,189], the induction of oxidative stress [101,190,191,192], a virus-specific mechanism that involves the HBx and HBs viral proteins [193,194,195,196], epigenetic modifications [197,198] and regulation of miRNA expression [186,199,200].
The role of immunity in inflammation, the progression of fibrosis and hepatocarcinogenesis has been also extensively studied. HBV is considered to be a non-cytopathic virus, and the hepatocellular damage observed during HBV infections is mediated by the host’s immune response to the virus [201]. Specifically, HBV induces little or no innate immune responses and is regulated—without being eliminated—by HBV-specific CD4+ and CD8+ T cell responses and neutralizing antibodies [202,203]. Dysregulation of these adaptive immune responses results in chronic liver injury and subsequently HCC. At the same time, there are findings that support that HBV may be directly cytopathic in conditions of severe immune suppression, albeit the underlying mechanisms of the cytopathic effect have not yet been defined [204,205].
Furthermore, recent studies suggest that core promoter mutations at T1762/A1764 of the HBV genome are associated with a higher risk for developing HCC [206], particularly in young and non-cirrhotic individuals infected with HBV genotype B or C [207], as well, in HBV genotype C infections, the development of HCC can be predicted by mutations/deletions in the preS region [208,209]. Another way that HCC can be promoted after HBV infection is through insertional mutagenesis induced by the integration of HBV DNA into the host genome.
Targeted viral genome integration may lead to the activation of cellular genes with oncogenic potential [210], while up-regulation of the target genes expression endows the carrier hepatocytes with a selective growth advantage and clonal expansion leading to malignancy [105,211,212]. Moreover, genetic instability triggered by HBV integration is considered an important contributor in the pathogenesis of HCC [213,214].
Recently, it has been suggested that HBV integrations serve as the relevant source of HBsAg in the context of chronic infection [215], while elevated levels of HBsAg (≥100 IU/mL) are associated with an increased risk of HCC development [216]. Additionally, elevated HBsAg levels lead to HBV-specific B and T cells exhaustion and weakening of their response [217,218] and, as a result, inflammation in the liver microenvironment that further induces HCC development.
Generalized genomic instability, gene and chromosomal deletions or translocations, the amplification of cellular DNA as well as the generation of fusion transcripts are some major genetic alterations that have been associated with HBV integration sites and have been found to be key points in the progression of HCC [14,111]. These alterations presumably lead to the selection of clones of hepatocytes with a growth advantage.
As a consequence, invasive neoplastic clones with multiple alterations arise, and this whole process results in the deregulated cell proliferation and suppressed apoptosis that is mandatory for autonomous cell expansion. This whole process is enhanced by the increased rate of liver cell turnover that occurs during chronic HBV infection leading to cellular senescence, telomere shortening and generalized genomic instability that contributes to the frequency of additional primary integration events as well as secondary genomic rearrangements occurring at the sites of HBV integrations [14].
HBV integration events could also result in a substantial enhancement of oncogenic or anti-apoptotic signals that possibly have a profound influence on the susceptibility of the liver to carcinogenesis [14]. The major cellular process for the development of cancer is unconstrained cell proliferation, which depends on the balance between cell growth activity and apoptotic signaling pathways. Oncogenic molecules with the ability to promote cell proliferation are capable of inducing apoptosis and, consequently, have the homeostatic capability to limit their own proliferative effects.
Moreover, these molecules have the ability to inhibit the pro-apoptotic programs and disrupt the innate homeostatic mechanism, in a process that results in unregulated proliferation [219]. Due to the presence of complex network molecules within both the oncogenic and anti-apoptotic programs, HBV integration sites into different genomic locations likely affect different molecules while having similar results on the overall cellular neoplastic phenotype [14].
Moreover, HBV integrations that occur within or near non-coding repetitive elements, including Long Interspersed Nuclear Elements (LINEs), Short Interspersed Nuclear Elements (SINEs), Alu elements (named after the restriction enzyme from which those sequences are cut) and Long Terminal Repeats (LTRs) of Endogenous Retroviruses (ERVs), have also been associated with the progression of HCC [15,82,107]. Specifically, HBx-LINE1 chimeric transcripts have been identified in HCC cases and have been associated with poor outcome of the individuals that carry them [96]. The HBx-LINE1 chimeric transcript acts as a long-non-coding RNA (lncRNA) with significant oncogenic properties additional to the protein production [96].
10. Conclusions
HBV, a 3200 bp-length hepatotropic virus of the Hepadnaviridae family is the main ‘culprit’ of viral hepatitis (along with the Hepatitis C Virus—HCV). In terms of mortality, viral hepatitis is on par with AIDS, malaria and tuberculosis or, in other words, among the top four global infectious diseases. In 2016, the Wold Health Organization (WHO) committed to eliminating viral hepatitis as a public health threat by 2030 (apps.who.int/iris/bitstream/handle/10665/206453/WHO-HIV-2016.04-eng.pdf), while on 25 June 2021, during the ongoing SARS-CoV-2 pandemic, guidance on how HBV elimination should be validated (www.who.int/publications/i/item/9789240028395) was first released.
With only 9 years to go until the 2030 deadline is reached, there are still some important key points that should be considered [220]. HBV DNA integration is a major key point regarding the elimination of the virus as well as a burden in the development of a sterilizing cure for HBV as viral proteins are continually expressed even when the HBV DNA is no longer replicating [221]. The cccDNA, the state in which the HBV DNA exists in the nucleus of the infected hepatocytes and serves as a template of HBV DNA replication is minimally inhibited by currently available treatments [220].
Moreover, the high percentage of maternal–fetal (or mother-to-child or vertical) transmission, the minimal screening of HBV infected mothers along with the inability to access medical care facilities and anti-HBV agents for HBV-infected mothers with high HBV DNA levels to prevent vertical transmission especially in developing countries [222,223], the low percentage of HepB3 immunization coverage among one-years-olds that is presented in some regions, as well as the gap in knowledge on whether HBV DNA integrations are inherited in a mendelian fashion—like the Endogenous Retroviruses (ERVs)—or not, further complicate the elimination process.
Additionally, the relationship between HCC and HBV DNA integrations in various host genomic locations is under thorough investigation, while the role of HBV genotypes and sub-genotypes in the integration process along with the role of HBV integration events in different subclasses of HCC should be further studied. Research on the HBV DNA integrations will further enlighten the multistep development of liver cancer and the cooperative mechanisms during neoplastic transformation and will provide evidence for treatment strategies targeting HBV integrations in order to eliminate chronic hepatitis and, thus, decrease the percentage of cirrhosis and HCC.
Supplementary Materials
The following are available at https://www.mdpi.com/article/10.3390/microorganisms9081787/s1, Figure S1: Cumulative number of HBV DNA integrations in the human genes.
Author Contributions
Conceptualization, M.B. and T.K.; methodology, M.B. and T.K.; software, M.B. and T.K.; validation, M.B., G.P., D.P. and T.K.; investigation, M.B. and T.K.; data curation, M.B. and T.K.; writing—original draft preparation, M.B., G.P., D.P. and T.K.; writing—review and editing, M.B., G.P., D.P. and T.K.; visualization, M.B. and T.K.; supervision, T.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| HBV | Hepatitis B Virus |
| CHB | Chronic HBV infection |
| HCC | Hepatocellular Carcinoma |
| HBeAg | Hepatitis B e antigen |
| HBcAg | Hepatitis B core antigen |
| HBsAg | Hepatitis B surface antigen |
| anti-HBs | Hepatitis B surface antibody |
| anti-HBc | Total hepatitis B core antibody |
| IgM anti-HBc | IgM antibody to hepatitis B core antigen |
| rcDNA | relaxed circular DNA |
| dslDNA | double-stranded linear DNA |
| cccDNA | covalently closed circular DNA |
| pgRNA | pre-genomic RNA |
| NHEJ | Non-Homologous End Joining |
| MMEJ | Microhomology-Mediated End Joining |
References
- Blumberg, B.S.; Alter, H.J.; Visnich, S. A “New” Antigen in Leukemia Sera. JAMA J. Am. Med. Assoc. 1984, 252, 252–257. [Google Scholar] [CrossRef]
- Block, T.M.; Alter, H.J.; London, W.T.; Bray, M. A historical perspective on the discovery and elucidation of the hepatitis B virus. Antivir. Res. 2016, 131, 109–123. [Google Scholar] [CrossRef]
