3.1. Chemistry
General Methods. Melting points were measured in glass capillary tubes on a Büchi SMP-20 apparatus and are uncorrected. 1H-NMR and 13C-NMR spectra were recorded in CDCl3, unless otherwise indicated, on a Varian Gemini spectrometer 400 MHz and 101 MHz, respectively. Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane (TMS), and spin multiplicities are given as s (singlet), d (doublet), t (triplet), m (multiplet) or br (broad). Direct infusion ES-MS spectra were recorded on a Waters Micromass ZQ 4000 apparatus (Waters Alliance, San Diego, CA, USA). Chromatographic separations were performed by flash chromatography on silica gel columns (Kieselgel 40, 0.0400–0.063 mm; Merck, Darmstadt, Germany). Organic solutions were dried over anhydrous sodium sulfate. All chemicals were purchased from Aldrich Chemistry, Milan (Italy), or from Alfa Aesar, Milan (Italy), and were of the highest purity. Compounds were named relying on the naming algorithm developed by CambridgeSoft Corporation (Waltham, MA, USA) and used in ChemDraw Professional 15.0 (PerkinElmer Inc., Waltham, MA, USA).
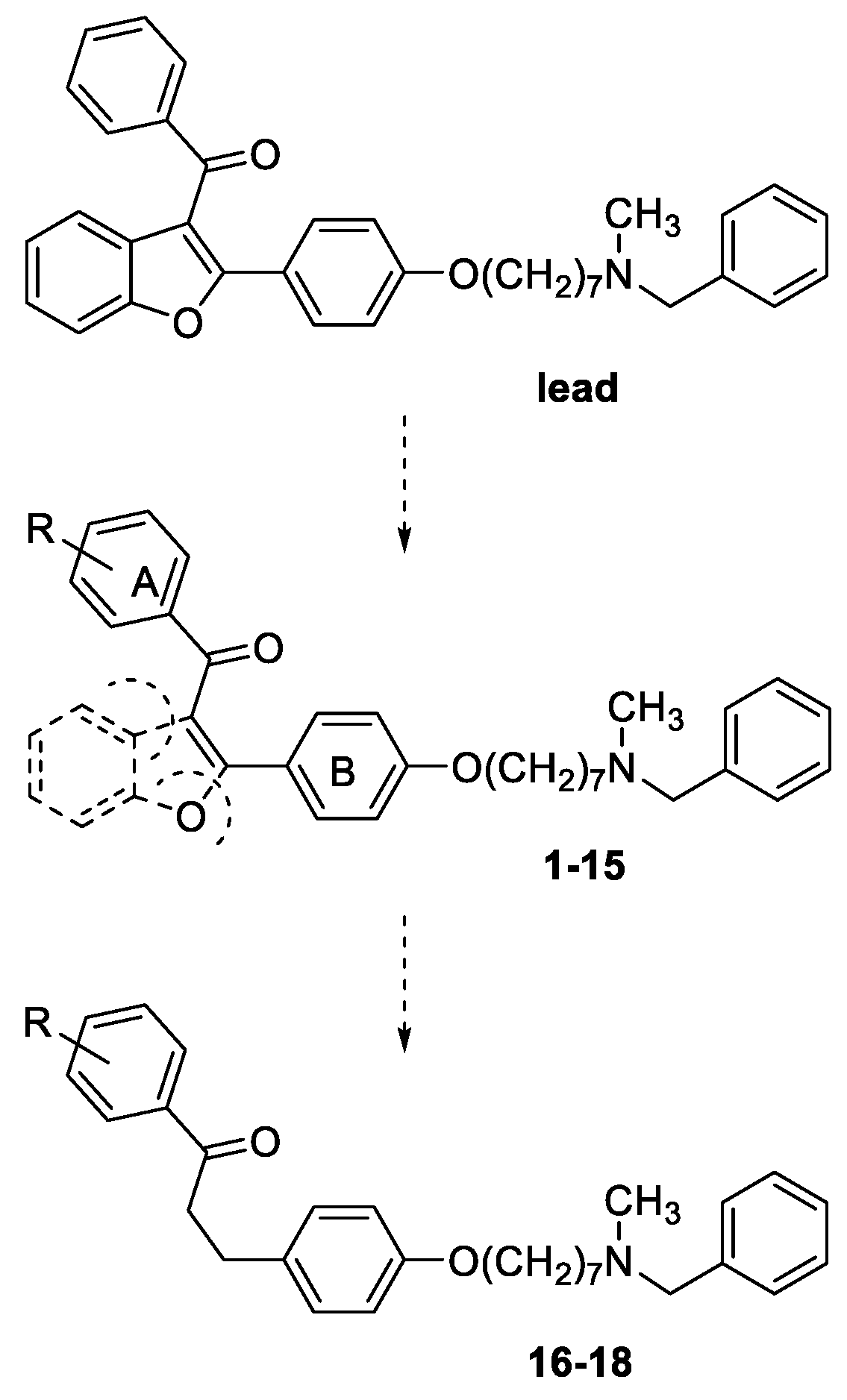
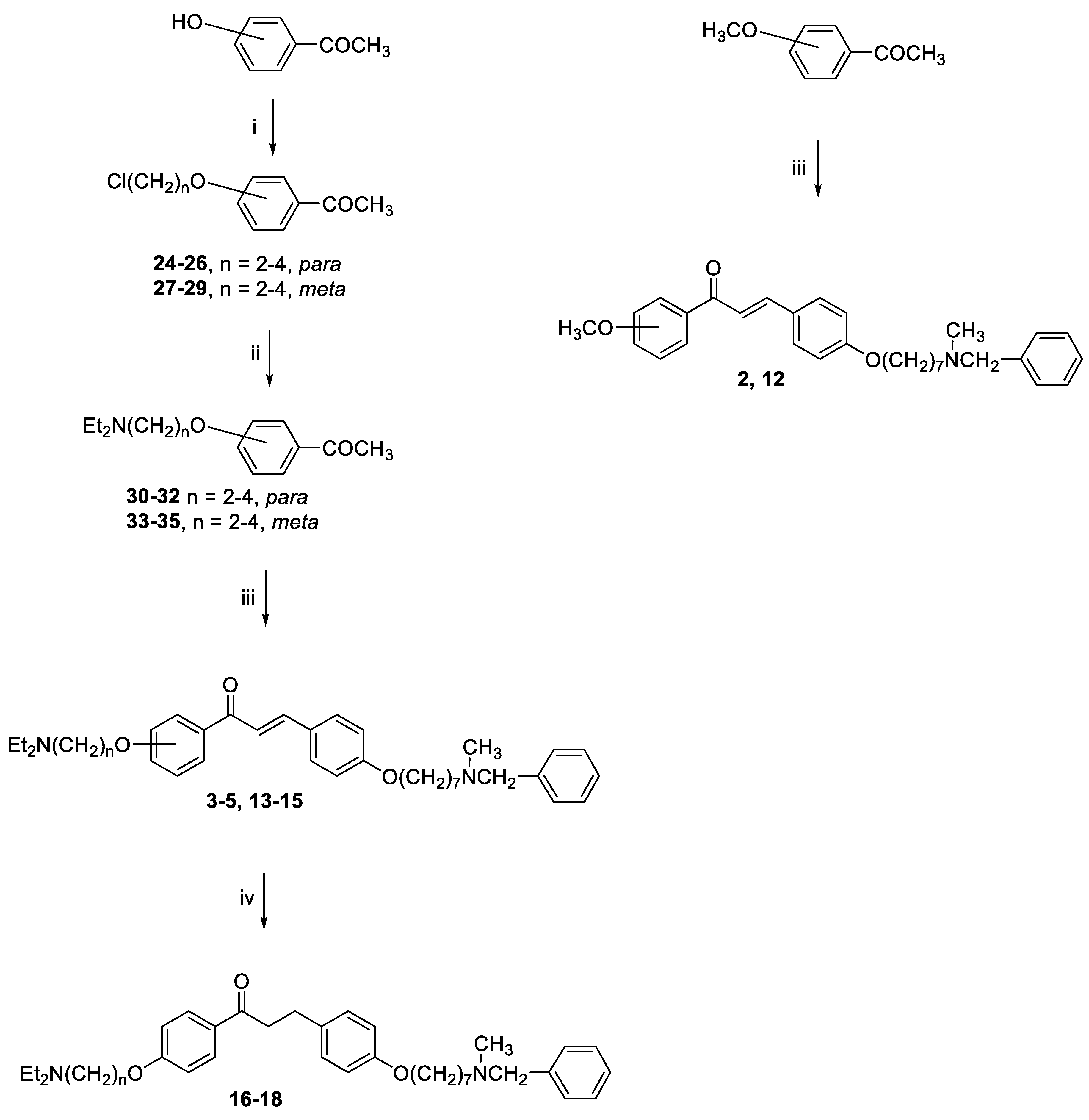
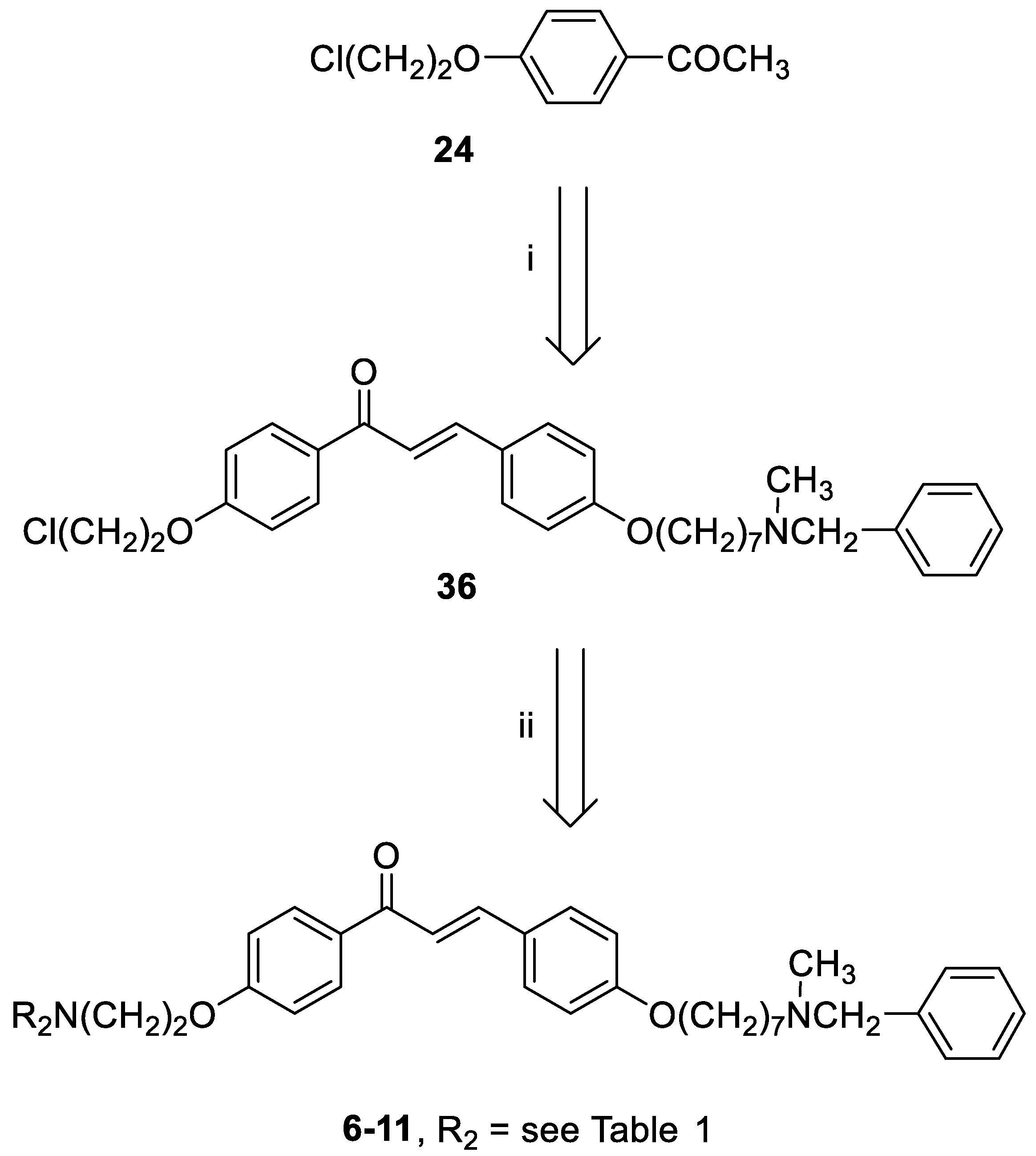
