
Role of Bile Acids in the Regulation of Food Intake, and Their Dysregulation in Metabolic Disease

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Effects of Bile Acids on Gastrointestinal Hormone Secretion
2.1. FXR
2.2. TGR5
3. Effects of Bile Acid Signaling on Energy Intake and Body Weight
4. Bile Acid Dysregulation in Obesity and T2D
5. Relevance of Bile Acids to Therapies for Metabolic Disorders
5.1. Bile Acid Sequestrants
5.2. Apical Sodium Bile Acid Co-Transporter (ASBT) Inhibitors
5.3. Metformin
5.4. Bariatric Surgery
6. Concluding Comments
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef]
- Setchell, K.D.; Rodrigues, C.M.; Clerici, C.; Solinas, A.; Morelli, A.; Gartung, C.; Boyer, J. Bile acid concentrations in human and rat liver tissue and in hepatocyte nuclei. Gastroenterology 1997, 112, 226–235. [Google Scholar] [CrossRef]
- Shiffman, M.L.; Sugerman, H.J.; Kellum, J.M.; Moore, E.W. Changes in gallbladder bile composition following gallstone formation and weight reduction. Gastroenterology 1992, 103, 214–221. [Google Scholar] [CrossRef]
- Northfield, T.C.; McColl, I. Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut 1973, 14, 513–518. [Google Scholar] [CrossRef] [Green Version]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihunnah, C.A.; Jiang, M.; Xie, W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim. Biophys. Acta 2011, 1812, 956–963. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Halilbasic, E.; Marschall, H.U.; Zollner, G.; Fickert, P.; Langner, C.; Zatloukal, K.; Denk, H.; Trauner, M. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 2005, 42, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Mertens, K.L.; Kalsbeek, A.; Soeters, M.R.; Eggink, H.M. Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front. Neurosci. 2017, 11, 617. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Li, J.-Y.; Lee, A.; Lu, Y.-X.; Zhou, S.-Y.; Owyang, C. Satiety induced by bile acids is mediated via vagal afferent pathways. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Jones, K.L.; Rayner, C.K.; Wu, T. Enteroendocrine hormone secretion and metabolic control: Importance of the region of the gut stimulation. Pharmaceutics 2020, 12, 790. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Mencarelli, A.; Palladino, G.; Cipriani, S. Bile-acid-activated receptors: Targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol. Sci. 2009, 30, 570–580. [Google Scholar] [CrossRef]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap—Bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.R.; Haeusler, R.A. Bile acids in glucose metabolism and insulin signalling–mechanisms and research needs. Nat. Rev. Endocrinol. 2019. [Google Scholar] [CrossRef]
- Bray, G.A.; Gallagher, T.F., Jr. Suppression of appetite by bile acids. Lancet 1968, 1, 1066–1067. [Google Scholar] [CrossRef]
- Roberts, R.E.; Glicksman, C.; Alaghband-Zadeh, J.; Sherwood, R.A.; Akuji, N.; le Roux, C.W. The relationship between postprandial bile acid concentration, GLP-1, PYY and ghrelin. Clin. Endocrinol. 2011, 74, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Nakatani, H.; Kasama, K.; Oshiro, T.; Watanabe, M.; Hirose, H.; Itoh, H. Serum bile acid along with plasma incretins and serum high-molecular weight adiponectin levels are increased after bariatric surgery. Metabolism 2009, 58, 1400–1407. [Google Scholar] [CrossRef]