- Gruber, W.; Virchow, R. Ueber das Vorkommen und den Nachweis des hepatogenen, insbesondere des katarrhalischen Icterus. Arch. Pathol. Anat. Physiol. Klin. Med. 1865, 32, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Papavramidou, N.; Fee, E.; Christopoulou-Aletra, H. Jaundice in the hippocratic corpus. J. Gastrointest. Surg. 2007, 11, 1728–1731. [Google Scholar] [CrossRef]
- Neefe, J.R.; Gellis, S.S.; Stokes, J. Homologous serum hepatitis and infectious (epidemic) hepatitis: Studies in volunteers bearing on immunological and other characteristics of the etiological agents. Am. J. Med. 1946, 1, 3–22. [Google Scholar] [CrossRef]
- Ratnoff, O.D.; Patek, A.J.J.R. The natural history of laennec’s cirrhosis of the liver an Analysis of 386 CASES. Medicine 1942, 21, 207–268. [Google Scholar] [CrossRef]
- Millman, I.; Eisenstein, T.; Blumberg, B.S. The Development of the Hepatitis B Vaccine; Springer: Jersey City, NJ, USA, 1984; pp. 137–147. [Google Scholar] [CrossRef]
- Hoofnagle, J.H.; Mullen, K.D.; Jones, D.B.; Rustgi, V.; Di Bisceglie, A.; Peters, M.; Waggoner, J.G.; Park, Y.; Jones, E.A. Treatment of Chronic Non-A, Non-B Hepatitis with Recombinant Human Alpha Interferon. N. Engl. J. Med. 1986, 315, 1575–1578. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.L.; Chien, R.N.; Leung, N.W.; Chang, T.T.; Guan, R.; Tai, D.I.; Ng, K.Y.; Wu, P.C.; Dent, J.C.; Barber, J.; et al. A One-Year Trial of Lamivudine for Chronic Hepatitis B. N. Engl. J. Med. 1998, 339, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Shafritz, D.A.; Shouval, D.; Sherman, H.I.; Hadziyannis, S.J.; Kew, M.C. Integration of Hepatitis B Virus DNA into the Genome of Liver Cells in Chronic Liver Disease and Hepatocellular Carcinoma. N. Engl. J. Med. 1981, 305, 1067–1073. [Google Scholar] [CrossRef]
- Koshy, R.; Koch, S.; Freytag von Loringhoven, A.; Kahmann, R.; Murray, K.; Hofschneider, P.H. Integration of hepatitis B virus DNA: Evidence for integration in the single-stranded gap. Cell 1983, 34, 215–223. [Google Scholar] [CrossRef]
- Brechot, C.; Pourcel, C.; Louise, A.; Rain, B.; Tiollais, P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature 1980, 286, 533–535. [Google Scholar] [CrossRef]
- Chakraborty, P.R.; Ruiz-Opazo, N.; Shouval, D.; Shafritz, D.A. Identification of integrated hepatitis B virus DNA and expression of viral RNA in an HBsAg-producing human hepatocellular carcinoma cell line. Nature 1980, 286, 531–533. [Google Scholar] [CrossRef]
- Guerrero, R.B.; Roberts, L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005, 42, 760–777. [Google Scholar] [CrossRef]
- Murakami, Y.; Saigo, K.; Takashima, H.; Minami, M.; Okanoue, T.; Bréchet, C.; Paterlini-Bréchot, P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut 2005, 54, 1162–1168. [Google Scholar] [CrossRef]
- Saigo, K.; Yoshida, K.; Ikeda, R.; Sakamoto, Y.; Murakami, Y.; Urashima, T.; Asano, T.; Kenmochi, T.; Inoue, I. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma. Hum. Mutat. 2008, 29, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Ferber, M.J.; Montoya, D.P.; Yu, C.; Aderca, I.; McGee, A.; Thorland, E.C.; Nagorney, D.M.; Gostout, B.S.; Burgart, L.J.; Boix, L.; et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene 2003, 22, 3813–3820. [Google Scholar] [CrossRef] [Green Version]
- Paterlini-Bréchot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Bréchot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Murakami, Y.; Saigo, K.; Chami, M.; Mugnier, C.; Lagorce, D.; Okanoue, T.; Urashima, T.; Bréchot, C.; Paterlini-Bréchot, P. Identification of human cancer-related genes by naturally occurring hepatitis B virus DNA tagging. Oncogene 2001, 20, 6233–6240. [Google Scholar] [CrossRef] [Green Version]
- Razavi-Shearer, D.; Gamkrelidze, I.; Nguyen, M.H.; Chen, D.S.; Van Damme, P.; Abbas, Z.; Abdulla, M.; Abou Rached, A.; Adda, D.; Aho, I.; et al. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef]
- Waheed, Y.; Siddiq, M.; Jamil, Z.; Najmi, M.H. Hepatitis elimination by 2030: Progress and challenges. World J. Gastroenterol. 2018, 24, 4959–4961. [Google Scholar] [CrossRef]
- Hutin, Y.J.F.; Bulterys, M.; Hirnschall, G.O. How far are we from viral hepatitis elimination service coverage targets? J. Int. AIDS Soc. 2018, 21, e25050. [Google Scholar] [CrossRef] [Green Version]
- Tatemichi, M.; Furuya, H.; Nagahama, S.; Takaya, N.; Shida, Y.; Fukai, K.; Owada, S.; Endo, H.; Kinoue, T.; Korenaga, M. A nationwide cross-sectional survey on hepatitis B and C screening among workers in Japan. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.; Locarnini, S.A.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Paraskevis, D.; Magiorkinis, G.; Magiorkinis, E.; Ho, S.Y.; Belshaw, R.; Allain, J.P.; Hatzakis, A. Dating the origin and dispersal of hepatitis B virus infection in humans and primates. Hepatology 2013, 57, 908–916. [Google Scholar] [CrossRef]
- Paraskevis, D.; Angelis, K.; Magiorkinis, G.; Kostaki, E.; Ho, S.Y.; Hatzakis, A. Dating the origin of hepatitis B virus reveals higher substitution rate and adaptation on the branch leading to F/H genotypes. Mol. Phylogenet. Evol. 2015, 93, 44–54. [Google Scholar] [CrossRef]
- Comas, I.; Coscolla, M.; Luo, T.; Borrell, S.; Holt, K.E.; Kato-Maeda, M.; Parkhill, J.; Malla, B.; Berg, S.; Thwaites, G.; et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 2013, 45, 1176–1182. [Google Scholar] [CrossRef]
- Linz, B.; Balloux, F.; Moodley, Y.; Manica, A.; Liu, H.; Roumagnac, P.; Falush, D.; Stamer, C.; Prugnolle, F.; van der Merwe, S.W.; et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature 2007, 445, 915–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahila Bar-Gal, G.; Kim, M.J.; Klein, A.; Shin, D.H.; Oh, C.S.; Kim, J.W.; Kim, T.H.; Kim, S.B.; Grant, P.R.; Pappo, O.; et al. Tracing hepatitis B virus to the 16th century in a Korean mummy. Hepatology 2012, 56, 1671–1680. [Google Scholar] [CrossRef]
- Patterson Ross, Z.; Klunk, J.; Fornaciari, G.; Giuffra, V.; Duchêne, S.; Duggan, A.T.; Poinar, D.; Douglas, M.W.; Eden, J.S.; Holmes, E.C.; et al. The paradox of HBV evolution as revealed from a 16th century mummy. PLoS Pathog. 2018, 14, e1006750. [Google Scholar] [CrossRef] [Green Version]
- Mühlemann, B.; Jones, T.C.; Damgaard, P.d.B.; Allentoft, M.E.; Shevnina, I.; Logvin, A.; Usmanova, E.; Panyushkina, I.P.; Boldgiv, B.; Bazartseren, T.; et al. Ancient hepatitis B viruses from the Bronze Age to the Medieval period. Nature 2018, 557, 418–423. [Google Scholar] [CrossRef]
- Krause-Kyora, B.; Susat, J.; Key, F.M.; Kühnert, D.; Bosse, E.; Immel, A.; Rinne, C.; Kornell, S.C.; Yepes, D.; Franzenburg, S.; et al. Neolithic and medieval virus genomes reveal complex evolution of hepatitis B. eLife 2018, 7, 1–15. [Google Scholar] [CrossRef]
- Dill, J.A.; Camus, A.C.; Leary, J.H.; Di Giallonardo, F.; Holmes, E.C.; Ng, T.F.F. Distinct Viral Lineages from Fish and Amphibians Reveal the Complex Evolutionary History of Hepadnaviruses. J. Virol. 2016, 90, 7920–7933. [Google Scholar] [CrossRef] [Green Version]
- Littlejohn, M.; Locarnini, S.; Yuen, L. Origins and Evolution of Hepatitis B Virus and Hepatitis D Virus. Cold Spring Harb. Perspect. Med. 2016, 6, a021360. [Google Scholar] [CrossRef]
- Velkov, S.; Ott, J.J.; Protzer, U.; Michler, T. The global hepatitis B virus genotype distribution approximated from available genotyping data. Genes 2018, 9, 495. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef]
- Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World J. Gastroenterol. 2014, 20, 5427–5434. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. MMBR 2012, 76, 159–216. [Google Scholar] [CrossRef] [Green Version]
- Raihan, R.; Akbar, S.M.F.; Mahtab, M.A.; Takahashi, K.; Masumoto, J.; Tabassum, S.; Tee, K.K.; Mohamed, R.B. Genomic analysis of Hepatitis B virus and its association with disease manifestations in Bangladesh. PLoS ONE 2019, 14, e0218744. [Google Scholar] [CrossRef]
- Sarma, M.P.; Bhattacharjee, M.; Kar, P.; Medhi, S. Detection of HBV Genotype C in Hepatocellular Carcinoma Patients from North East India: A Brief Report. Asian Pac. J. Cancer Prev. APJCP 2018, 19, 1741–1746. [Google Scholar] [CrossRef]
- Livingston, S.E.; Simonetti, J.P.; Bulkow, L.R.; Homan, C.E.; Snowball, M.M.; Cagle, H.H.; Negus, S.E.; McMahon, B.J. Clearance of Hepatitis B e Antigen in Patients with Chronic Hepatitis B and Genotypes A, B, C, D, and F. Gastroenterology 2007, 133, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Giovanna, F.; Bortolotti, F.; Francesco, D. Natural history of chronic hepatitis B: Special emphasis on disease progression and prognostic factors. J. Hepatol. 2008, 48, 335–352. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef] [Green Version]
- Kuipery, A.; Gehring, A.J.; Isogawa, M. Mechanisms of HBV immune evasion. Antivir. Res. 2020, 179, 104816. [Google Scholar] [CrossRef]
- McMahon, B.J. The natural history of chronic hepatitis B virus infection. Hepatology 2009, 49, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, V.; Lo Iacono, O.; Cammà, C.; Vaccaro, A.; Giunta, M.; Martorana, G.; Fuschi, P.; Almasio, P.L.; Craxì, A. The long-term course of chronic hepatitis B. Hepatology 1999, 30, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Iloeje, U.H.; Yang, H.I.; Su, J.; Jen, C.L.; You, S.L.; Chen, C.J. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006, 130, 678–686. [Google Scholar] [CrossRef] [PubMed]
- European Association For The Study Of The Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [Green Version]