General method for the synthesis of final compounds 1–5, 12–15 and of intermediate 36: To a mixture of the selected acetophenone (0.001 mol) and 21 (0.001 mol) in EtOH (10 mL), a solution of KOH (1 g, 0.018 mol, in 5 mL of H2O) was added, and the reaction mixture was stirred at rt overnight. The mixture was poured into ice and the resulting yellow solid was filtered off. The residue was purified by flash chromatography on silica gel (toluene/acetone 3:2, then MeOH).
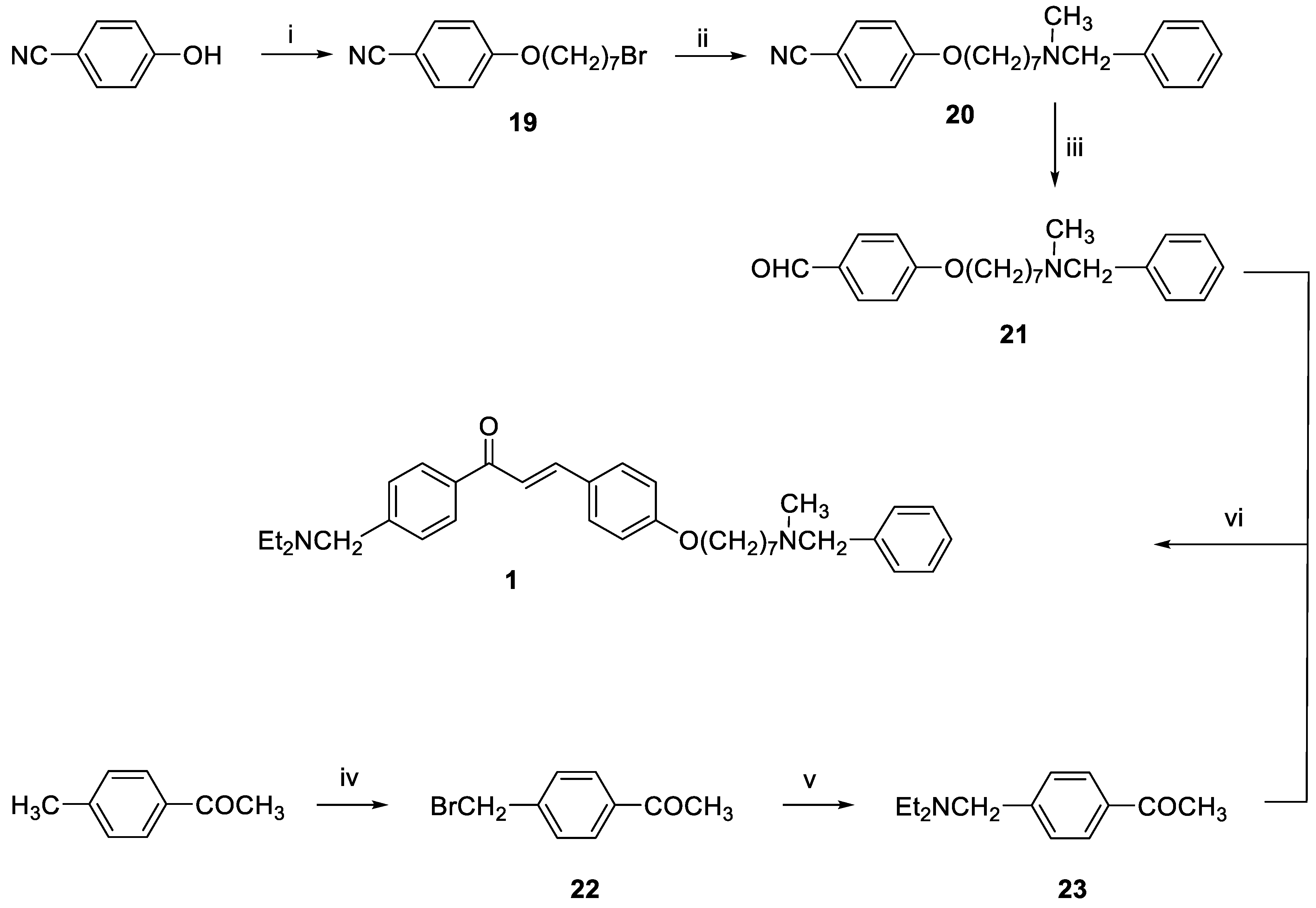
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-((diethylamino)methyl)phenyl)prop-2-en-1-one (1). Using the previous procedure and starting from 21 and 23, 1 (yield 55%) was obtained as an oil. 1H-NMR δ 1.03 (t, J = 7.2 Hz, 6H, 2CH3), 1.25–1.61 (m, 8H, 4CH2), 1.64–1.85 (m, 2H, CH2), 2.19 (s, 3H, NCH3), 2.35 (t, J = 7.2 Hz, 2H, CH2N), 2.42–2.60 (m, 4H, 2CH2), 3.46 (s, 2H, CH2), 3.62 (s, 2H, NCH2), 3.99 (t, J = 6.8 Hz, 2H, OCH2), 6.86–7.99 (m, 15H, Ar). 13C-NMR δ 11.88, 26.08, 27.31, 27.42, 29.22, 29.37, 42.25, 47.05, 57.45, 57.50, 62.32, 68.25, 115.02, 119.81, 127.06, 127.60, 128.31, 128.53, 128.62, 129.00, 129.25, 130.31, 137.30, 139.03, 144.59, 145.49, 161.36, 190.35. MS (ES) m/z: 527 [M + H]+. C35H46N2O2.