- Kuhre, R.E.; Wewer Albrechtsen, N.J.; Larsen, O.; Jepsen, S.L.; Balk-Moller, E.; Andersen, D.B.; Deacon, C.F.; Schoonjans, K.; Reimann, F.; Gribble, F.M.; et al. Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol. Metab. 2018, 11, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Gomez, G.; Upp, J.R., Jr.; Lluis, F.; Alexander, R.W.; Poston, G.J.; Greeley, G.H., Jr.; Thompson, J.C. Regulation of the release of cholecystokinin by bile salts in dogs and humans. Gastroenterology 1988, 94, 1036–1046. [Google Scholar] [CrossRef]
- Koop, I.; Schindler, M.; Bosshammer, A.; Scheibner, J.; Stange, E.; Koop, H. Physiological control of cholecystokinin release and pancreatic enzyme secretion by intraduodenal bile acids. Gut 1996, 39, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonne, D.P.; Hansen, M.; Knop, F.K. Bile acid sequestrants in type 2 diabetes: Potential effects on GLP1 secretion. Eur. J. Endocrinol. 2014, 171, R47–R65. [Google Scholar] [CrossRef]
- Guida, C.; Ramracheya, R. PYY, a therapeutic option for type 2 diabetes? Clin. Med. Insights Endocrinol. Diabetes 2020, 13, 1179551419892985. [Google Scholar] [CrossRef]
- Wu, T.; Bound, M.J.; Standfield, S.D.; Jones, K.L.; Horowitz, M.; Rayner, C.K. Effects of taurocholic acid on glycemic, glucagon-like peptide-1, and insulin responses to small intestinal glucose infusion in healthy humans. J. Clin. Endocrinol. Metab. 2013, 98, E718–E722. [Google Scholar] [CrossRef]
- Trabelsi, M.S.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 2015, 6, 7629. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Chen, T.; Jiang, R.; Zhao, A.; Wu, Q.; Kuang, J.; Sun, D.; Ren, Z.; Li, M.; Zhao, M.; et al. Hyocholic acid species improve glucose homeostasis through a distinct TGR5 and FXR signaling mechanism. Cell Metab. 2020. [Google Scholar] [CrossRef]
- Li, P.; Zhu, L.; Yang, X.; Li, W.; Sun, X.; Yi, B.; Zhu, S. Farnesoid X receptor interacts with cAMP response element binding protein to modulate glucagon-like peptide-1 (7-36) amide secretion by intestinal L cell. J. Cell Physiol. 2019, 234, 12839–12846. [Google Scholar] [CrossRef]
- Ducastel, S.; Touche, V.; Trabelsi, M.S.; Boulinguiez, A.; Butruille, L.; Nawrot, M.; Peschard, S.; Chavez-Talavera, O.; Dorchies, E.; Vallez, E.; et al. The nuclear receptor FXR inhibits Glucagon-Like Peptide-1 secretion in response to microbiota-derived Short-Chain Fatty Acids. Sci. Rep. 2020, 10, 174. [Google Scholar] [CrossRef]
- Xie, C.; Jiang, C.; Shi, J.; Gao, X.; Sun, D.; Sun, L.; Wang, T.; Takahashi, S.; Anitha, M.; Krausz, K.W.; et al. An intestinal farnesoid X receptor-ceramide signaling axis modulates hepatic gluconeogenesis in mice. Diabetes 2017, 66, 613–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albaugh, V.L.; Banan, B.; Antoun, J.; Xiong, Y.; Guo, Y.; Ping, J.; Alikhan, M.; Clements, B.A.; Abumrad, N.N.; Flynn, C.R. Role of bile acids and GLP-1 in mediating the metabolic improvements of bariatric surgery. Gastroenterology 2019, 156, 1041–1051.e1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018, 68, 1574–1588. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.E.; Wallis, K.; le Roux, C.W.; Wong, K.Y.; Reimann, F.; Gribble, F.M. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br. J. Pharmacol. 2012, 165, 414–423. [Google Scholar] [CrossRef] [Green Version]
- Brighton, C.A.; Rievaj, J.; Kuhre, R.E.; Glass, L.L.; Schoonjans, K.; Holst, J.J.; Gribble, F.M.; Reimann, F. Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G protein-coupled bile acid receptors. Endocrinology 2015, 156, 3961–3970. [Google Scholar] [CrossRef] [Green Version]
- Hodge, R.J.; Lin, J.; Vasist Johnson, L.S.; Gould, E.P.; Bowers, G.D.; Nunez, D.J.; Team, S.B.P. Safety, pharmacokinetics, and pharmacodynamic effects of a selective TGR5 agonist, SB-756050, in type 2 diabetes. Clin. Pharmacol. Drug Dev. 2013, 2, 213–222. [Google Scholar] [CrossRef]