- Hui, C.K.; Leung, N.; Yuen, S.T.; Zhang, H.Y.; Leung, K.W.; Lu, L.; Cheung, S.K.; Wong, W.M.; Lau, G.K. Natural history and disease progression in Chinese chronic hepatitis B patients in immune-tolerant phase. Hepatology 2007, 46, 395–401. [Google Scholar] [CrossRef]
- McMahon, B.J. Epidemiology and natural history of hepatitis B. Semin. Liver Dis. 2005, 25, 3–8. [Google Scholar] [CrossRef]
- Lok, A.S.F.; McMahon, B.J. Chronic hepatitis B. Hepatology (Baltimore Md.) 2007, 45, 507–539. [Google Scholar] [CrossRef] [Green Version]
- Gish, R.G.; Given, B.D.; Lai, C.L.; Locarnini, S.A.; Lau, J.Y.; Lewis, D.L.; Schluep, T. Chronic hepatitis B: Virology, natural history, current management and a glimpse at future opportunities. Antivir. Res. 2015, 121, 47–58. [Google Scholar] [CrossRef]
- Thomas, E.; Yoneda, M.; Schiff, E.R. Viral hepatitis: Past and future of HBV and HDV. Cold Spring Harb. Perspect. Med. 2015, 5, a021345. [Google Scholar] [CrossRef] [Green Version]
- Iannacone, M.; Guidotti, L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844. [Google Scholar] [CrossRef]
- Hu, J.; Liu, K. Complete and incomplete hepatitis B virus particles: Formation, function, and application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef]
- Dane, D.S.; Cameron, C.H.; Briggs, M. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet 1970, 1, 695–698. [Google Scholar] [CrossRef]
- Luckenbaugh, L.; Kitrinos, K.M.; Delaney, W.E.t.; Hu, J. Genome-free hepatitis B virion levels in patient sera as a potential marker to monitor response to antiviral therapy. J. Viral Hepat. 2015, 22, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Barrera, A.; Guerra, B.; Notvall, L.; Lanford, R.E. Mapping of the Hepatitis B Virus Pre-S1 Domain Involved in Receptor Recognition. J. Virol. 2005, 79, 9786–9798. [Google Scholar] [CrossRef] [Green Version]
- Rabe, B.; Glebe, D.; Kann, M. Lipid-Mediated Introduction of Hepatitis B Virus Capsids into Nonsusceptible Cells Allows Highly Efficient Replication and Facilitates the Study of Early Infection Events. J. Virol. 2006, 80, 5465–5473. [Google Scholar] [CrossRef] [Green Version]
- Blondot, M.L.; Bruss, V.; Kann, M. Intracellular transport and egress of hepatitis B virus. J. Hepatol. 2016, 64, S49–S59. [Google Scholar] [CrossRef]
- KöNiger, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; McAllister, R.; Boregowda, R.; Sohn, J.A.; Ledesma, F.C.; Caldecott, K.W.; Seeger, C.; Hu, J. Does tyrosyl DNA phosphodiesterase-2 play a role in hepatitis B virus genome repair? PLoS ONE 2015, 10, e0128401. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA Polymerase κ Is a Key Cellular Factor for the Formation of Covalently Closed Circular DNA of Hepatitis B Virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Sheraz, M.; McGrane, M.; Chang, J.; Guo, J.T. DNA Polymerase alpha is essential for intracellular amplification of hepatitis B virus covalently closed circular DNA. PLoS Pathog. 2019, 15, e1007742. [Google Scholar] [CrossRef]
- Long, Q.; Yan, R.; Hu, J.; Cai, D.; Mitra, B.; Kim, E.S.; Marchetti, A.; Zhang, H.; Wang, S.; Liu, Y.; et al. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog. 2017, 13, e1006784. [Google Scholar] [CrossRef]
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap endonuclease 1 is involved in cccDNA formation in the hepatitis B virus. PLoS Pathog. 2018, 14, e1007124. [Google Scholar] [CrossRef]
- Gerlich, W.H.; Robinson, W.S. Hepatitis B virus contains protein attached to the 5’ terminus of its complete DNA strand. Cell 1980, 21, 801–809. [Google Scholar] [CrossRef]
- Guo, H.; Jiang, D.; Zhou, T.; Cuconati, A.; Block, T.M.; Guo, J.T. Characterization of the Intracellular Deproteinized Relaxed Circular DNA of Hepatitis B Virus: An Intermediate of Covalently Closed Circular DNA Formation. J. Virol. 2007, 81, 12472–12484. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [Green Version]
- Rall, L.B.; Standring, D.N.; Laub, O.; Rutter, W.J. Transcription of hepatitis B virus by RNA polymerase II. Mol. Cell. Biol. 1983, 3, 1766–1773. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Summers, J. Integration of hepadnavirus DNA in infected liver: Evidence for a linear precursor. J. Virol. 1999, 73, 9710–9717. [Google Scholar] [CrossRef] [Green Version]
- Mason, W.S.; Jilbert, A.R.; Summers, J. Clonal expansion of hepatocytes during chronic woodchuck hepatitis virus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 1139–1144. [Google Scholar] [CrossRef] [Green Version]
- Summers, J.; Mason, W.S. Residual integrated viral DNA after hepadnavirus clearance by nucleoside analog therapy. Proc. Natl. Acad. Sci. USA 2004, 101, 638–640. [Google Scholar] [CrossRef] [Green Version]
- Mason, W.S.; Liu, C.; Aldrich, C.E.; Litwin, S.; Yeh, M.M. Clonal expansion of normal-appearing human hepatocytes during chronic hepatitis B virus infection. J. Virol. 2010, 84, 8308–8315. [Google Scholar] [CrossRef] [Green Version]
- Tu, T.; Mason, W.S.; Clouston, A.D.; Shackel, N.A.; McCaughan, G.W.; Yeh, M.M.; Schiff, E.R.; Ruszkiewicz, A.R.; Chen, J.W.; Harley, H.A.; et al. Clonal expansion of hepatocytes with a selective advantage occurs during all stages of chronic hepatitis B virus infection. J. Viral Hepat. 2015, 22, 737–753. [Google Scholar] [CrossRef]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology 2016, 151, 986–998.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an In Vitro Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92, e02007-17. [Google Scholar] [CrossRef] [Green Version]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Zhao, L.H.; Liu, X.; Yan, H.X.; Li, W.Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jhunjhunwala, S.; Liu, J.; Haverty, P.M.; Kennemer, M.I.; Guan, Y.; Lee, W.; Carnevali, P.; Stinson, J.; Johnson, S.; et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012, 22, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Liu, A.; Xia, Y. Insights into Hepatitis B Virus DNA Integration-55 Years after Virus Discovery. Innov. States 2020, 1, 100034. [Google Scholar] [CrossRef]
- Dejean, A.; Sonigo, P.; Wain-Hobson, S.; Tiollais, P. Specific hepatitis B virus integration in hepatocellular carcinoma DNA through a viral 11-base-pair direct repeat. Proc. Natl. Acad. Sci. USA 1984, 81, 5350–5354. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Yang, Y.; Zhang, L.; Tang, G.; Wang, Y.; Xue, G.; Zhou, W.; Sun, S. Characterization of the genotype and integration patterns of hepatitis B virus in early- and late-onset hepatocellular carcinoma. Hepatology 2015, 61, 1821–1831. [Google Scholar] [CrossRef]
- Chauhan, R.; Churchill, N.D.; Mulrooney-Cousins, P.M.; Michalak, T.I. Initial sites of hepadnavirus integration into host genome in human hepatocytes and in the woodchuck model of hepatitis B-associated hepatocellular carcinoma. Oncogenesis 2017, 6. [Google Scholar] [CrossRef]
- Mason, W.S.; Low, H.C.; Xu, C.; Aldrich, C.E.; Scougall, C.A.; Grosse, A.; Clouston, A.; Chavez, D.; Litwin, S.; Peri, S.; et al. Detection of Clonally Expanded Hepatocytes in Chimpanzees with Chronic Hepatitis B Virus Infection. J. Virol. 2009, 83, 8396–8408. [Google Scholar] [CrossRef] [Green Version]
- Koch, S.; Freytag von Loringhoven, A.; Kahmann, R.; Hofschneider, P.H.; Koshy, R. The genetic organization of integrated hepatitis B virus DNA in the human hepatoma cell line PLC/PRF/5. Nucleic Acids Res. 1984, 12, 6871–6886. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.; Burke, K.; Chou, M.J.; Zeldis, J.B.; Yang, C.S.; Lee, C.S.; Isselbacher, K.J.; Wands, J.R.; Goodman, H.M. Tight clustering of human hepatitis B virus integration sites in hepatomas near a triple-stranded region. J. Virol. 1987, 61, 3491–3498. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.H.; Robinson, W.S. Hepatitis B virus DNA forms in nuclear and cytoplasmic fractions of infected human liver. Virology 1984, 137, 390–399. [Google Scholar] [CrossRef]
- Yaginuma, K.; Kobayashi, M.; Yoshida, E.; Koike, K. Hepatitis B virus integration in hepatocellular carcinoma DNA: Duplication of cellular flanking sequences at the integration site. Proc. Natl. Acad. Sci. USA 1985, 82, 4458–4462. [Google Scholar] [CrossRef] [Green Version]
- Hino, O.; Ohtake, K.; Rogler, C.E. Features of two hepatitis B virus (HBV) DNA integrations suggest mechanisms of HBV integration. J. Virol. 1989, 63, 2638–2643. [Google Scholar] [CrossRef] [Green Version]
- Hino, O.; Shows, T.B.; Rogler, C.E. Hepatitis B virus integration site in hepatocellular carcinoma at chromosome 17;18 translocation. Proc. Natl. Acad. Sci. USA 1986, 83, 8338–8342. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.C.; Sun, T.; Ching, A.K.; He, M.; Li, J.W.; Wong, A.M.; Co, N.N.; Chan, A.W.; Li, P.S.; Lung, R.W.; et al. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef] [Green Version]
- Budzinska, M.A.; Shackel, N.A.; Urban, S.; Tu, T. Sequence analysis of integrated hepatitis B virus DNA during HBeAg-seroconversion. Emerg. Microbes Infect. 2018, 7. [Google Scholar] [CrossRef]
- Mcvey, M.; Lee, S.E.; Avenue, H.; Antonio, S. MMEJ repair of double-strand breaks: Deleted sequences and alternative endings. Trends Genet. 2017, 24, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lau, S.H.; Sham, J.S.T.; Wu, M.C.; Wang, T.; Guan, X.Y. Characterization of HBV integrants in 14 hepatocellular carcinomas: Association of truncated X gene and hepatocellular carcinogenesis. Oncogene 2004, 23, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Tarocchi, M.; Polvani, S.; Marroncini, G.; Galli, A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J. Gastroenterol. 2014, 20, 11630–11640. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Schneider, R.J. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef] [Green Version]