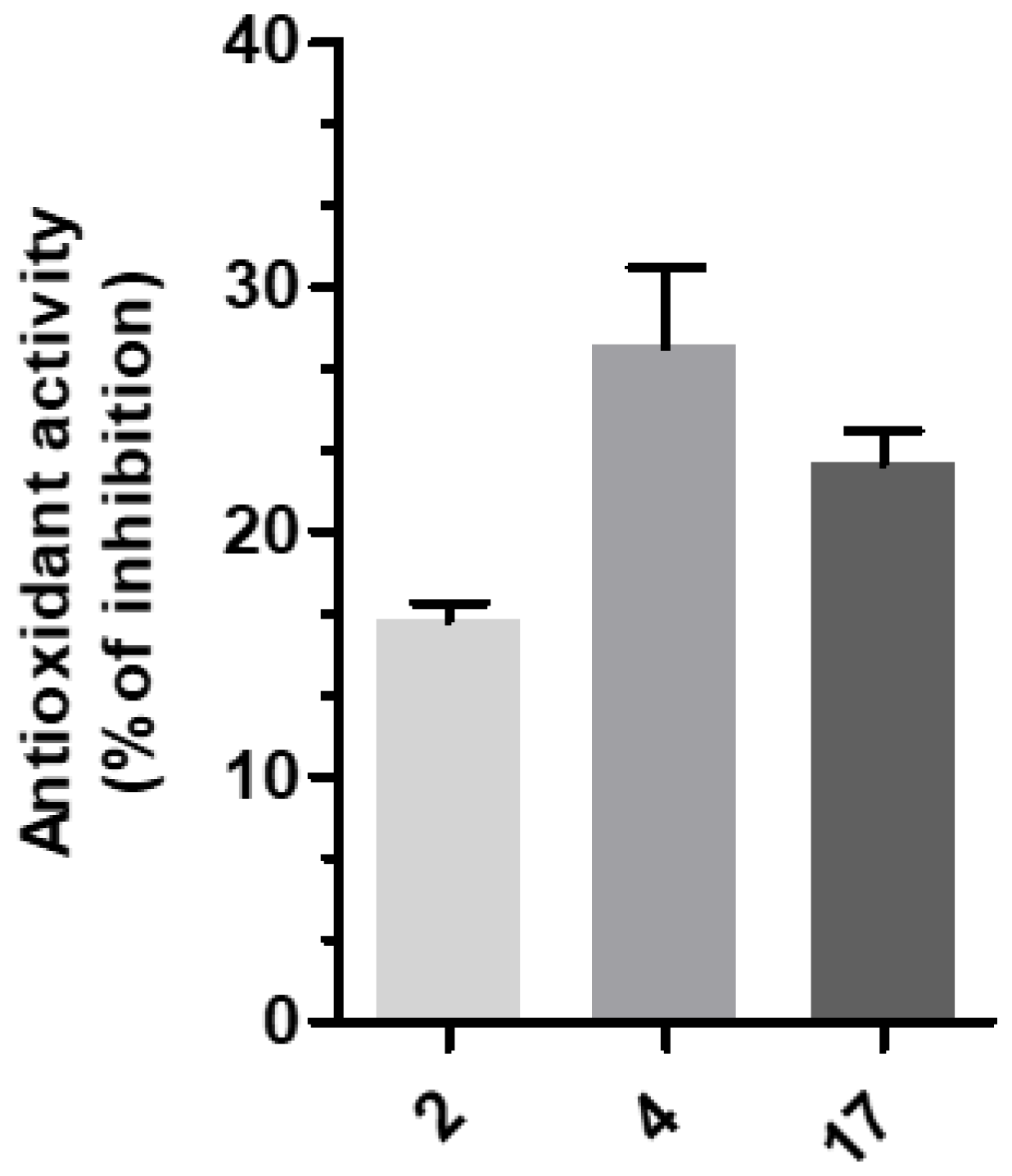
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-methoxyphenyl)prop-2-en-1-one (2). Using the previous procedure and starting from 4-methoxyacetophenone and 21, 2 (yield 75%) was obtained as an oil. 1H-NMR δ 1.22–1.59 (m, 8H), 1.64–1.82 (m, 2H, CH2), 2.20 (s, 3H, NCH3), 2.36 (t, J = 7.2 Hz, 2H, CH2N), 3.48 (s, 2H, CH2), 3.88 (s, 3H, OCH3), 4.00 (t, J = 6.8 Hz, 2H, OCH2), 6.88 (m, 3H, Ar), 7.20–7.84 (m, 10H, Ar), 8.02 (d, J = 8.8 Hz, 2H, Ar). 13C-NMR δ 26.18, 27.35, 27.41, 29.28, 29.43, 42.30, 55.72, 57.46, 62.38, 68.22, 112.94, 114.65, 115.36, 119.18, 119.77, 120.96, 121.11, 127.16, 127.56, 128.34, 128.49, 129.37, 129.64, 129.72, 130.39, 138.96, 140.11, 144.96, 160.13, 161.44, 190.76. MS (ES) m/z: 472 [M + H]+. C31H37NO3.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(2-(diethylamino)ethoxy)phenyl)-prop-2-en-1-one (3). Using the previous procedure and starting from 30 and 21, 3 (yield 70%) was obtained as a solid, mp 40–41 °C (ligroin). 1H-NMR δ 1.09 (t, J = 7.2 Hz, 6H, 2CH3), 1.45–1.55 (m, 4H, 2CH2), 1.61–1.82 (m, 2H, CH2), 2.19 (s, 3H, NCH3), 2.37 (t, J = 7.2 Hz, 2H, NCH2), 2.63–2.69 (m, 4H, 2CH2), 2.91 (t, J = 6.4 Hz, 2H, NCH2), 3.48 (s, 2H, CH2), 4.00 (t, J = 6.8 Hz, 2H, OCH2), 4.13 (t, J = 5.6 Hz, 2H, OCH2), 6.92 (d, J = 8.8 Hz, 2H, Ar), 6.98 (d, J = 9.2 Hz, 2H, Ar), 7.24–7.45 (m, 4H, Ar), 6.92 (d, J = 8.8 Hz, 2H, Ar), 7.59 (d, J = 8.8 Hz, 2H, Ar), 7.78 (d, J = 15.6 Hz, 1H), 8.02 (d, J = 8.8 Hz, 2H, Ar). 13C-NMR δ 12.37, 26.07, 27.37, 29.20, 29.37, 42.31, 46.51, 46.69, 52.31, 57.51, 62.39, 68.05, 68.24, 113.69, 114.65, 114.84, 115.01, 119.49, 119.75, 120.92, 126.99, 127.49, 127.77, 128.27, 129.18, 129.59, 130.35, 139.19, 140.01, 144.90, 159.40, 161.42, 190.43. MS (ES) m/z: 557 [M + H]+. C36H48N2O3.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(3-(diethylamino)propoxy)phenyl)-prop-2-en-1-one (4). Using the previous procedure and starting from 31 and 21, 4 (yield 20%) was obtained as an oil. 1H-NMR δ 1.01 (t, J = 7.2 Hz, 6H, 2CH3), 1.45–1.58 (m, 4H, 2CH2), 1.61–1.81 (m, 2H, CH2), 1.83–2.01 (m, 2H, CH2), 2.18 (s, 3H, NCH3), 2.36 (t, J = 7.2 Hz, 2H, NCH2), 2.41–2.65 (m, 6H, 2CH2), 3.45 (s, 2H, CH2), 3.99 (t, J = 6.8 Hz, 2H, OCH2), 4.10 (t, J = 5.6 Hz, 2H, OCH2), 6.92–7.01 (m, 4H, Ar), 7.19–7.42 (m, 6H, Ar), 7.59 (d, J = 8.8 Hz, 2H, Ar), 7.78 (d, J = 15.6 Hz, 1H), 8.02 (d, J = 8.8 Hz, 2H, Ar). 13C-NMR δ 11.49, 26.38, 27.51, 28.47, 29.28, 29.45, 42.41, 46.56, 46.82, 52.33, 57.60, 62.96, 68.22, 68.63, 113.74, 114.55, 114.99, 115.36, 119.14, 119.85, 120.92, 126.98, 127.52, 127.78, 128.29, 129.18, 129.61, 130.45, 139.29, 140.22, 144.96, 159.40, 161.02, 190.55. MS (ES) m/z: 571 [M + H]+. HRMS Esi + [M + 1]: calcd for C37H51N2O3, 571.3900. Found: 571.3902.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(4-(diethylamino)butoxy)phenyl)prop-2-en-1-one (5). Using the previous procedure and starting from 32 and 21, 5 (yield 25%) was obtained as an oil. 1H-NMR δ 1.02 (t, J = 7.2 Hz, 6H, 2CH3), 1.351–1.82 (m, 14H, 7CH2), 2.18 (s, 3H, NCH3), 2.37 (t, J = 7.2 Hz, 2H, NCH2), 2.42–2.64 (m, 6H, 2CH2), 3.45 (s, 2H, CH2), 3.99 (t, J = 6.8 Hz, 2H, OCH2), 4.10 (t, J = 5.6 Hz, 2H, OCH2), 6.84–7.03 (m, 4H, Ar), 7.21–7.42 (m, 6H, Ar), 7.60 (d, J = 8.8 Hz, 2H, Ar), 7.78 (d, J = 15.6 Hz, 1H), 8.01 (d, J = 8.8 Hz, 2H, Ar). 13C-NMR δ 11.34, 23.27, 26.05, 27.37, 27.48, 29.20, 29.45, 42.37, 46.58, 46.71, 52.36, 57.54, 62.42, 68.25, 68.44, 113.66, 114.65, 114.92, 115.03, 119.48, 119.76, 120.92, 126.99, 127.47, 127.79, 128.27, 129.19, 129.61, 130.37, 139.03, 140.34, 144.92, 159.43, 161.42, 190.55. MS (ES) m/z: 585 [M + H]+. C38H52N2O3.