- Adrian, T.; Ballantyne, G.; Longo, W.; Bilchik, A.; Graham, S.; Basson, M.; Tierney, R.; Modlin, I. Deoxycholate is an important releaser of peptide YY and enteroglucagon from the human colon. Gut 1993, 34, 1219–1224. [Google Scholar] [CrossRef] [Green Version]
- Adrian, T.E.; Gariballa, S.; Parekh, K.A.; Thomas, S.A.; Saadi, H.; Al Kaabi, J.; Nagelkerke, N.; Gedulin, B.; Young, A.A. Rectal taurocholate increases L cell and insulin secretion, and decreases blood glucose and food intake in obese type 2 diabetic volunteers. Diabetologia 2012, 55, 2343–2347. [Google Scholar] [CrossRef]
- Wu, T.; Bound, M.J.; Standfield, S.D.; Gedulin, B.; Jones, K.L.; Horowitz, M.; Rayner, C.K. Effects of rectal administration of taurocholic acid on glucagon-like peptide-1 and peptide YY secretion in healthy humans. Diabetes Obes. Metab. 2013, 15, 474–477. [Google Scholar] [CrossRef]
- Tough, I.R.; Schwartz, T.W.; Cox, H.M. Synthetic G protein-coupled bile acid receptor agonists and bile acids act via basolateral receptors in ileal and colonic mucosa. Neurogastroenterol. Motil. 2020, 32, e13943. [Google Scholar] [CrossRef]
- Christiansen, C.B.; Trammell, S.A.J.; Wewer Albrechtsen, N.J.; Schoonjans, K.; Albrechtsen, R.; Gillum, M.P.; Kuhre, R.E.; Holst, J.J. Bile acids drive colonic secretion of glucagon-like-peptide 1 and peptide-YY in rodents. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G574–G584. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Sato, H.; Macchiarulo, A.; Thomas, C.; Gioiello, A.; Une, M.; Hofmann, A.F.; Saladin, R.; Schoonjans, K.; Pellicciari, R.; Auwerx, J. Novel potent and selective bile acid derivatives as TGR5 agonists: Biological screening, structure-activity relationships, and molecular modeling studies. J. Med. Chem. 2008, 51, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Bertaggia, E.; Jensen, K.K.; Castro-Perez, J.; Xu, Y.; Di Paolo, G.; Chan, R.B.; Wang, L.; Haeusler, R.A. Cyp8b1 ablation prevents Western diet-induced weight gain and hepatic steatosis because of impaired fat absorption. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E121–E133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, S.; Ahmad, T.R.; Argueta, D.A.; Perez, P.A.; Zhao, C.; Schwartz, G.J.; DiPatrizio, N.V.; Haeusler, R.A. Bile acid composition regulates GPR119-dependent intestinal lipid sensing and food intake regulation in mice. Gut 2020, 69, 1620–1628. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Huang, F.; Zhao, L.; Zhang, Y.; Yang, W.; Wang, S.; Li, M.; Han, X.; Ge, K.; Qu, C.; et al. A dysregulated bile acid-gut microbiota axis contributes to obesity susceptibility. EBioMedicine 2020, 55, 102766. [Google Scholar] [CrossRef]
- Zietak, M.; Kozak, L.P. Bile acids induce uncoupling protein 1-dependent thermogenesis and stimulate energy expenditure at thermoneutrality in mice. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E346–E354. [Google Scholar] [CrossRef] [Green Version]
- Zaborska, K.E.; Lee, S.A.; Garribay, D.; Cha, E.; Cummings, B.P. Deoxycholic acid supplementation impairs glucose homeostasis in mice. PLoS ONE 2018, 13, e0200908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Liu, G.; Zhang, X.; Bi, D.; Hu, S. Improvement of glucose metabolism following long-term taurocholic acid gavage in a diabetic rat model. Med. Sci. Monit. 2018, 24, 7206–7212. [Google Scholar] [CrossRef] [PubMed]