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artaman, A.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.M.; Hwang, L.Y.; Hatten, C.J.; Swaim, M.; Li, D.; Abbruzzese, J.L.; Beasley, P.; Patt, Y.Z. Risk factors for hepatocellular carcinoma: Synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology 2002, 36, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.A.; Wu, D.M.; Lin, C.C.; Lu, S.N.; You, S.L.; Wang, L.Y.; Wu, M.H.; Chen, C.J. Incidence and cofactors of hepatitis C virus-related hepatocellular carcinoma: A prospective study of 12,008 men in Taiwan. Am. J. Epidemiol. 2003, 157, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Yang, Z.; Li, W.; Li, X.; Wang, Y.; Zhang, J.; Xu, C.; Chen, P.J.; Hou, J.; McCrae, M.A.; et al. Re-evaluation of the Carcinogenic Significance of Hepatitis B Virus Integration in Hepatocarcinogenesis. PLoS ONE 2012, 7, e40363. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Chan, J.Y.; Yeo, W.; Tam, J.S.; Johnson, P.J. Frequent integration of precore/core mutants of hepatitis B virus in human hepatocellular carcinoma tissues. J. Viral Hepat. 2000, 7, 115–123. [Google Scholar] [CrossRef]
- Ding, D.; Lou, X.; Hua, D.; Yu, W.; Li, L.; Wang, J.; Gao, F.; Zhao, N.; Ren, G.; Li, L.; et al. Recurrent Targeted Genes of Hepatitis B Virus in the Liver Cancer Genomes Identified by a Next-Generation Sequencing-Based Approach. PLoS Genet. 2012, 8, e1003065. [Google Scholar] [CrossRef] [Green Version]
- Minami, M.; Daimon, Y.; Mori, K.; Takashima, H.; Nakajima, T.; Itoh, Y.; Okanoue, T. Hepatitis B virus-related insertional mutagenesis in chronic hepatitis B patients as an early drastic genetic change leading to hepatocarcinogenesis. Oncogene 2005, 24, 4340–4348. [Google Scholar] [CrossRef] [Green Version]
- Tokino, T.; Matsubara, K. Chromosomal sites for hepatitis B virus integration in human hepatocellular carcinoma. J. Virol. 1991, 65, 6761–6764. [Google Scholar] [CrossRef] [Green Version]
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659. [Google Scholar] [CrossRef] [Green Version]
- Feitelson, M.A.; Lee, J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007, 252, 157–170. [Google Scholar] [CrossRef]
- Tang, D.; Li, B.; Xu, T.; Hu, R.; Tan, D.; Song, X.; Jia, P.; Zhao, Z. VISDB: A manually curated database of viral integration sites in the human genome. Nucleic Acids Res. 2019, 48, D633–D641. [Google Scholar] [CrossRef]
- Horikawa, I.; Barrett, J.C. Transcriptional regulation of the telomerase hTERT gene as a target for cellular and viral oncogenic mechanisms. Carcinogenesis 2003, 24, 1167–1176. [Google Scholar] [CrossRef]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Tannir, N.M.; Williams, M.D.; Chen, Y.; Yao, H.; Zhang, J.; Thompson, E.J.; Meric-Bernstam, F.; Medeiros, L.J.; Weinstein, J.N.; et al. Landscape of DNA Virus Associations across Human Malignant Cancers: Analysis of 3775 Cases Using RNA-Seq. J. Virol. 2013, 87, 8916–8926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zeng, X.; Lee, N.P.; Liu, X.; Chen, S.; Guo, B.; Yi, S.; Zhuang, X.; Chen, F.; Wang, G.; et al. HIVID: An efficient method to detect HBV integration using low coverage sequencing. Genomics 2013, 102, 338–344. [Google Scholar] [CrossRef]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2016, 51, 473–486. [Google Scholar] [CrossRef]
- Chiu, Y.T.; Wong, J.K.; Choi, S.W.; Sze, K.M.; Ho, D.W.; Chan, L.K.; Lee, J.M.; Man, K.; Cherny, S.; Yang, W.L.; et al. Novel pre-mRNA splicing of intronically integrated HBV generates oncogenic chimera in hepatocellular carcinoma. J. Hepatol. 2016, 64, 1256–1264. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, Z.C.; Duan, M.; Lin, Y.H.; Zhou, X.Y.; Worthley, D.L.; Wang, X.Y.; Niu, G.; Xia, Y.; Deng, M.; et al. Cell Culture System for Analysis of Genetic Heterogeneity Within Hepatocellular Carcinomas and Response to Pharmacologic Agents; Elsevier: Amsterdam, The Netherlands, 2017; pp. 232–242.e4. Volume 152. [Google Scholar] [CrossRef] [Green Version]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.; Wang, W.; Wang, Q.; Fiel, M.I.; Lee, E.; Hiotis, S.P.; Zhu, J. A pilot systematic genomic comparison of recurrence risks of hepatitis B virus-associated hepatocellular carcinoma with low- and high-degree liver fibrosis. BMC Med. 2017, 15, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, M.; Tanaka, H.; Shiraishi, Y.; Unida, T.; Imamura, M.; Fujimoto, A.; Fujita, M.; Sasaki-Oku, A.; Maejima, K.; Nakano, K.; et al. Characterization of HBV integration patterns and timing in liver cancer and HBV-infected livers. Oncotarget 2018, 9, 25075–25088. [Google Scholar] [CrossRef] [Green Version]
- Li, C.L.; Li, C.Y.; Lin, Y.Y.; Ho, M.C.; Chen, D.S.; Chen, P.J.; Yeh, S.H. Androgen Receptor Enhances Hepatic Telomerase Reverse Transcriptase Gene Transcription After Hepatitis B Virus Integration or Point Mutation in Promoter Region. Hepatology 2019, 69, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ye, S.; Zhao, X.; Ji, L.; Zhang, Y.; Zhou, P.; Sun, J.; Guan, Y.; Han, Y.; Ni, C.; et al. Molecular characterization of HBV DNA integration in patients with hepatitis and hepatocellular carcinoma. J. Cancer 2018, 9, 3225–3235. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.W.; Alaei-Mahabadi, B.; Samuelsson, T.; Lindh, M.; Larsson, E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 2013, 4, 2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhang, L.; Qian, Z.; Zhu, X.; Zhu, G.; Chen, Y.; Xie, X.; Ye, Q.; Zang, J.; Ren, Z.; et al. Identification of HBV-MLL4 integration and its molecular basis in Chinese hepatocellular carcinoma. PLoS ONE 2015, 10, e0123175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liu, S.; Shen, C.; Wu, Y.; Zhang, L.; Chen, X.; Lu, F. DNA methylation consistency implicates the primary tumor cell origin of recurrent hepatocellular carcinoma. Epigenomics 2015, 7, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Wendl, M.C.; Wyczalkowski, M.A.; Wylie, K.; Ye, K.; Jayasinghe, R.; Xie, M.; Wu, S.; Niu, B.; Grubb, R.; et al. Divergent viral presentation among human tumors and adjacent normal tissues. Sci. Rep. 2016, 6, 28294. [Google Scholar] [CrossRef] [Green Version]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V.; Shinde, J.; Nault, J.C.; Mami, I.; Couchy, G.; Amaddeo, G.; Tubacher, E.; et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Bertoli, C.; Skotheim, J.M.; De Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Deng, Q.; Wang, Q.; Li, K.Y.; Dai, J.H.; Li, N.; Zhu, Z.D.; Zhou, B.; Liu, X.Y.; Liu, R.F.; et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat. Genet. 2012, 44, 1117–1121. [Google Scholar] [CrossRef]
- Toh, S.T.; Jin, Y.; Liu, L.; Wang, J.; Babrzadeh, F.; Gharizadeh, B.; Ronaghi, M.; Toh, H.C.; Chow, P.K.H.; Chung, A.Y.; et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis 2013, 34, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Hunter, T.; Pines, J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell 1994, 79, 573–582. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.I.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 2015, 6, 6–13. [Google Scholar] [CrossRef]
- Furler, R.L.; Uittenbogaart, C.H. GLI2 regulates TGF-β1 in human CD4+ T cells: Implications in cancer and HIV pathogenesis. PLoS ONE 2012, 7, e40874. [Google Scholar] [CrossRef] [PubMed]
- Swartling, F.J.; Savov, V.; Persson, A.I.; Chen, J.; Hackett, C.S.; Northcott, P.A.; Grimmer, M.R.; Lau, J.; Chesler, L.; Perry, A.; et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 2012, 21, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wang, L.; Nandy, D.; Zhang, Y.; Basu, A.; Radisky, D.; Mukhopadhyay, D. Neuropilin-1 upholds dedifferentiation and propagation phenotypes of renal cell carcinoma cells by activating Akt and sonic hedgehog axes. Cancer Res. 2008, 68, 8667–8672. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Joon, W.Y.; Xiao, X.; Dean, N.M.; Monia, B.P.; Marcusson, E.G. Selective down-regulation of glioma-associated oncogene 2 inhibits the proliferation of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 3583–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Han, C.; Lu, L.; Magliato, S.; Wu, T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology 2013, 58, 995–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Cao, L.; Li, Y.; Lu, H.; Yang, X.; Xue, P. Expression of glioma-associated oncogene 2 (Gli 2) is correlated with poor prognosis in patients with hepatocellular carcinoma undergoing hepatectomy. World J. Surg. Oncol. 2013, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Chen, L.H.; Huang, Y.; Chang, C.C.; Wang, P.; Pirozzi, C.J.; Qin, X.; Bao, X.; Greer, P.K.; McLendon, R.E.; et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget 2013, 4, 2144–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsubo, T.; Akiyama, Y.; Hashimoto, Y.; Shimada, S.; Goto, K.; Yuasa, Y. Microrna-126 inhibits sox2 expression and contributes to gastric carcinogenesis. PLoS ONE 2011, 6, e16617. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Han, S.; Wang, X.; Peng, R.; Li, X. SOX5 promotes epithelial–mesenchymal transition and cell invasion via regulation of Twist1 in hepatocellular carcinoma. Med. Oncol. 2015, 32, 10. [Google Scholar] [CrossRef]