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(3-methoxyphenyl)prop-2-en-1-one (12). Using the previous procedure and starting from 3-methoxyacetophenone and 21, 12 (yield 68%) was obtained as an oil. 1H-NMR δ 1.24–1.58 (m, 8H), 1.69–1.82 (m, 2H, CH2), 2.19 (s, 3H, NCH3), 2.37 (t, J = 7.2 Hz, 2H, NCH2), 3.44 (s, 2H, CH2), 3.88 (s, 3H, OCH2), 4.00 (t, J = 6.8 Hz, 2H, OCH2), 6.88–7.80 (m, 15H, Ar). 13C-NMR δ 26.11, 27.30, 27.45, 29.25, 29.40, 42.23, 55.63, 57.44, 62.30, 68.28, 112.94, 114.68, 115.06, 119.19, 119.79, 120.96, 121.09, 127.11, 127.53, 128.34, 128.48, 129.30, 129.64, 129.70, 130.39, 138.96, 140.11, 144.98, 160.01, 161.47, 190.46. MS (ES) m/z: 472 [M + H]+. C31H37NO3.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(3-(2-(diethylamino)ethoxy)phenyl)-prop-2-en-1-one (13). Using the previous procedure and starting from 33 and 21, 13 (yield 20%) was obtained as an oil. 1H-NMR δ 1.10 (t, J = 7.2 Hz, 6H, 2CH3), 1.20–1.90 (m, 10H, 5CH2), 2.20 (s, 3H, NCH3), 2.38 (t, J = 7.2 Hz, 2H, NCH2), 2.60–2.73 (m, 4H, 2CH2), 2.90 (t, J = 6.4 Hz, 2H, NCH2), 3.50 (s, 2H, CH2), 4.01 (t, J = 6.8 Hz, 2H, OCH2), 4.10 (t, J = 5.4 Hz, 2H, OCH2), 6.98 (d, J = 8.8 Hz, 2H, Ar), 7.15–7.82 (m, 13H, Ar). 13C-NMR δ 12.37, 26.07, 27.37, 29.20, 29.37, 42.31, 46.51, 46.69, 52.31, 57.51, 62.39, 68.05, 68.24, 113.69, 114.65, 114.84, 115.01, 119.49, 119.75, 120.92, 126.99, 127.49, 127.77, 128.27, 129.18, 129.59, 130.35, 139.19, 140.01, 144.90, 159.40, 161.42, 190.43. MS (ES) m/z: 557 [M + H]+. HRMS Esi + [M + 1]: calcd for C36H49N2O3, 557.3743. Found: 557.3741.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(3-(3-(diethylamino)propoxy)phenyl)-prop-2-en-1-one (14). Using the previous procedure and starting from 34 and 21, 14 (yield 45%) was obtained as an oil. 1H-NMR δ 1.01 (t, J = 7.2 Hz, 6H, 2CH3), 1.20–1.81 (m, 10H, 5CH2), 1.82–2.01 (m, 2H, CH2), 2.18 (s, 3H, NCH3), 2.36 (t, J = 7.2 Hz, 2H, NCH2), 2.41–2.66 (m, 6H, 3CH2), 3.45 (s, 2H, CH2), 3.95 (t, J = 6.8 Hz, 2H, OCH2), 4.05 (t, J = 5.8 Hz, 2H, OCH2), 6.79–7.80 (m, 15H, Ar). 13C-NMR δ 11.47, 26.35, 27.37, 28.01, 29.24, 29.37, 42.38, 46.51, 46.76, 52.29, 57.52, 62.93, 68.15, 68.24, 113.55, 114.47, 114.91, 115.19, 119.04, 119.75, 120.92, 126.99, 127.49, 127.77, 128.27, 129.18, 129.59, 130.38, 139.19, 140.14, 144.90, 159.40, 160.98, 190.36. MS (ES) m/z: 571 [M + H]+. C37H50N2O3.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(4-(diethylamino)butoxy)phenyl)prop-2-en-1-one (15). Using the previous procedure and starting from 35 and 21, 15 (yield 55%) was obtained as an oil. 1H-NMR δ 1.02 (t, J = 7.2 Hz, 6H, 2CH3), 1.22–1.60 (m, 10H, 5CH2), 1.61–1.90 (m, 4H, 2CH2), 2.19 (s, 3H, NCH3), 2.38 (t, J = 7.2 Hz, 2H, NCH2), 2.42–2.59 (m, 6H, 3CH2), 3.42 (s, 2H, CH2), 3.99–4.13 (m, 4H, 2OCH2), 6.85 (d, J = 8.8 Hz, 2H, Ar), 7.10–7.82 (m, 13H, Ar). 13C-NMR δ 11.35, 23.21, 26.07, 27.37, 27.41, 29.20, 29.37, 42.31, 46.51, 46.69, 52.31, 57.51, 62.39, 68.05, 68.24, 113.69, 114.65, 114.84, 115.01, 119.49, 119.75, 120.92, 126.99, 127.49, 127.77, 128.27, 129.18, 129.59, 130.35, 139.19, 140.01, 144.90, 159.40, 161.42, 190.43. MS (ES) m/z: 585 [M + H]+. C38H52N2O3.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(2-chloroethoxy)phenyl)prop-2-en-1-one (36). Using the previous procedure and starting from 24 and 21, 36 (yield 77%) was obtained as yellow oil. 1H-NMR δ 1.22–1.80 (m, 10H), 2.18 (s, 3H), 2.35 (t, J = 7.2 Hz, 2H), 3.45 (s, 2H), 3.80 (t, 2H), 3.99 (t, 2H), 4.30 (t, 2H), 6.85–7.80 (m, 13H, Ar), 8.02 (d, 2H).