- Calderon, G.; McRae, A.; Rievaj, J.; Davis, J.; Zandvakili, I.; Linker-Nord, S.; Burton, D.; Roberts, G.; Reimann, F.; Gedulin, B.; et al. Ileo-colonic delivery of conjugated bile acids improves glucose homeostasis via colonic GLP-1-producing enteroendocrine cells in human obesity and diabetes. EBioMedicine 2020, 55, 102759. [Google Scholar] [CrossRef]
- Fang, S.; Suh, J.M.; Reilly, S.M.; Yu, E.; Osborn, O.; Lackey, D.; Yoshihara, E.; Perino, A.; Jacinto, S.; Lukasheva, Y.; et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 2015, 21, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Zou, J.; Li, S.; Guo, J.; Shi, W.; Wang, B.; Han, X.; Zhang, H.; Zhang, P.; Miao, Z.; et al. Farnesoid X receptor contributes to body weight-independent improvements in glycemic control after Roux-en-Y gastric bypass surgery in diet-induced obese mice. Mol. Metab. 2020, 37, 100980. [Google Scholar] [CrossRef]
- McGavigan, A.K.; Garibay, D.; Henseler, Z.M.; Chen, J.; Bettaieb, A.; Haj, F.G.; Ley, R.E.; Chouinard, M.L.; Cummings, B.P. TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut 2017, 66, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: A phase 2 randomized controlled trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef]
- Steiner, C.; Othman, A.; Saely, C.H.; Rein, P.; Drexel, H.; von Eckardstein, A.; Rentsch, K.M. Bile acid metabolites in serum: Intraindividual variation and associations with coronary heart disease, metabolic syndrome and diabetes mellitus. PLoS ONE 2011, 6, e25006. [Google Scholar] [CrossRef] [Green Version]
- Prinz, P.; Hofmann, T.; Ahnis, A.; Elbelt, U.; Goebel-Stengel, M.; Klapp, B.F.; Rose, M.; Stengel, A. Plasma bile acids show a positive correlation with body mass index and are negatively associated with cognitive restraint of eating in obese patients. Front. Neurosci. 2015, 9, 199. [Google Scholar] [CrossRef] [Green Version]
- Cariou, B.; Chetiveaux, M.; Zair, Y.; Pouteau, E.; Disse, E.; Guyomarc’h-Delasalle, B.; Laville, M.; Krempf, M. Fasting plasma chenodeoxycholic acid and cholic acid concentrations are inversely correlated with insulin sensitivity in adults. Nutr. Metab. 2011, 8, 48. [Google Scholar] [CrossRef] [Green Version]
- Straniero, S.; Rosqvist, F.; Edholm, D.; Ahlstrom, H.; Kullberg, J.; Sundbom, M.; Riserus, U.; Rudling, M. Acute caloric restriction counteracts hepatic bile acid and cholesterol deficiency in morbid obesity. J. Intern. Med. 2017, 281, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeusler, R.A.; Camastra, S.; Nannipieri, M.; Astiarraga, B.; Castro-Perez, J.; Xie, D.; Wang, L.; Chakravarthy, M.; Ferrannini, E. Increased Bile Acid Synthesis and Impaired Bile Acid Transport in Human Obesity. J. Clin. Endocrinol. Metab. 2016, 101, 1935–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, O.; Harsch, S.; Matysik, S.; Lutjohann, D.; Schmitz, G.; Stange, E.F. Upregulation of hepatic bile acid synthesis via fibroblast growth factor 19 is defective in gallstone disease but functional in overweight individuals. United Eur. Gastroenterol. J. 2014, 2, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Ambrosi, J.; Gallego-Escuredo, J.M.; Catalan, V.; Rodriguez, A.; Domingo, P.; Moncada, R.; Valenti, V.; Salvador, J.; Giralt, M.; Villarroya, F.; et al. FGF19 and FGF21 serum concentrations in human obesity and type 2 diabetes behave differently after diet- or surgically-induced weight loss. Clin. Nutr. 2017, 36, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Glicksman, C.; Pournaras, D.J.; Wright, M.; Roberts, R.; Mahon, D.; Welbourn, R.; Sherwood, R.; Alaghband-Zadeh, J.; le Roux, C.W. Postprandial plasma bile acid responses in normal weight and obese subjects. Ann. Clin. Biochem. 2010, 47, 482–484. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.N.; Pfalzer, A.; Kaplan, L.M. Roux-en-Y gastric bypass normalizes the blunted postprandial bile acid excursion associated with obesity. Int. J. Obes. 2013, 37, 1553–1559. [Google Scholar] [CrossRef] [Green Version]