- You, J.; Zhao, Q.; Fan, X.; Wang, J. SOX5 promotes cell invasion and metastasis via activation of twist-mediated epithelial– mesenchymal transition in gastric cancer. OncoTargets Ther. 2019, 12, 2465–2476. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, Y.; Fujimoto, A.; Furuta, M.; Tanaka, H.; Chiba, K.I.; Boroevich, K.A.; Abe, T.; Kawakami, Y.; Ueno, M.; Gotoh, K.; et al. Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers. PLoS ONE 2014, 9, e114263. [Google Scholar] [CrossRef]
- Tamkun, J.W.; Hynes, R.O. Plasma fibronectin is synthesized and secreted by hepatocytes. J. Biol. Chem. 1983, 258, 4641–4647. [Google Scholar] [CrossRef]
- Wang, F.; Denison, S.; Lai, J.P.; Philips, L.A.; Montoya, D.; Kock, N.; Schüle, B.; Klein, C.; Shridhar, V.; Roberts, L.R.; et al. Parkin Gene Alterations in Hepatocellular Carcinoma. Genes Chromosom. Cancer 2004, 40, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Kähkönen, M. Population cytogenetics of folate-sensitive fragile sites—I. Common fragile sites. Hum. Genet. 1988, 80, 344–348. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Iny Stein, T.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 2016, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.L.; Du, H.; Gregor, P.D.; Novina, C.D.; Martinez, E.; Roeder, R.G. Cloning of an inr- and E-box-binding protein, TFII-I, that interacts physically and functionally with USF1. EMBO J. 1997, 16, 7091–7104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarrini, G.; Ciabatti, E.; Pacini, S.; Galimberti, S.; Petrini, I. GTF2I mutations are common in thymic epithelial tumors but not in hematological malignancies. Anticancer Res. 2017, 37, 5459–5462. [Google Scholar] [CrossRef] [PubMed]
- Müllauer, L. GTF2I gene mutation—A driver of thymoma pathogenesis. Mediastinum 2017, 1, 18. [Google Scholar] [CrossRef]
- Petrini, I.; Meltzer, P.S.; Kim, I.K.; Lucchi, M.; Park, K.S.; Fontanini, G.; Gao, J.; Zucali, P.A.; Calabrese, F.; Favaretto, A.; et al. A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors. Nat. Genet. 2014, 46, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, R.; Goto, T.; Hirotsu, Y.; Yokoyama, Y.; Nakagomi, T.; Otake, S.; Amemiya, K.; Oyama, T.; Mochizuki, H.; Omata, M. Primary Driver Mutations in GTF2I Specific to the Development of Thymomas. Cancers 2020, 12, 2032. [Google Scholar] [CrossRef]
- Meacci, E.; Taira, M.; Moos, M.; Smith, C.J.; Movsesian, M.A.; Degerman, E.; Belfrage, P.; Manganiello, V. Molecular cloning and expression of human myocardial cGMP-inhibited cAMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 1992, 89, 3721–3725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, N.; Shen, W.; Du, R.; Jiang, S.; Zhu, J.; Chen, Y.; Huang, C.; Shi, Y.; Xiang, R.; Luo, Y. Phosphodiesterase 3A Represents a Therapeutic Target that Drives Stem Cell-like Property and Metastasis in Breast Cancer. Mol. Cancer Ther. 2020, 19, 868–881. [Google Scholar] [CrossRef] [PubMed]
- De Waal, L.; Lewis, T.A.; Rees, M.G.; Tsherniak, A.; Wu, X.; Choi, P.S.; Gechijian, L.; Hartigan, C.; Faloon, P.W.; Hickey, M.J.; et al. Identification of cancer-cytotoxic modulators of PDE3A by predictive chemogenomics. Nat. Chem. Biol. 2016, 12, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.G.; Nguyen, L.; Samadzadeh, S.; Masouminia, M.; Mendoza, A.; Sweeney, O.; Tillman, B.; Afifyan, N.; Morgan, T.; French, B.A.; et al. Expression of proteins upregulated in hepatocellular carcinoma in patients with alcoholic hepatitis (AH) compared to non-alcoholic steatohepatitis (NASH): An immunohistochemical analysis of candidate proteins. Exp. Mol. Pathol. 2018, 104, 125–129. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Wen, S.; Yang, X.; Zhang, Y.; Wang, Z.; Zhang, Z. CHRM3 is a novel prognostic factor of poor prognosis in patients with endometrial carcinoma. Am. J. Transl. Res. 2015, 7, 902–911. [Google Scholar]
- Felton, J.; Hu, S.; Raufman, J.P. Targeting M3 Muscarinic Receptors for Colon Cancer Therapy. Curr. Mol. Pharmacol. 2018, 11, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Cheng, K.; Shant, J.; Raufman, J.P. Acetylcholine-induced activation of M3 muscarinic receptors stimulates robust matrix metalloproteinase gene expression in human colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Shin, V.Y.; Cheuk, I.; Siu, J.; Kwong, A. Abstract P1-07-01: Cholinergic receptor muscarinic 3 (CHRM3) contributes to breast cancer tumorigenesis through angiogenesis regulation. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Yin, Q.Q.; Xu, L.H.; Zhang, M.; Xu, C. Muscarinic acetylcholine receptor M1 mediates prostate cancer cell migration and invasion through hedgehog signaling. Asian J. Androl. 2018, 20, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, J.; Yao, L.; Lang, Y.; Liang, Y.; Chen, L.; Zhang, J.; Wang, F.; Wang, Y.; Chen, H.; et al. High expression of M3 muscarinic acetylcholine receptor is a novel biomarker of poor prognostic in patients with non-small cell lung cancer. Tumor Biol. 2013, 34, 3939–3944. [Google Scholar] [CrossRef]
- Kodaira, M.; Kajimura, M.; Takeuchi, K.; Lin, S.; Hanai, H.; Kaneko, E. Functional muscarinic m3 receptor expressed in gastric cancer cells stimulates tyrosine phosphorylation and MAP kinase. J. Gastroenterol. 1999, 34, 163–171. [Google Scholar] [CrossRef]
- Celiktas, M.; Tanaka, I.; Tripathi, S.C.; Fahrmann, J.F.; Aguilar-Bonavides, C.; Villalobos, P.; Delgado, O.; Dhillon, D.; Dennison, J.B.; Ostrin, E.J.; et al. Role of CPS1 in cell growth, metabolism, and prognosis in LKB1-inactivated lung adenocarcinoma. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kärkkäinen, S.; Hiipakka, M.; Wang, J.H.; Kleino, I.; Vähä-Jaakkola, M.; Renkema, G.H.; Liss, M.; Wagner, R.; Saksela, K. Identification of preferred protein interactions by phage-display of the human Src homology-3 proteome. EMBO Rep. 2006, 7, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Mi, J.Q.; Debernardi, A.; Vitte, A.L.; Emadali, A.; Meyer, J.A.; Charmpi, K.; Ycart, B.; Callanan, M.B.; Carroll, W.L.; et al. A six gene expression signature defines aggressive subtypes and predicts outcome in childhood and adult acute lymphoblastic leukemia. Oncotarget 2015, 6, 16527–16542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, P.; Zhao, Z.; Hulgan, T.; Bush, W.S.; Samuels, D.C.; Bloss, C.S.; Heaton, R.K.; Ellis, R.J.; Schork, N.; Marra, C.M.; et al. Genome-wide association study of HIV-associated neurocognitive disorder (HAND): A CHARTER group study. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2017, 174, 413–426. [Google Scholar] [CrossRef]
- Lee, J.H.; Cheng, R.; Vardarajan, B.; Lantigua, R.; Reyes-Dumeyer, D.; Ortmann, W.; Graham, R.R.; Bhangale, T.; Behrens, T.W.; Medrano, M.; et al. Genetic Modifiers of Age at Onset in Carriers of the G206A Mutation in PSEN1 with Familial Alzheimer Disease Among Caribbean Hispanics. JAMA Neurol. 2015, 72, 1043–1051. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Liu, Y.; Lian, C.; Cao, X.; Wang, Y.; Li, X.; Cong, M.; Tian, P.; Zhang, X.; Wei, G.; et al. SH3RF3 promotes breast cancer stem-like properties via JNK activation and PTX3 upregulation. Nat. Commun. 2020, 11, 2487. [Google Scholar] [CrossRef]
- Mao, X.; Bruneau, N.; Gao, Q.; Becq, H.; Jia, Z.; Xi, H.; Shu, L.; Wang, H.; Szepetowski, P.; Aniksztejn, L. The Epilepsy of Infancy with Migrating Focal Seizures: Identification of de novo Mutations of the KCNT2 Gene That Exert Inhibitory Effects on the Corresponding Heteromeric KNa1.1/KNa1.2 Potassium Channel. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Hines, I.N.; Hartwell, H.J.; Feng, Y.; Theve, E.J.; Hall, G.A.; Hashway, S.; Connolly, J.; Fecteau, M.; Fox, J.G.; Rogers, A.B. Insulin resistance and metabolic hepatocarcinogenesis with parent-of-origin effects in A×B mice. Am. J. Pathol. 2011, 179, 2855–2865. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.C.; Couchy, G.; Balabaud, C.; Morcrette, G.; Caruso, S.; Blanc, J.F.; Bacq, Y.; Calderaro, J.; Paradis, V.; Ramos, J.; et al. Molecular Classification of Hepatocellular Adenoma Associates with Risk Factors, Bleeding, and Malignant Transformation. Gastroenterology 2017, 152, 880–894.e6. [Google Scholar] [CrossRef] [Green Version]
- Bérubé, J.C.; Gaudreault, N.; Lavoie-Charland, E.; Sbarra, L.; Henry, C.; Madore, A.M.; Paré, P.D.; Van Den Berge, M.; Nickle, D.; Laviolette, M.; et al. Identification of Susceptibility Genes of Adult Asthma in French Canadian Women. Can. Respir. J. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel, N.N.; Pare, P.D.; Rafaels, N.; Sin, D.D.; Sandford, A.; Daley, D.; Vergara, C.; Huang, L.; Mark Elliott, W.; Pascoe, C.D.; et al. Genome-wide association study identification of novel loci associated with airway responsiveness in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2015, 53, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.Q.; Pfeiffer, R.M.; Hyland, P.L.; Shi, J.; Gu, F.; Wang, Z.; Bhattacharjee, S.; Luo, J.; Xiong, X.; Yeager, M.; et al. Genetic polymorphisms in the 9p21 region associated with risk of multiple cancers. Carcinogenesis 2014, 35, 2698–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, J.H.; Park, S.B.; Park, S.; Lee, H.S.; Kim, C.; Jung, D.E.; Song, S.Y. Novel gastric cancer stem cell-related marker LINGO2 is associated with cancer cell phenotype and patient outcome. Int. J. Mol. Sci. 2019, 20, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belle, N.M.; Ji, Y.; Herbine, K.; Wei, Y.; Park, J.H.; Zullo, K.; Hung, L.Y.; Srivatsa, S.; Young, T.; Oniskey, T.; et al. TFF3 interacts with LINGO2 to regulate EGFR activation for protection against colitis and gastrointestinal helminths. Nat. Commun. 