General method for the synthesis of final compounds 6–11: A stirred solution of 36 (0.5 mmol) and the selected amine (1 mmol) in toluene (50 mL) was refluxed for 24 h. The mixture was washed with water (3 × 25 mL) and the organic layer was dried. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (toluene/acetone 4:1, then only MeOH).
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(2-morpholinoethoxy)phenyl)prop-2-en-1-one (6). Using the previous procedure and starting from morpholine, 6 (yield 15%) was obtained as an oil. 1H-NMR δ 1.25–1.90 (m, 10H, 5CH2), 2.18 (s, 3H, NCH3), 2.35 (t, J = 7.2 Hz, 2H, NCH2), 2.43–2.61 (m, 4H, 2NCH2), 2.83 (t, 2H, NCH2), 3.42 (s, 2H, CH2), 3.70–3.80 (m, 4H, 2OCH2), 3.99 (t, J = 6.8 Hz, 2H, OCH2), 4.15 (t, J = 6.8 Hz, 2H, OCH2), 6.80–8.03 (m, 15H, Ar). 13C-NMR δ 26.42, 27.33, 27.45, 29.25, 29.40, 42.32, 56.21, 57.54, 57.56, 57.86, 62.38, 66.71, 66.73, 67.98, 68.26, 114.47, 115.21, 114.84, 119.49, 119.55, 127.16, 127.74, 127.77, 128.37, 129.28, 130.24, 130.82, 138.69, 140.34, 144.23, 159.62, 162.65, 189.97. MS (ES) m/z: 571 [M + H]+. HRMS Esi + [M + 1]: calcd for C36H47N2O4, 571.3536. Found: 571.3535.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(2-(piperidin-1-yl)ethoxy)phenyl)prop-2-en-1-one (7). Using the previous procedure and starting from piperidine, 7 (yield 95%) was obtained as a solid, mp 58–59 °C (ligroin). 1H-NMR δ 1.20–1.90 (m, 16H, 8CH2), 2.18 (s, 3H, NCH3), 2.33 (t, J = 7.2 Hz, 2H, NCH2), 2.41–2.59 (m, 4H, 2NCH2), 2.79 (t, J = 6.8 Hz, 2H, NCH2), 3.43 (s, 2H, CH2), 3.99 (t, J = 6.8 Hz, 2H, OCH2), 4.18 (t, J = 6.8 Hz, 2H, OCH2), 6.82–7.90 (m, 13H, Ar), 8.01 (d, J = 8.8 Hz, 2H, Ar). 13C-NMR δ 24.26, 26.02, 26.12, 27.01, 27.39, 27.45, 29.26, 29.41, 42.34, 55.21, 57.54, 57.86, 62.41, 66.35, 68.26, 114.49, 115.01, 114.84, 119.49, 119.54, 127.06, 127.74, 127.77, 128.33, 129.24, 130.24, 130.80, 138.69, 140.11, 144.03, 159.61, 162.65, 188.91. MS (ES) m/z: 569 [M + H]+. HRMS Esi + [M + 1]: calcd for C37H49N2O3, 569.3743. Found: 569.3744.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(2-(3,4-dihydroisoquinolin-2(1H)-yl)ethoxy)phenyl)prop-2-en-1-one (8). Using the previous procedure and starting from 1,2,3,4-tetrahydroisoquinoline, 8 (yield 30%) was obtained as an oil. 1H-NMR δ 1.22–1.80 (m, 10H, 5CH2), 2.18 (s, 3H, NCH3), 2.31 (t, J = 7.2 Hz, 2H, NCH2), 2.80–3.02 (m, 6H, 3NCH2), 3.45 (s, 2H, CH2), 3.79 (s, 2H, CH2), 3.99 (t, J = 6.8 Hz, 2H, OCH2), 4.26 (t, J = 6.6 Hz, 2H, OCH2), 6.86–7.85 (m, 17H, Ar), 8.03 (d, J = 8.8 Hz, 2H, Ar). 13C-NMR δ 25.91, 26.74, 27.22, 29.06, 29.17, 41.74, 51.39, 56.38, 57.01, 61.83, 67.04, 68.07, 114.18, 114.35, 119.29, 125.68, 126.25, 126.57, 126.95, 127.04, 127.27, 128.29, 129.37, 130.11, 130.68, 130.73, 131.81, 132.17, 138.27, 145.19, 158.42, 162.20, 192.42. MS (ES) m/z: 617 [M + H]+. C41H48N2O3.
1-(1-(2-(4-(3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)acryloyl)phenoxy)ethyl)-piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (9). Using the previous procedure and starting from 1-(piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one, 9 (yield 70%) was obtained as an oil. 1H-NMR δ 1.20–1.60 (m, 8H, 4CH2), 1.60–1.95 (m, 2H, CH2), 2.18 (s, 3H, NCH3), 2.24–2.60 (m, 4H, 2NCH2), 2.90 (t, J = 6.8 Hz, 2H, NCH2), 3.10–3.20 (m, 2H, NCH2), 3.43 (s, 2H, CH2), 3.99 (t, J = 6.8 Hz, 2H, OCH2), 4.20 (t, J = 6.8 Hz, 2H, OCH2), 4.25–4.38 (m, 1H), 6.82–7.82 (m, 17H, Ar), 8.02 (d, J = 8.8 Hz, 2H, Ar), 9.10 (br, 1H, NH). 13C-NMR δ 26.02, 26.18, 27.11, 27.38, 27.46, 29.26, 29.41, 42.34, 55.21, 57.54, 57.86, 59.01, 62.41, 66.34, 68.26, 111.76, 114.49, 115.13, 114.84, 119.49, 119.54, 124.51, 124.56, 126.81, 127.06, 127.74, 127.77, 128.33, 128.89, 129.24, 130.24, 130.80, 138.69, 140.11, 144.03, 152.62, 159.61, 162.65, 188.91. MS (ES) m/z: 701 [M + H]+. C44H52N4O4.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(2-(4-phenylpiperidin-1-yl)ethoxy)-phenyl)prop-2-en-1-one (10). Using the previous procedure and starting from phenylpiperidine, 10 (yield 65%) was obtained as a solid, mp 81–82 °C (ligroin). 1H-NMR δ 1.20–1.61 (m, 10H, 5CH2), 1.62–1.97 (m, 4H, 2CH2), 2.18 (s, 3H, NCH3), 2.20–2.61 (m, 5H), 2.90 (t, J = 6.8 Hz, 2H, NCH2), 3.05–3.20 (m, 2H), 3.46 (s, 2H, CH2), 3.99 (t, J = 6.8 Hz, 2H, OCH2), 4.20 (t, J = 6.8 Hz, 2H, OCH2), 6.86–7.82 (m, 18H, Ar). 8.02 (d, J = 8.8 Hz, 2H, Ar). 13C-NMR δ 26.18, 27.11, 27.46, 29.26, 29.41, 30.21, 30.26, 42.07, 42.44, 55.21, 57.54, 57.86, 62.41, 66.34, 68.26, 111.76, 114.49, 115.13, 114.84, 119.49, 119.54, 124.51, 124.56, 126.81, 127.06, 127.74, 127.77, 128.36, 128.89, 129.24, 130.25, 130.80, 138.69, 140.11, 144.03, 159.66, 162.65, 189.75. MS (ES) m/z: 645 [M + H]+. C43H52N2O3.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(2-(4-(4-chlorophenyl)-4-hydroxypiperidin-1-yl)ethoxy)phenyl)prop-2-en-1-one (11). Using the previous procedure and starting from 4-(4-chlorophenyl)piperidin-4-ol, 11 (yield 15%) was obtained as an oil. 1H-NMR δ 1.20–1.61 (m, 10H, 5CH2), 1.62–1.90 (m, 4H, 2CH2), 2.05–2.15 (m, 2H), 2.18 (s, 3H, NCH3), 2.30 (t, J = 6.8 Hz, 2H, NCH2), 2.54–2.61 (m, 2H), 2.91 (t, 2H, NCH2), 3.41 (s, 2H, CH2), 3.99 (t, 2H, OCH2), 4.20 (t, 2H, OCH2), 6.86–7.79 (m, 17H, Ar), 8.02 (d, J = 8.8 Hz, 2H, Ar). 13C-NMR δ 26.78, 27.12, 27.48, 29.26, 29.44, 30.24, 30.30, 42.44, 55.28, 57.53, 57.87, 62.41, 66.37, 68.26, 69.88, 111.66, 114.51, 115.14, 114.84, 119.49, 119.54, 124.51, 124.56, 127.06, 127.74, 127.77, 128.36, 128.89, 129.24, 130.25, 130.80, 131.63, 138.69, 140.11, 144.03, 159.66, 162.65, 189.75. MS (ES) m/z: 696 [M + H]+. C43H51ClN2O4.