- Aleman, J.O.; Bokulich, N.A.; Swann, J.R.; Walker, J.M.; De Rosa, J.C.; Battaglia, T.; Costabile, A.; Pechlivanis, A.; Liang, Y.; Breslow, J.L.; et al. Fecal microbiota and bile acid interactions with systemic and adipose tissue metabolism in diet-induced weight loss of obese postmenopausal women. J. Transl. Med. 2018, 16, 244. [Google Scholar] [CrossRef]
- Kudchodkar, B.J.; Sodhi, H.S.; Mason, D.T.; Borhani, N.O. Effects of acute caloric restriction on cholesterol metabolism in man. Am. J. Clin. Nutr. 1977, 30, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.C.; Hoffmann, C.; Mota, J.F. The human gut microbiota: Metabolism and perspective in obesity. Gut Microbes 2018, 9, 308–325. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Distrutti, E. Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Legry, V.; Francque, S.; Haas, J.T.; Verrijken, A.; Caron, S.; Chavez-Talavera, O.; Vallez, E.; Vonghia, L.; Dirinck, E.; Verhaegen, A.; et al. Bile acid alterations are associated with insulin resistance, but not with NASH, in obese subjects. J. Clin. Endocrinol. Metab. 2017, 102, 3783–3794. [Google Scholar] [CrossRef] [PubMed]
- De Vuono, S.; Ricci, M.A.; Nulli Migliola, E.; Monti, M.C.; Morretta, E.; Boni, M.; Ministrini, S.; Carino, A.; Fiorucci, S.; Distrutti, E.; et al. Serum bile acid levels before and after sleeve gastrectomy and their correlation with obesity-related comorbidities. Obes. Surg. 2019, 29, 2517–2526. [Google Scholar] [CrossRef]
- Sonne, D.P.; van Nierop, F.S.; Kulik, W.; Soeters, M.R.; Vilsboll, T.; Knop, F.K. Postprandial plasma concentrations of individual bile acids and FGF-19 in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 3002–3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeusler, R.A.; Astiarraga, B.; Camastra, S.; Accili, D.; Ferrannini, E. Human insulin resistance is associated with increased plasma levels of 12alpha-hydroxylated bile acids. Diabetes 2013, 62, 4184–4191. [Google Scholar] [CrossRef] [Green Version]
- Wewalka, M.; Patti, M.E.; Barbato, C.; Houten, S.M.; Goldfine, A.B. Fasting serum taurine-conjugated bile acids are elevated in type 2 diabetes and do not change with intensification of insulin. J. Clin. Endocrinol. Metab. 2014, 99, 1442–1451. [Google Scholar] [CrossRef] [Green Version]
- Brufau, G.; Stellaard, F.; Prado, K.; Bloks, V.W.; Jonkers, E.; Boverhof, R.; Kuipers, F.; Murphy, E.J. Improved glycemic control with colesevelam treatment in patients with type 2 diabetes is not directly associated with changes in bile acid metabolism. Hepatology 2010, 52, 1455–1464. [Google Scholar] [CrossRef]
- Bennion, L.J.; Grundy, S.M. Effects of diabetes mellitus on cholesterol metabolism in man. N. Engl. J. Med. 1977, 296, 1365–1371. [Google Scholar] [CrossRef]
- Wu, H.; Tremaroli, V.; Schmidt, C.; Lundqvist, A.; Olsson, L.M.; Kramer, M.; Gummesson, A.; Perkins, R.; Bergstrom, G.; Backhed, F. The gut microbiota in prediabetes and diabetes: A population-based cross-sectional study. Cell Metab. 2020, 32, 379–390.e373. [Google Scholar] [CrossRef]
- Annaba, F.; Ma, K.; Kumar, P.; Dudeja, A.K.; Kineman, R.D.; Shneider, B.L.; Saksena, S.; Gill, R.K.; Alrefai, W.A. Ileal apical Na+-dependent bile acid transporter ASBT is upregulated in rats with diabetes mellitus induced by low doses of streptozotocin. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G898–G906. [Google Scholar] [CrossRef] [Green Version]