2019, 10, 4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhry, H.; Albukhari, A.; Morotti, M.; Haider, S.; Moralli, D.; Smythies, J.; Schödel, J.; Green, C.M.; Camps, C.; Buffa, F.; et al. Tumor hypoxia induces nuclear paraspeckle formation through HIF-2α dependent transcriptional activation of NEAT1 leading to cancer cell survival. Oncogene 2015, 34, 4482–4490. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Sboner, A.; Nair, S.S.; Giannopoulou, E.; Li, R.; Hennig, S.; Mosquera, J.M.; Pauwels, J.; Park, K.; Kossai, M.; et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat. Commun. 2014, 5, 5383. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Tanigawa, A.; Naganuma, T.; Ohkawa, Y.; Souquere, S.; Pierron, G.; Hirose, T.; Steitz, J.A. SWI/SNF chromatin-remodeling complexes function in noncoding RNA-dependent assembly of nuclear bodies. Proc. Natl. Acad. Sci. USA 2015, 112, 4304–4309. [Google Scholar] [CrossRef] [Green Version]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jiang, L.; Ji, X.; Yang, B.; Zhang, Y.; Fu, X.D. Hepatitis B viral RNA directly mediates down-regulation of the tumor suppressor microRNA miR-15a/miR-16-1 in hepatocytes. J. Biol. Chem. 2013, 288, 18484–18493. [Google Scholar] [CrossRef] [Green Version]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Li, Q.J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-β-miR-34a-CCL22 Signaling-Induced Treg Cell Recruitment Promotes Venous Metastases of HBV-Positive Hepatocellular Carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.H.; Mei, M.H.; Fei, R.; Liu, F.; Wang, J.H.; Liao, W.J.; Qin, L.L.; Wei, L.; Chen, H.S. Regulatory T cells in chronic hepatitis B patients affect the immunopathogenesis of hepatocellular carcinoma by suppressing the anti-tumour immune responses. J. Viral Hepat. 2010, 17, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Sugimoto, R.; Ma, N.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Kawanishi, S.; Watanabe, S.; Kaito, M.; Takei, Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J. Viral Hepat. 2008, 15, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.L.; Shin, H.J.; Feitelson, M.A.; Yu, D.Y. Oxidative stress and antioxidants in hepatic pathogenesis. World J. Gastroenterol. 2010, 16, 6035–6043. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Navas-Martin, S. Hepatitis B and C virus hepatocarcinogenesis: Lessons learned and future challenges. Cancer Lett. 2011, 305, 123–143. [Google Scholar] [CrossRef]
- Chun, E.; Lee, J.; Cheong, H.S.; Lee, K.Y. Tumor Eradication by Hepatitis B Virus X Antigen-Specific CD8 + T Cells in Xenografted Nude Mice. J. Immunol. 2003, 170, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Malmassari, S.L.; Deng, Q.; Fontaine, H.; Houitte, D.; Rimlinger, F.; Thiers, V.; Maillere, B.; Pol, S.; Michel, M.L. Impact of hepatitis B virus basic core promoter mutations on T cell response to an immunodominant HBx-derived epitope. Hepatology 2007, 45, 1199–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, G.Y.; Lin, C.Y.; Huang, L.M.; Wang, Y.H.; Wang, J.C.; Hsu, C.T.; Yang, S.S.; Wu, C.C. Detection of the Hepatitis B Virus X Protein (HBx) Antigen and Anti-HBx Antibodies in Cases of Human Hepatocellular Carcinoma. J. Clin. Microbiol. 2003, 41, 5598–5603. [Google Scholar] [CrossRef] [Green Version]
- Martín-Vílchez, S.; Sanz-Cameno, P.; Rodríguez-Muñoz, Y.; Majano, P.L.; Molina-Jiménez, F.; López-Cabrera, M.; Moreno-Otero, R.; Lara-Pezzi, E. The hepatitis B virus X protein induces paracrine activation of human hepatic stellate cells. Hepatology 2008, 47, 1872–1883. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Lee, J.S.; Conner, E.A.; Schroeder, I.; Factor, V.M.; Thorgeirsson, S.S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Investig. 2007, 117, 2713–2722. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wu, G.; Bu, F.; Lu, B.; Liang, A.; Cao, L.; Tong, X.; Lu, X.; Wu, M.; Guo, Y. Epigenetic silence of ankyrin-repeat-containing, SH3-domain-containing, and proline-rich-region-containing protein 1 (ASPP1) and ASPP2 genes promotes tumor growth in hepatitis B virus-positive hepatocellular carcinoma. Hepatology 2010, 51, 142–153. [Google Scholar] [CrossRef]
- Zhang, Z.Z.; Liu, X.; Wang, D.Q.; Teng, M.K.; Niu, L.W.; Huang, A.L.; Liang, Z. Hepatitis B virus and hepatocellular carcinoma at the miRNA level. World J. Gastroenterol. 2011, 17, 3353–3358. [Google Scholar] [CrossRef]
- Wei, X.; Tan, C.; Tang, C.; Ren, G.; Xiang, T.; Qiu, Z.; Liu, R.; Wu, Z. Epigenetic repression of miR-132 expression by the hepatitis B virus x protein in hepatitis B virus-related hepatocellular carcinoma. Cell. Signal. 2013, 25, 1037–1043. [Google Scholar] [CrossRef]
- Chisari, F.; Ferrari, C. Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 1995, 13, 29–60. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Isogawa, M.; Chisari, F.V. Host–virus interactions in hepatitis B virus infection. Curr. Opin. Immunol. 2015, 36, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Bertoletti, A.; Ferrari, C. Adaptive immunity in HBV infection. J. Hepatol. 2016, 64, S71–S83. [Google Scholar] [CrossRef]
- Meuleman, P.; Libbrecht, L.; Wieland, S.; De Vos, R.; Habib, N.; Kramvis, A.; Roskams, T.; Leroux-Roels, G. Immune suppression uncovers endogenous cytopathic effects of the hepatitis B virus. J. Virol. 2006, 80, 2797–2807. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Y.; Hu, K.Q. Rethinking the pathogenesis of hepatitis B virus (HBV) infection. J. Med. Virol. 2015, 87, 1989–1999. [Google Scholar] [CrossRef]
- Raihan, R.; Akbar, S.M.F.; Al Mahtab, M.; Khan, M.S.I.; Tabassum, S.; Tee, K.K.; Mohamed, R.B. Increased Proinflammatory Cytokine Production by Chronic Hepatitis B Patients with Mutant Hepatitis B Virus: Plausible Mechanisms Underlying Severe Liver Diseases in These Patients. Viral Immunol. 2020, 33, 530–534. [Google Scholar] [CrossRef]
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Chen, D.S. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology 2003, 124, 327–334. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: A meta-analysis. J. Natl. Cancer Inst. 2009, 101, 1066–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elpek, G.O. Molecular pathways in viral hepatitis-associated liver carcinogenesis: An update. World J. Clin. Cases 2021, 9, 4890–4917. [Google Scholar] [CrossRef]
- Enrietto, P.J.; Wyke, J.A. The pathogenesis of oncogenic avian retroviruses. Adv. Cancer Res. 1983, 39, 269–314. [Google Scholar] [CrossRef]
- Bruni, R.; D’Ugo, E.; Giuseppetti, R.; Argentini, C.; Rapicetta, M. Activation of the N-myc2 oncogene by woodchuck hepatitis virus integration in the linked downstream b3n locus in woodchuck hepatocellular carcinoma. Virology 1999, 257, 483–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, J.R.; Sterczer, A.; Toshkov, I.A.; Yeager, A.E.; Korba, B.E.; Cote, P.J.; Buendia, M.A.; Gerin, J.L.; Tennant, B.C. Integration of Woodchuck Hepatitis and N-myc Rearrangement Determine Size and Histologic Grade of Hepatic Tumors. Hepatology 2004, 39, 1008–1016. [Google Scholar] [CrossRef]
- Nakajima, T.; Moriguchi, M.; Mitsumoto, Y.; Sekoguchi, S.; Nishikawa, T.; Takashima, H.; Watanabe, T.; Katagishi, T.; Kimura, H.; Okanoue, T.; et al. Centrosome aberration accompanied with p53 mutation can induce genetic instability in hepatocellular carcinoma. Mod. Pathol. 2004, 17, 722–727. [Google Scholar] [CrossRef] [Green Version]
- Wilkens, L.; Bredt, M.; Flemming, P.; Kubicka, S.; Klempnauer, J.; Kreipe, H. Cytogenetic aberrations in primary and recurrent fibrolamellar hepatocellular carcinoma detected by comparative genomic hybridization. Am. J. Clin. Pathol. 2000, 114, 867–874. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Yuen, M.F.; Chan, H.L.Y.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thi Vo, T.; Poovorawan, K.; Charoen, P.; Soonthornworasiri, N.; Nontprasert, A.; Kittitrakul, C.; Phumratanaprapin, W.; Tangkijvanich, P. Association between Hepatitis B Surface Antigen Levels and the Risk of Hepatocellular Carcinoma in Patients with Chronic Hepatitis B Infection: Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. APJCP 2019, 20, 2239–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar] [CrossRef] [Green Version]
- Bertoletti, A.; Ferrari, C. Innate and adaptive immune responses in chronic hepatitis B virus infections: Towards restoration of immune control of viral infection. Postgrad. Med. J. 2013, 89, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Evan, G.I. A matter of life and death. Cancer Cell 2002, 1, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.L.; El-Sayed, M.H.; Kao, J.H.; Lazarus, J.V.; Lemoine, M.; Lok, A.S.; Zoulim, F. Progress towards elimination goals for viral hepatitis. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 533–542. [Google Scholar] [CrossRef]
- Cornberg, M.; Lok, A.S.F.; Terrault, N.A.; Zoulim, F.; Berg, T.; Brunetto, M.R.; Buchholz, S.; Buti, M.; Chan, H.L.; Chang, K.M.; et al. Guidance for Design and Endpoints of Clinical Trials in Chronic Hepatitis B—Report From the 2019 EASL-AASLD HBV Treatment Endpoints Conference. Hepatology 2020, 71, 1070–1092. [Google Scholar] [CrossRef]
- Lin, C.L.; Kao, J.H. Perspectives and control of hepatitis B virus infection in Taiwan. J. Formos. Med. Assoc. Taiwan Yi Zhi 2015, 114, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Kao, J.H. Hepatitis B: From control to cure. J. Formos. Med. Assoc. 2018, 117, 868–870. [Google Scholar] [CrossRef]
Figure 1.