General method for the synthesis of final compounds 16–18: A solution of 2 or 3 or 4 (0.1 mmol) in THF (50 mL) was hydrogenated at room temperature and pressure over Pd/CaCO3. The solution was filtered from catalyst and evaporated to dryness. The residue was purified by flash chromatography on silica gel (only MeOH).
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(2-(diethylamino)ethoxy)phenyl)-propan-1-one (16). Using the previous procedure and starting from 2, 16 (yield 95%) was obtained as an oil. 1H-NMR δ 1.05 (t, J = 7.2 Hz, 6H, 2CH3), 1.20–1.61 (m, 8H, 4CH2), 1.63–1.82 (m, 2H, CH2), 2.19 (s, 3H, NCH3), 2.38 (t, J = 7.2 Hz, 2H, NCH2), 2.58–2.69 (m, 4H, 2CH2), 2.85–3.05 (m, 4H, 2CH2), 3.18 (t, J = 8.6 Hz, 2H, NCH2), 3.46 (s, 2H, CH2), 3.97 (t, J = 8.6 Hz, 2H, OCH2), 4.15 (t, J = 8.6 Hz, 2H, OCH2), 6.63 (s, 1H), 6.80–7.35 (m, 10H, Ar), 7.98 (d, J = 8.8 Hz, 2H, Ar). 13C-NMR δ 12.37, 26.07, 27.37, 29.20, 29.37, 35.08, 40.65, 42.31, 46.51, 46.69, 52.31, 57.51, 62.39, 68.05, 68.24, 113.69, 114.65, 114.84, 115.01, 119.49, 119.75, 126.99, 127.49, 127.77, 128.27, 129.18, 129.59, 130.35, 139.19, 140.01, 159.40, 161.42, 197.83. MS (ES) m/z: 560 [M + H]+. C36H50N2O3.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(3-(diethylamino)propoxy)phenyl)-propan-1-one (17). Using the previous procedure and starting from 3, 17 (yield 95%) was obtained as an oil. 1H-NMR δ 1.01 (t, J = 7.2 Hz, 6H, 2CH3), 1.20–1.61 (m, 8H, 4CH2), 1.63–1.81 (m, 2H, CH2), 1.85–2.02 (m, 2H, CH2), 2.18 (s, 3H, NCH3), 2.35 (t, J = 7.2 Hz, 2H, NCH2), 2.41–2.64 (m, 6H), 2.99 (t, J = 8.6 Hz, 2H), 3.19 (t, J = 8.6 Hz, 2H), 3.45 (s, 2H, CH2), 3.90–4.08 (m, 4H, 2OCH2), 6.78–7.83 (m, 10H, Ar), 7.95 (d, J = 12.6 Hz, 2H), 8.02 (d, J = 8.8 Hz, 1H, Ar). 13C-NMR δ 11.37, 26.05, 27.37, 27.84, 29.24, 29.37, 35.15, 40.77, 42.38, 46.51, 46.76, 52.29, 57.52, 62.41, 68.15, 68.26, 113.55, 114.47, 114.91, 115.19, 119.04, 119.75, 126.99, 127.49, 127.77, 128.27, 129.18, 129.59, 130.41, 139.24, 140.14, 159.40, 160.98, 197.86. MS (ES) m/z: 573 [M + H]+. C37H52N2O3.
3-(4-((7-(Benzyl(methyl)amino)heptyl)oxy)phenyl)-1-(4-(4-(diethylamino)butoxy)phenyl)-propan-1-one (18). Using the previous procedure and starting from 4, 18 (yield 90%) was obtained as an oil. 1H-NMR δ 1.05 (t, J = 7.2 Hz, 6H, 2CH3), 1.20–1.85 (m, 14H, 7CH2), 2.19 (s, 3H, NCH3), 2.38 (t, 2H, NCH2), 2.58–2.69 (m, 6H), 2.95–3.05 (m, 2H), 3.18–3.24 (m, 2H), 3.46 (s, 2H, CH2), 4.00 (t, J = 8.6 Hz, 2H, OCH2), 4.11 (t, J = 8.6 Hz, 2H, OCH2), 6.80–7.21 (m, 11H, Ar), 7.98 (d, J = 8.8 Hz, 2H, Ar). 13C-NMR δ 11.69, 23.21, 26.17, 27.32, 29.46, 29.65, 35.04, 40.54, 42.39, 46.89, 46.69, 52.57, 57.63, 62.47, 68.12, 68.24, 113.72, 114.31, 114.67, 115.22, 119.39, 119.65, 126.98, 127.51, 127.84, 128.30, 129.20, 129.42, 130.43, 139.29, 140.11, 156.20, 160.42, 198.22. MS (ES) m/z: 587 [M + H]+. C38H54N2O3.
4-(7-Bromoheptyloxy)benzonitrile (19). A stirred mixture of 4-hydroxybenzonitrile (2 g, 0.017 mol), 1,7-dibromoheptane (4.3 mL, 0.025 mol) and K2CO3 (4 g) was refluxed in acetone (150 mL) for 20 h. The suspension was filtered while hot, and the solvent was removed under reduced pressure. After adding petroleum ether, the residue was kept in the freezer overnight and the white solid that formed was filtered off, affording 19 (1.41 g, 70%). mp 47–49 °C (ligroin). 1H-NMR δ 1.46–1.98 (m, 10H), 3.41 (t, J = 6.8 Hz, 2H), 3.99 (t, J = 6.8 Hz, 2H), 6.5 (d, J = 8.8 Hz, 2H, Ar), 7.61 (d, 2H, J = 6.8 Hz, Ar).
4-((7-(Benzyl(methyl)amino)heptyl)oxy)benzonitrile (20). A stirred solution of 19 (1.14 g, 3.85 mmol), N-benzyl-N-methylamine (1.5 mL, 3.8 mmol) and TEA (0.4 mL, 3.8 mmol) in toluene (100 mL) was refluxed for 20 h. The mixture was washed with water (3 × 25 mL) and the organic layer was dried. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate 9:1), affording 20 as an oil (0.9 g, 70%). 1H-NMR δ 1.30–1.58 (m, 8H), 1.71–1.82 (m, 2H), 2.18 (s, 3H), 2.37 (t, J = 7.2 Hz, 2H), 3.45 (s, 2H), 3.97 (t, J = 6.8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H, Ar), 7.19–7.30 (m, 5H, Ar), 7.55 (d, J = 8.8 Hz, 2H, Ar).