- Sonne, D.P.; Rehfeld, J.F.; Holst, J.J.; Vilsboll, T.; Knop, F.K. Postprandial gallbladder emptying in patients with type 2 diabetes: Potential implications for bile-induced secretion of glucagon-like peptide 1. Eur. J. Endocrinol. 2014, 171, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Sansome, D.J.; Xie, C.; Veedfald, S.; Horowitz, M.; Rayner, C.K.; Wu, T. Mechanism of glucose-lowering by metformin in type 2 diabetes: Role of bile acids. Diabetes Obes. Metab. 2020, 22, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Lee, Y.H.; Choi, E.; Cho, Y.; Kim, J.H. Fasting serum bile acids concentration is associated with insulin resistance independently of diabetes status. Clin. Chem. Lab. Med. 2019, 57, 1218–1228. [Google Scholar] [CrossRef]
- Lu, J.; Wang, S.; Li, M.; Gao, Z.; Xu, Y.; Zhao, X.; Hu, C.; Zhang, Y.; Liu, R.; Hu, R.; et al. Association of serum bile acids profile and pathway dysregulation with the risk of developing diabetes among normoglycemic chinese adults: Findings from the 4C study. Diabetes Care 2021, 44, 499–510. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, C.D.; Paumgartner, G.; Mlitz, V.; Kunczer, V.; Halilbasic, E.; Leditznig, N.; Wahlstrom, A.; Stahlman, M.; Thuringer, A.; Kashofer, K.; et al. Colesevelam attenuates cholestatic liver and bile duct injury in Mdr2(−/−) mice by modulating composition, signalling and excretion of faecal bile acids. Gut 2018, 67, 1683–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Morimoto, K.; Houten, S.M.; Kaneko-Iwasaki, N.; Sugizaki, T.; Horai, Y.; Mataki, C.; Sato, H.; Murahashi, K.; Arita, E.; et al. Bile acid binding resin improves metabolic control through the induction of energy expenditure. PLoS ONE 2012, 7, e38286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schadt, H.S.; Wolf, A.; Mahl, J.A.; Wuersch, K.; Couttet, P.; Schwald, M.; Fischer, A.; Lienard, M.; Emotte, C.; Teng, C.H.; et al. Bile acid sequestration by cholestyramine mitigates FGFR4 inhibition-induced ALT elevation. Toxicol. Sci. 2018, 163, 265–278. [Google Scholar] [CrossRef]
- Herrema, H.; Meissner, M.; van Dijk, T.H.; Brufau-Dones, G.; Boverhof, R.; Müller, M.; Stellaard, F.; Kuipers, F. Bile salt sequestration induces the hepatic lipogenic pathway without altering bile salt pool size and transhepatic bile salt flux in mice. Chem. Phys. Lipids 2008, 154, S59. [Google Scholar] [CrossRef]
- Beysen, C.; Murphy, E.J.; Deines, K.; Chan, M.; Tsang, E.; Glass, A.; Turner, S.M.; Protasio, J.; Riiff, T.; Hellerstein, M.K. Effect of bile acid sequestrants on glucose metabolism, hepatic de novo lipogenesis, and cholesterol and bile acid kinetics in type 2 diabetes: A randomised controlled study. Diabetologia 2012, 55, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Karhus, M.L.; Bronden, A.; Sonne, D.P.; Vilsboll, T.; Knop, F.K. Evidence connecting old, new and neglected glucose-lowering drugs to bile acid-induced GLP-1 secretion: A review. Diabetes Obes. Metab. 2017, 19, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Shang, Q.; Saumoy, M.; Holst, J.J.; Salen, G.; Xu, G. Colesevelam improves insulin resistance in a diet-induced obesity (F-DIO) rat model by increasing the release of GLP-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G419–G424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smushkin, G.; Sathananthan, M.; Piccinini, F.; Dalla Man, C.; Law, J.H.; Cobelli, C.; Zinsmeister, A.R.; Rizza, R.A.; Vella, A. The effect of a bile acid sequestrant on glucose metabolism in subjects with type 2 diabetes. Diabetes 2013, 62, 1094–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, M.; Scheltema, M.J.; Sonne, D.P.; Hansen, J.S.; Sperling, M.; Rehfeld, J.F.; Holst, J.J.; Vilsboll, T.; Knop, F.K. Effect of chenodeoxycholic acid and the bile acid sequestrant colesevelam on glucagon-like peptide-1 secretion. Diabetes Obes. Metab. 