Geographical distribution of liver cancer incidence and mortality rates (Age-Standarized Rate-ASR per 100,000—data derived from WHO-Global Cancer Observatory: Available online: gco.iarc.fr (accessed on 8 May 2012)), percentage of Hepatitis B (HepB3) immunization coverage among one-years-olds (data derived from WHO–Global Health Observatory data repository: apps.who.int/gho/data/view.main.80300 accessed on 18 August 2021) and HBV genotypes (data derived from Velkov et al. [36]). The world map has been colored with respect to the Liver Cancer Incidence Rate (ASR per 100,000). The ASR Incidence and Mortality as well as the percentage of the HepB3 immunization coverage among one-years-olds are depicted as a heatmap, while the pie charts represent the proportional (%) HBV genotype distributions in the respective countries. All plots were generated using the ggplot2 R package [37].
Figure 1.
Geographical distribution of liver cancer incidence and mortality rates (Age-Standarized Rate-ASR per 100,000—data derived from WHO-Global Cancer Observatory: Available online: gco.iarc.fr (accessed on 8 May 2012)), percentage of Hepatitis B (HepB3) immunization coverage among one-years-olds (data derived from WHO–Global Health Observatory data repository: apps.who.int/gho/data/view.main.80300 accessed on 18 August 2021) and HBV genotypes (data derived from Velkov et al. [36]). The world map has been colored with respect to the Liver Cancer Incidence Rate (ASR per 100,000). The ASR Incidence and Mortality as well as the percentage of the HepB3 immunization coverage among one-years-olds are depicted as a heatmap, while the pie charts represent the proportional (%) HBV genotype distributions in the respective countries. All plots were generated using the ggplot2 R package [37].

Figure 2.
The natural history of chronic hepatitis B is described by five phases—which are not necessarily sequential: the HBeAg positive chronic HBV infection or immune tolerant phase (phase 1), the HBeAg positive chronic hepatitis or immune reactive HBeAg positive phase (phase 2), the HBeAg chronic infection or inactive carrier phase (phase 3), the HBeAg negative or HBeAg negative chronic hepatitis (phase 4) and the Occult HBV infection or HBsAg negative phase (phase 5) [50]. During Phase 1, the HBV DNA levels are very high (> IU/mL), and HBV integrates parts or the whole of its genome into the host’s hepatocyte genomic DNA as a result of the reverse transcription mediated by the HBV polymerase. The alanine transaminase (ALT) levels are within the normal range, and there is minimal or no liver necroinflammation or fibrosis. Phase 2 is described by high levels of HBV DNA and elevated ALT levels, while there is moderate fibrosis that is accelerating. From this phase most patients transit to Phase 3 after HBeAg seroconversion, while others progress to Phase 4 for many years. Phase 3 is characterized by low (<2000 IU/mL) HBV DNA levels and normal ALT levels. Patients who remain in this phase have a low risk of progression to cirrhosis or HCC but progression to CHB and transition to Phase 4 usually occurs. Phase 4 is described by high levels of serum HBV DNA and elevated ALT levels, while liver necroinflammation and fibrosis are present. Phase 5 is characterized by undetectable levels of HBV DNA and normal ALT levels, while the liver histology and the risk of HCC are sequelae of the previous phases. The general shape of the curves that describe HBV DNA and ALT levels has beeen adapted from [57].
Figure 2.
The natural history of chronic hepatitis B is described by five phases—which are not necessarily sequential: the HBeAg positive chronic HBV infection or immune tolerant phase (phase 1), the HBeAg positive chronic hepatitis or immune reactive HBeAg positive phase (phase 2), the HBeAg chronic infection or inactive carrier phase (phase 3), the HBeAg negative or HBeAg negative chronic hepatitis (phase 4) and the Occult HBV infection or HBsAg negative phase (phase 5) [50]. During Phase 1, the HBV DNA levels are very high (> IU/mL), and HBV integrates parts or the whole of its genome into the host’s hepatocyte genomic DNA as a result of the reverse transcription mediated by the HBV polymerase. The alanine transaminase (ALT) levels are within the normal range, and there is minimal or no liver necroinflammation or fibrosis. Phase 2 is described by high levels of HBV DNA and elevated ALT levels, while there is moderate fibrosis that is accelerating. From this phase most patients transit to Phase 3 after HBeAg seroconversion, while others progress to Phase 4 for many years. Phase 3 is characterized by low (<2000 IU/mL) HBV DNA levels and normal ALT levels. Patients who remain in this phase have a low risk of progression to cirrhosis or HCC but progression to CHB and transition to Phase 4 usually occurs. Phase 4 is described by high levels of serum HBV DNA and elevated ALT levels, while liver necroinflammation and fibrosis are present. Phase 5 is characterized by undetectable levels of HBV DNA and normal ALT levels, while the liver histology and the risk of HCC are sequelae of the previous phases. The general shape of the curves that describe HBV DNA and ALT levels has beeen adapted from [57].

Figure 3.
The molecular mechanisms of HBV DNA integration into the host genome with respect to the relaxed circular DNA (rcDNA) and double-stranded linear DNA (dslDNA) structure. (A) The ‘single-stranded-gap’ model [11], implies that the DNA polymerase switches from the human genomic DNA strand to the 5′ single-stranded region of the nearby HBV genome, followed by a recombination event. (B) The HBV ‘DR-mediated integration’ model [86] implies that HBV DNA integration occurs via a site-specific recombination event between host genomic DNA and either in DR1 or DR2. (C) The ‘partial-sequence homology’ model [90] implies that a microhomology between the host DNA and the HBV genome triggers the switch of the DNA polymerase from the host DNA to the single-stranded region of the HBV rcDNA, which is followed by a recombination event in the site of the microhomology that determines the site of integration. (D) The ‘roll-in’ model [14] is based on HBV dslDNA where the free end at a single-strand break in cellular DNA invades into the HBV genome and forms the host–viral junction. (E) The minus strand of the HBV rcDNA is released from the replication complex before HBcAg synthesis, while a staggered break at the host DNA leaves sticky ends in which the incomplete HBV genome is inserted, resulting in the formation of 12 bp DRs that are neither viral nor cellular on either side of the viral integration site [93]. (F) The ‘NNEJ/MMEJ-based model’ implies that the HBV dslDNA is integrated at a site of DSB after its repair and removal of the Pol protein and the 5’cap via Non-Homologous End Joining in both directions or via Microhomology-Mediated End Joining in a direction determined based on the microhomology. (G) The relaxed-circular HBV DNA structure and genome organization. The inner region depicts the organization of the HBV genes, the dashed blue circle represents the (+) strand, the solid blue circle represents the (−) strand and the outer petrol-blue line represents the HBV pgRNA.
Figure 3.