4-((7-(Benzyl(methyl)amino)heptyl)oxy)benzaldehyde (21). A mixture of 20 (2.18 g, 6.48 mmol) and Ni/Raney alloy (3.06 g, 13 mmol) in 75% HCOOH (44 mL) was refluxed for 7 h, and then hot filtered. The residue was diluted with 100–150 mL of water, basified by K2CO3 and extracted with DCM (3 × 25 mL). The combined organic extracts were dried, and concentrated under reduced pressure to afford 21 (1.9 g, 90%) as yellow oil (purified by flash chromatography with toluene/acetone 9:1 as eluent). 1H-NMR δ 1.22–1.59 (m, 8H), 1.65–1.83 (m, 2H), 2.18 (s, 3H), 2.38 (t, J = 7.2 Hz, 2H), 3.43 (s, 2H), 4.01 (t, J = 6.8 Hz, 2H), 6.94–6.99 (m, 2H, Ar), 7.18–7.35 (m, 5H, Ar), 7.79 (d, J = 8.8 Hz, 2H, Ar), 9.85 (broad, 1H).
1-(4-(Bromomethyl)phenyl)ethan-1-one (22). A mixture of 4-methylacetophenone (2 g, 0.015 mol), N-bromosuccinimide (NBS, 2.66 g, 0.015 mol) and a catalytic amount of benzoyl peroxide in CCl4 was refluxed for 4 h. The mixture was hot filtered and evaporated to dryness to afford 22 (2.9 g, 95%) as brown oil. 1H-NMR δ 2.51 (s, 3H), 4.52 (s, 2H), 7.40 (d, J = 8.8 Hz, 2H, Ar), 7.91 (d, J = 8.8 Hz, 2H, Ar).
1-(4-((Diethylamino)methyl)phenyl)ethan-1-one (23). A stirred solution of 22 (1.5 g, 0.007 mol) and diethylamine (2.2 mL, 0.021 mol) in toluene (30 mL) was refluxed for 24 h. The mixture was washed with water (3 × 25 mL) and the organic layer was dried. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate 9:1), affording 23 as an oil used for the next step without further purification. 1H-NMR δ 1.01 (t, J = 6.8 Hz, 6H), 2.43–2.59 (m, 7H), 3.60 (s, 2H), 7.40 (d, J = 8.8 Hz, 2H, Ar), 7.90 (d, J = 8.8 Hz, 2H, Ar).
General method for the synthesis of compounds 24–29: A stirred mixture of 4-hydroxyacetophenone (0.022 mol), selected bromochloroalkane (0.044 mol) and K2CO3 (6 g) was refluxed in acetone (150 mL) for 20 h. The suspension was filtered while hot, and the solvent was removed under reduced pressure. After adding petroleum ether, the residue was kept in the freezer overnight and the white solid that formed was filtered off and purified by flash chromatography on silica gel (petroleum ether/ethyl acetate 9:1).
1-(4-(2-Chloroethoxy)phenyl)ethan-1-one (24). Using the previous procedure and starting from 1-bromo-2-chloroethane, 24 (yield 50%) was obtained as a white solid, mp 59–60 °C (ligroin). 1H-NMR δ 2.58 (s, 3H), 3.85 (t, J = 6.8 Hz, 2H), 4.25 (t, 2H), 6.95 (d, J = 8.8 Hz, 2H, Ar), 7.95 (d, J = 8.8 Hz, 2H, Ar).
1-(4-(3-Chloropropoxy)phenyl)ethan-1-one (25). Using the previous procedure and starting from 1-chloro-3-bromopropane, 25 (yield 78%) was obtained as a white solid, mp 25–26 °C (ligroin). 1H-NMR δ 2.11–2.35 (m, 2H), 2.57 (s, 3H), 3.72 (t, J = 6.8 Hz, 2H), 4.18 (t, J = 6.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H, Ar), 7.95 (d, J = 8.8 Hz, 2H, Ar).
1-(4-(4-Chlorobutoxy)phenyl)ethan-1-one (26). Using the previous procedure and starting from 1-chloro-4-bromobutane, 26 (yield 80%) was obtained as a white solid, mp 39–40 °C (ligroin). 1H-NMR δ 1.98–2.05 (m, 4H), 2.58 (s, 3H), 3.62 (t, J = 6.8 Hz, 2H), 4.17 (t, J = 6.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H, Ar), 7.91 (d, J = 8.8 Hz, 2H, Ar).
1-(3-(2-Chloroethoxy)phenyl)ethan-1-one (27). Using the previous procedure and starting from 3-hydroxyacetophenone and 1-bromo-2-chloroethane, 27 (yield 45%) was obtained as an oil. 1H-NMR δ 2.59 (s, 3H), 3.81 (t, J = 6.8 Hz, 2H), 4.27 (t, J = 6.8 Hz, 2H), 7.05–7.60 (m, 4H, Ar).
1-(3-(3-Chloropropoxy)phenyl)ethan-1-one (28). Using the previous procedure and starting from 3-hydroxyacetophenone and 1-chloro-3-bromopropane, 28 (yield 70%) was obtained as yellow oil. 1H-NMR δ 2.12–2.36 (m, 2H), 2.57 (s, 3H), 3.71 (t, J = 6.8 Hz, 2H), 4.17 (t, J = 6.8 Hz, 2H), 7.05–7.58 (m, 4H, Ar).
1-(3-(4-Chlorobutoxy)phenyl)ethan-1-one (29). Using the previous procedure and starting from 3-hydroxyacetophenone and 1-chloro-4-bromobutane, 29 (yield 50%) was obtained as a white solid, mp 25–26 °C (ligroin). 1H-NMR δ 1.98–2.05 (m, 4H), 2.58 (s, 3H), 3.60 (t, J = 6.8 Hz, 2H), 4.06 (t, J = 6.8 Hz, 2H), 7.06–7.56 (m, 4H, Ar).
General method for the synthesis of compounds 30–35. A stirred solution of selected chloroderivative (6.5 mmol) and diethylamine (13 mmol) in toluene (100 mL) was refluxed for 24 h. The mixture was washed with water (3 × 25 mL) and the organic layer was dried. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (toluene/acetone 4:1).
1-(4-(2-(Diethylamino)ethoxy)phenyl)ethan-1-one (30). Using the previous procedure and starting from 24, 30 (yield 40%) was obtained as an oil. 1H-NMR δ 1.03 (t, J = 7.2 Hz, 6H), 2.48–2.70 (m, 7H), 2.89 (t, J = 6.8 Hz, 2H), 4.05 (t, J = 6.8 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H, Ar), 7.90 (d, J = 8.8 Hz, 2H, Ar).
1-(4-(3-(Diethylamino)propoxy)phenyl)ethan-1-one (31). Using the previous procedure and starting from 25, 31 (yield 70%) was obtained as an oil. 1H-NMR δ 1.03 (t, J = 7.2 Hz, 6H), 1.85–2.02 (m, 2H), 2.45–2.64 (m, 9H), 4.09 (t, J = 6.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H, Ar), 7.90 (d, J = 8.8 Hz, 2H, Ar).
1-(4-(4-(Diethylamino)butoxy)phenyl)ethan-1-one (32). Using the previous procedure and starting from 26, 32 (yield 90%) was obtained as an oil. 1H-NMR δ 1.03 (t, J = 7.2 Hz, 6H), 1.61–1.89 (m, 4H), 2.43–2.61 (m, 9H), 4.02 (t, J = 6.8 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H, Ar), 7.90 (d, J = 8.8 Hz, 2H, Ar).
1-(3-(2-(Diethylamino)ethoxy)phenyl)ethan-1-one (33). Using the previous procedure and starting from 27, 33 (yield 40%) was obtained as yellow oil. 1H-NMR δ 1.06 (t, J = 7.2 Hz, 6H), 2.59–2.72 (m, 7H), 2.95 (t, J = 6.8 Hz, 2H), 4.08 (t, J = 6.8 Hz, 2H), 7.08–7.58 (m, 4H, Ar).