2016, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Bronden, A.; Alber, A.; Rohde, U.; Gasbjerg, L.S.; Rehfeld, J.F.; Holst, J.J.; Vilsboll, T.; Knop, F.K. The bile acid-sequestering resin sevelamer eliminates the acute GLP-1 stimulatory effect of endogenously released bile acids in patients with type 2 diabetes. Diabetes Obes. Metab. 2018, 20, 362–369. [Google Scholar] [CrossRef]
- Sugimoto-Kawabata, K.; Shimada, H.; Sakai, K.; Suzuki, K.; Kelder, T.; Pieterman, E.J.; Cohen, L.H.; Havekes, L.M.; Princen, H.M.; van den Hoek, A.M. Colestilan decreases weight gain by enhanced NEFA incorporation in biliary lipids and fecal lipid excretion. J. Lipid Res. 2013, 54, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, V.A.; Rosenstock, J.; Wang, A.C.; Truitt, K.E.; Jones, M.R. Colesevelam HCl improves glycemic control and reduces LDL cholesterol in patients with inadequately controlled type 2 diabetes on sulfonylurea-based therapy. Diabetes Care 2008, 31, 1479–1484. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, R.B.; Fonseca, V.A.; Truitt, K.E.; Jones, M.R. Efficacy and safety of colesevelam in patients with type 2 diabetes mellitus and inadequate glycemic control receiving insulin-based therapy. Arch. Intern. Med. 2008, 168, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Bays, H.E.; Goldberg, R.B.; Truitt, K.E.; Jones, M.R. Colesevelam hydrochloride therapy in patients with type 2 diabetes mellitus treated with metformin: Glucose and lipid effects. Arch. Intern. Med. 2008, 168, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Graffner, H.; Gillberg, P.G.; Rikner, L.; Marschall, H.U. The ileal bile acid transporter inhibitor A4250 decreases serum bile acids by interrupting the enterohepatic circulation. Aliment. Pharmacol. Ther. 2016, 43, 303–310. [Google Scholar] [CrossRef]
- Kitayama, K.; Nakai, D.; Kono, K.; van der Hoop, A.G.; Kurata, H.; de Wit, E.C.; Cohen, L.H.; Inaba, T.; Kohama, T. Novel non-systemic inhibitor of ileal apical Na+-dependent bile acid transporter reduces serum cholesterol levels in hamsters and monkeys. Eur. J. Pharmacol. 2006, 539, 89–98. [Google Scholar] [CrossRef]
- Wu, Y.; Aquino, C.J.; Cowan, D.J.; Anderson, D.L.; Ambroso, J.L.; Bishop, M.J.; Boros, E.E.; Chen, L.; Cunningham, A.; Dobbins, R.L.; et al. Discovery of a highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor (GSK2330672) for treatment of type 2 diabetes. J. Med. Chem. 2013, 56, 5094–5114. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Kosters, A.; Mells, J.E.; Zhang, W.; Setchell, K.D.; Amanso, A.M.; Wynn, G.M.; Xu, T.; Keller, B.T.; Yin, H.; et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci. Transl. Med. 2016, 8, 357ra122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Dury, S.; Marschall, H.U. Ileal bile acid transporter inhibition for the treatment of chronic constipation, cholestatic pruritus, and NASH. Front. Pharmacol. 2018, 9, 931. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yao, X.; Young, A.; McNulty, J.; Anderson, D.; Liu, Y.; Nystrom, C.; Croom, D.; Ross, S.; Collins, J.; et al. Inhibition of apical sodium-dependent bile acid transporter as a novel treatment for diabetes. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E68–E76. [Google Scholar] [CrossRef] [PubMed]