The molecular mechanisms of HBV DNA integration into the host genome with respect to the relaxed circular DNA (rcDNA) and double-stranded linear DNA (dslDNA) structure. (A) The ‘single-stranded-gap’ model [11], implies that the DNA polymerase switches from the human genomic DNA strand to the 5′ single-stranded region of the nearby HBV genome, followed by a recombination event. (B) The HBV ‘DR-mediated integration’ model [86] implies that HBV DNA integration occurs via a site-specific recombination event between host genomic DNA and either in DR1 or DR2. (C) The ‘partial-sequence homology’ model [90] implies that a microhomology between the host DNA and the HBV genome triggers the switch of the DNA polymerase from the host DNA to the single-stranded region of the HBV rcDNA, which is followed by a recombination event in the site of the microhomology that determines the site of integration. (D) The ‘roll-in’ model [14] is based on HBV dslDNA where the free end at a single-strand break in cellular DNA invades into the HBV genome and forms the host–viral junction. (E) The minus strand of the HBV rcDNA is released from the replication complex before HBcAg synthesis, while a staggered break at the host DNA leaves sticky ends in which the incomplete HBV genome is inserted, resulting in the formation of 12 bp DRs that are neither viral nor cellular on either side of the viral integration site [93]. (F) The ‘NNEJ/MMEJ-based model’ implies that the HBV dslDNA is integrated at a site of DSB after its repair and removal of the Pol protein and the 5’cap via Non-Homologous End Joining in both directions or via Microhomology-Mediated End Joining in a direction determined based on the microhomology. (G) The relaxed-circular HBV DNA structure and genome organization. The inner region depicts the organization of the HBV genes, the dashed blue circle represents the (+) strand, the solid blue circle represents the (−) strand and the outer petrol-blue line represents the HBV pgRNA.

Figure 4.
HBV DNA integration density plots for the chromosomes 1–7 and 19 in which the recurrently affected genes are localized. The yellow density plots represent the density of integrations per 1 Mbp (1,000,000 bp) through the chromosome that are referring to coding genomic regions, while the blue density plots represent the density of integrations per 1 Mbp that are referring to all genomic regions (coding and non-coding). The mean values are depicted via the horizontal dashed lines, and the locations of the main target genes are also present. The plots were generated with the karyoploteR R package [187].
Figure 4.
HBV DNA integration density plots for the chromosomes 1–7 and 19 in which the recurrently affected genes are localized. The yellow density plots represent the density of integrations per 1 Mbp (1,000,000 bp) through the chromosome that are referring to coding genomic regions, while the blue density plots represent the density of integrations per 1 Mbp that are referring to all genomic regions (coding and non-coding). The mean values are depicted via the horizontal dashed lines, and the locations of the main target genes are also present. The plots were generated with the karyoploteR R package [187].


{kind=link}
{kind=link}
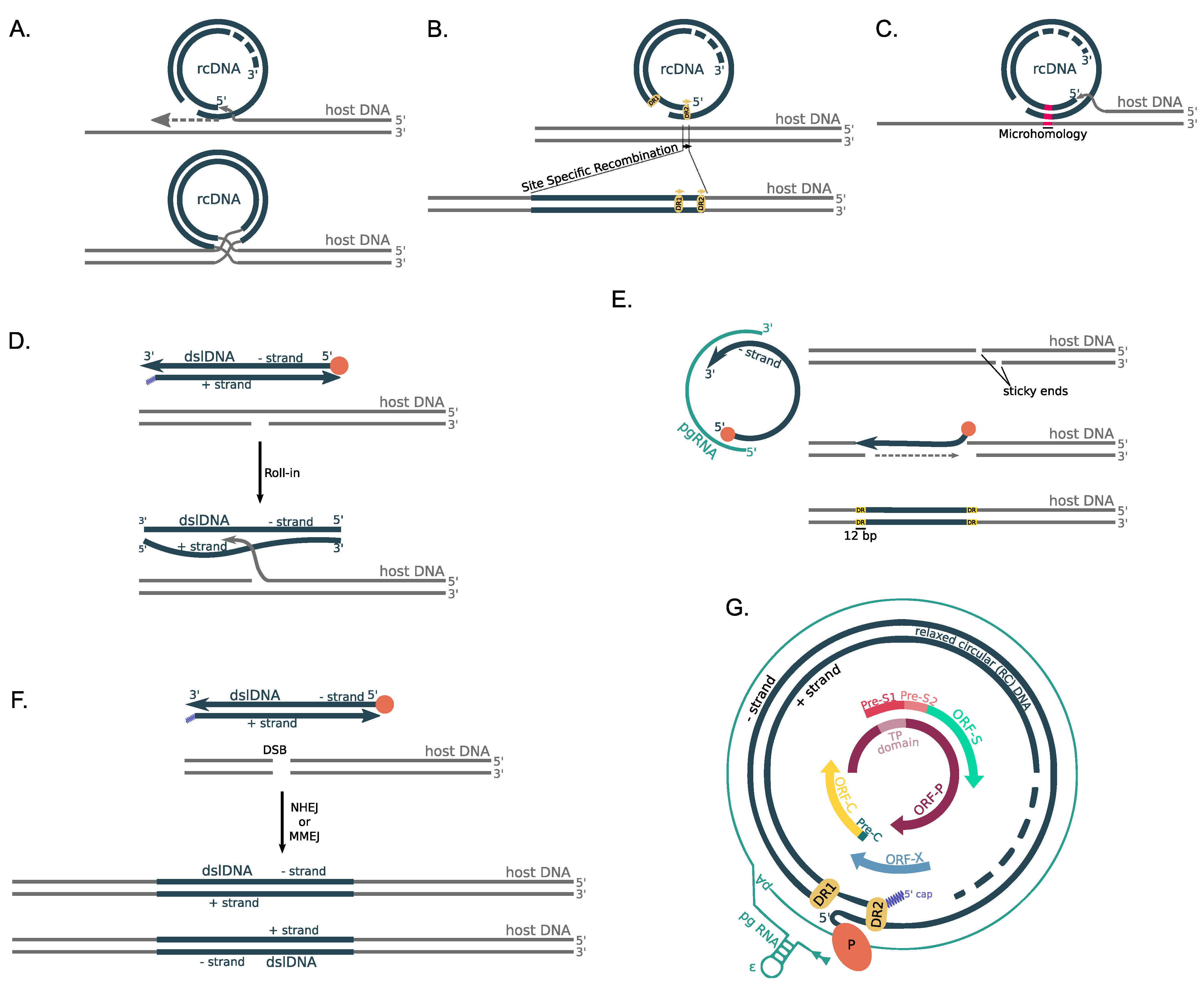{kind=link}
{kind=link}
Table 1.
The main target human genes affected by >20 reported HBV integrations.
| Gene Symbol | Gene Full Name | Gene Type | External Link | Genomic Location | Times Reported | References |
|---|---|---|---|---|---|---|
| TERT | Telomerase reverse transciptase | protein coding | HGNC:11730 | 5p15.33 | 457 | [82,83,87,96,105,107,114,115,116,117,118,119,120,121,122,123,124,125] |
| FN1 | Fibronectin 1 | protein coding | HGNC:3778 | 2q35 | 283 | [82,83,84,87,105,107,115,118,119,121,122,123,125,126,127] |
| KMT2B | Lysine methyltransferase 2B | protein coding | HGNC:15840 | 19q13.12 | 243 | [82,83,84,87,107,115,117,118,119,121,122,123,124,125,126,128,129] |
| ALB | Albumin | protein coding | HGNC:399 | 4q13.3 | 70 | [83,84,87,107,117,118,119,122,123,125,129] |
| CCNA2 | Cyclin A2 | protein coding | HGNC:1578 | 4q27 | 36 | [83,107,118,119,122,123,124,125,130] |
| LINC00486 | Long intergenic non-protein coding RNA 486 | ncRNA | HGNC:42946 | 2p22.3 | 36 | [82,83,118,125] |
| CPS1 | Carbamoyl-phosphate synthase 1 | protein coding | HGNC:2323 | 2q34 | 34 | [82,83,107,118,119,121,122,123,125,126,129] |
| SH3RF3 | SH3 domain containing ring finger 3 | protein coding | HGNC:24699 | 2q13 | 29 | [84,125] |
| CCNE1 | Cyclin E1 | protein coding | HGNC:1589 | 19q12 | 27 | [82,83,119,121,124,127] |
| GLI2 | GLI family zinc finger 2 | protein coding | HGNC:4318 | 2q14.2 | 27 | [84,115,119,121,126] |
| KCNT2 | Potassium sodium-activated channel subfamily T member 2 | protein coding | HGNC:18866 | 1q31.3 | 26 | [118,119,121,122,123,125] |
| LINGO2 | Leucine rich repeat and Ig domain containing 2 | protein coding | HGNC:21207 | 9p21.1 | 25 | [83,105,107,123,125] |
| PRKN | Parkin RBR E3 ubiquitin protein ligase | protein coding | HGNC:8607 | 6q26 | 24 | [81,119,123,125] |
| FAM157A | Family with sequence similarity 157 member A | ncRNA | HGNC:34079 | 3q29 | 23 | [82,83,87,122,125] |
| SOX5 | SRY-box transcription factor 5 | protein coding | HGNC:11201 | 12p12.1 | 23 | [81,83,105,107,119,122,123,124,125] |
| GTF2I | General transcription factor IIi | protein coding | HGNC:4659 | 7q11.23 | 22 | [84,105,118,122,123,125,127] |
| PDE3A | Phosphodiesterase 3A | protein coding | HGNC:8778 | 12q12.2 | 22 | [82,84,118,122,125] |
| CHRM3 | Cholinergic receptor muscarinic 3 | protein coding | HGNC:1952 | 1q43 | 21 | [83,122,125,129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bousali, M.; Papatheodoridis, G.; Paraskevis, D.; Karamitros, T. Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma. Microorganisms 2021, 9, 1787. https://doi.org/10.3390/microorganisms9081787
AMA Style
Bousali M, Papatheodoridis G, Paraskevis D, Karamitros T. Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma. Microorganisms. 2021; 9(8):1787. https://doi.org/10.3390/microorganisms9081787
Chicago/Turabian StyleBousali, Maria, George Papatheodoridis, Dimitrios Paraskevis, and Timokratis Karamitros. 2021. "Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma" Microorganisms 9, no. 8: 1787. https://doi.org/10.3390/microorganisms9081787
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.