1-(3-(3-(Diethylamino)propoxy)phenyl)ethan-1-one (34). Using the previous procedure and starting from 28, 34 (yield 90%) was obtained as an oil. 1H-NMR δ 1.02 (t, J = 7.2 Hz, 6H), 1.88–1.99 (m, 2H), 2.58–2.64 (m, 9H), 4.01 (t, J = 6.8 Hz, 2H), 7.06–7.55 (m, 4H, Ar).
1-(3-(4-(Diethylamino)butoxy)phenyl)ethan-1-one (35). Using the previous procedure and starting from 29, 35 (yield 50%) was obtained as an oil. 1H-NMR δ 1.01 (t, J = 7.2 Hz, 6H), 1.52–1.90 (m, 4H), 2.40–2.60 (m, 9H), 4.01 (t, J = 6.8 Hz, 2H), 7.07–7.57 (m, 4H, Ar).


 ,
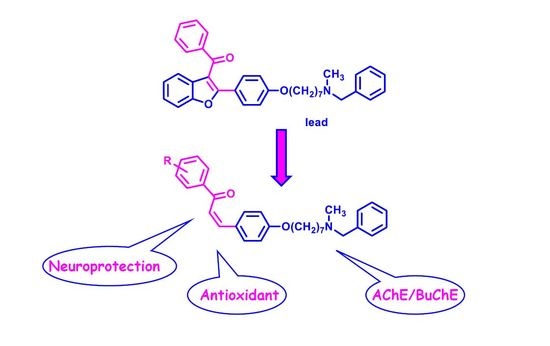
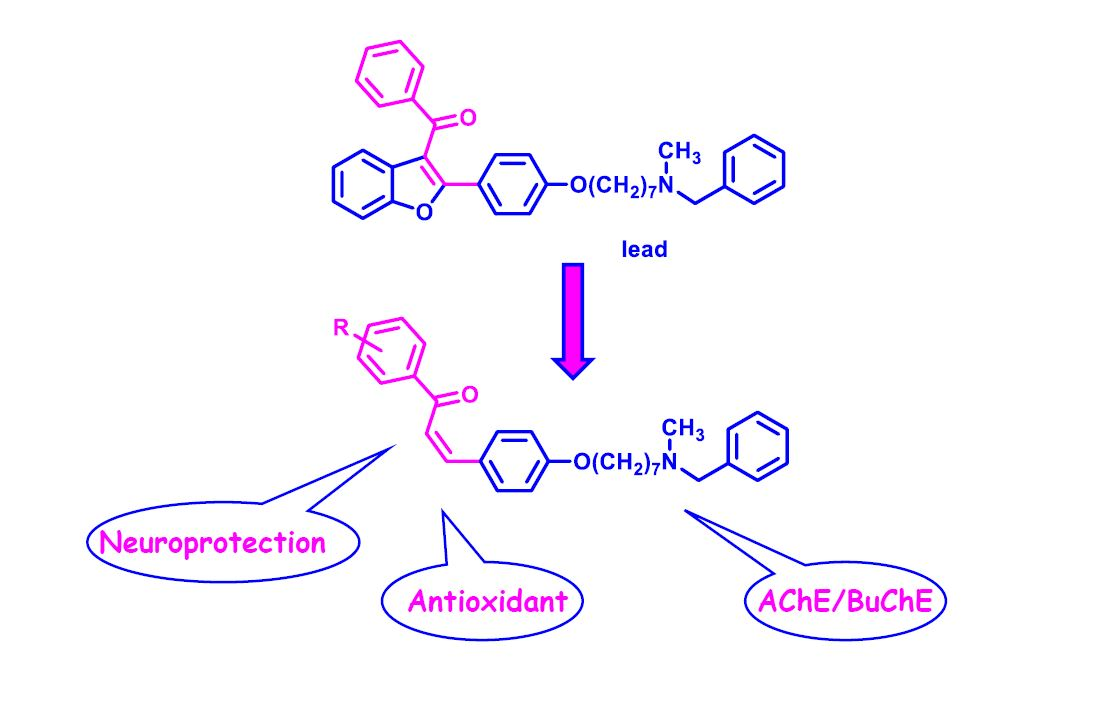
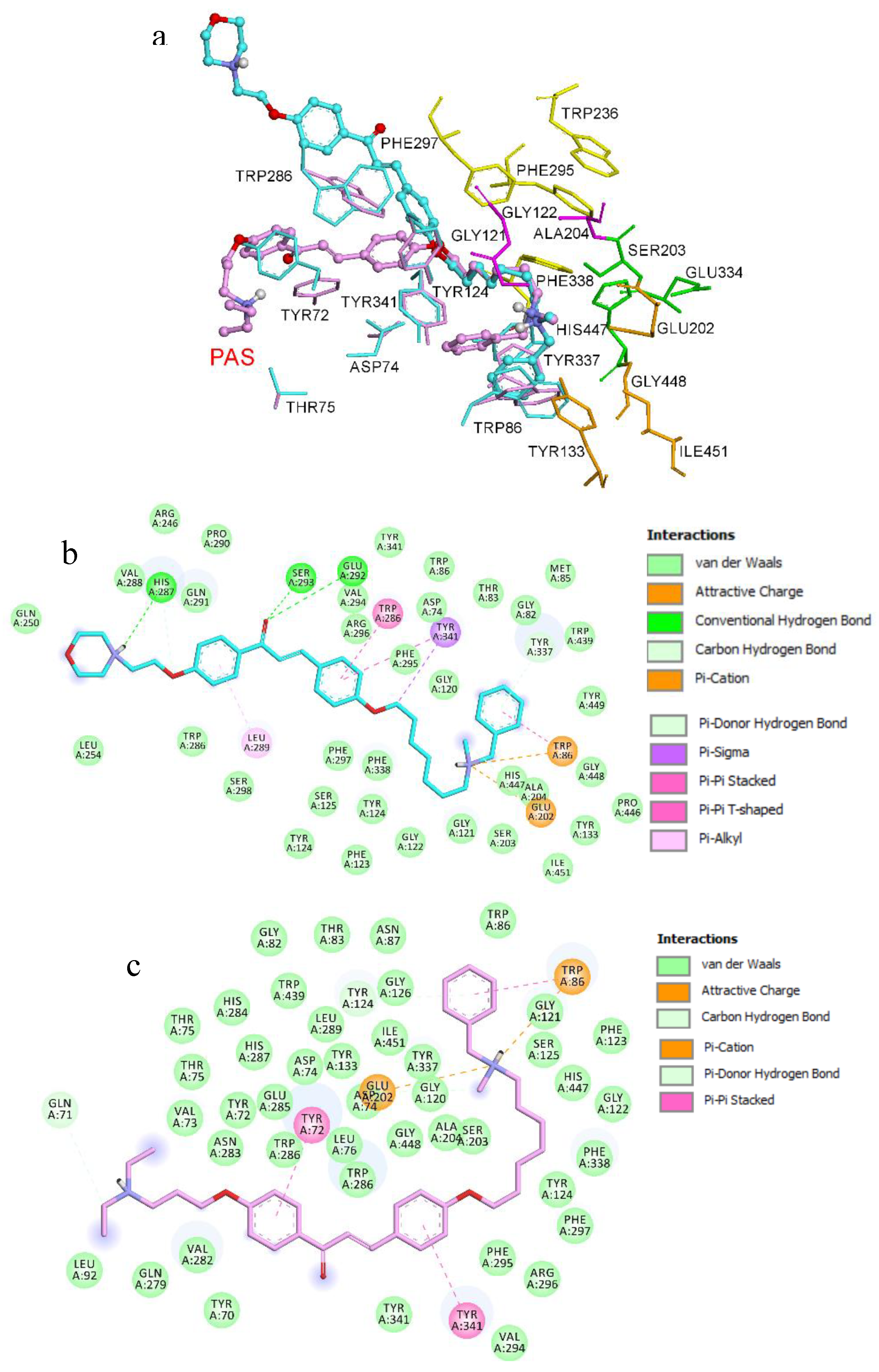
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