- Rudling, M.; Camilleri, M.; Graffner, H.; Holst, J.J.; Rikner, L. Specific inhibition of bile acid transport alters plasma lipids and GLP-1. BMC Cardiovasc. Disord. 2015, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Day, E.A.; Ford, R.J.; Smith, B.K.; Mohammadi-Shemirani, P.; Morrow, M.R.; Gutgesell, R.M.; Lu, R.; Raphenya, A.R.; Kabiri, M.; McArthur, A.G.; et al. Metformin-induced increases in GDF15 are important for suppressing appetite and promoting weight loss. Nat. Metab. 2019, 1, 1202–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, A.P.; Chen, M.; Taskar, P.; Rimmington, D.; Patel, S.; Tadross, J.A.; Cimino, I.; Yang, M.; Welsh, P.; Virtue, S.; et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature 2020, 578, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Adeyemo, M.A.; McDuffie, J.R.; Kozlosky, M.; Krakoff, J.; Calis, K.A.; Brady, S.M.; Yanovski, J.A. Effects of metformin on energy intake and satiety in obese children. Diabetes Obes. Metab. 2015, 17, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Thazhath, S.S.; Bound, M.J.; Jones, K.L.; Horowitz, M.; Rayner, C.K. Mechanism of increase in plasma intact GLP-1 by metformin in type 2 diabetes: Stimulation of GLP-1 secretion or reduction in plasma DPP-4 activity? Diabetes Res. Clin. Pract. 2014, 106, e3–e6. [Google Scholar] [CrossRef]
- Bahne, E.; Sun, E.W.L.; Young, R.L.; Hansen, M.; Sonne, D.P.; Hansen, J.S.; Rohde, U.; Liou, A.P.; Jackson, M.L.; de Fontgalland, D.; et al. Metformin-induced glucagon-like peptide-1 secretion contributes to the actions of metformin in type 2 diabetes. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, M.J.; Bound, M.; Grivell, J.; Sun, Z.; Jones, K.L.; Horowitz, M.; Rayner, C.K.; Wu, T. Comparative effects of proximal and distal small intestinal administration of metformin on plasma glucose and glucagon-like peptide-1, and gastric emptying after oral glucose, in type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 640–647. [Google Scholar] [CrossRef]
- Sun, E.W.; Martin, A.M.; Wattchow, D.A.; de Fontgalland, D.; Rabbitt, P.; Hollington, P.; Young, R.L.; Keating, D.J. Metformin triggers PYY secretion in human gut mucosa. J. Clin. Endocrinol. Metab. 2019, 104, 2668–2674. [Google Scholar] [CrossRef]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Fruhbeck, G. Bariatric and metabolic surgery: A shift in eligibility and success criteria. Nat. Rev. Endocrinol. 2015, 11, 465–477. [Google Scholar] [CrossRef]
- Rubino, F.; Schauer, P.R.; Kaplan, L.M.; Cummings, D.E. Metabolic surgery to treat type 2 diabetes: Clinical outcomes and mechanisms of action. Annu. Rev. Med. 2010, 61, 393–411. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, T.; Wang, Y.; Zhong, M.; Zhang, G.; Liu, S.; Wu, T.; Rayner, C.K.; Hu, S. Comparative effects of bile diversion and duodenal-jejunal bypass on glucose and lipid metabolism in male diabetic rats. Obes. Surg. 2016, 26, 1565–1575. [Google Scholar] [CrossRef]
- Flynn, C.R.; Albaugh, V.L.; Cai, S.; Cheung-Flynn, J.; Williams, P.E.; Brucker, R.M.; Bordenstein, S.R.; Guo, Y.; Wasserman, D.H.; Abumrad, N.N. Bile diversion to the distal small intestine has comparable metabolic benefits to bariatric surgery. Nat. Commun. 2015, 6, 7715. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cheng, Z.; Wang, Y.; Dai, Y.; Zhang, X.; Hu, S. Role of bile acids in bariatric surgery. Front. Physiol. 2019, 10, 374. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]



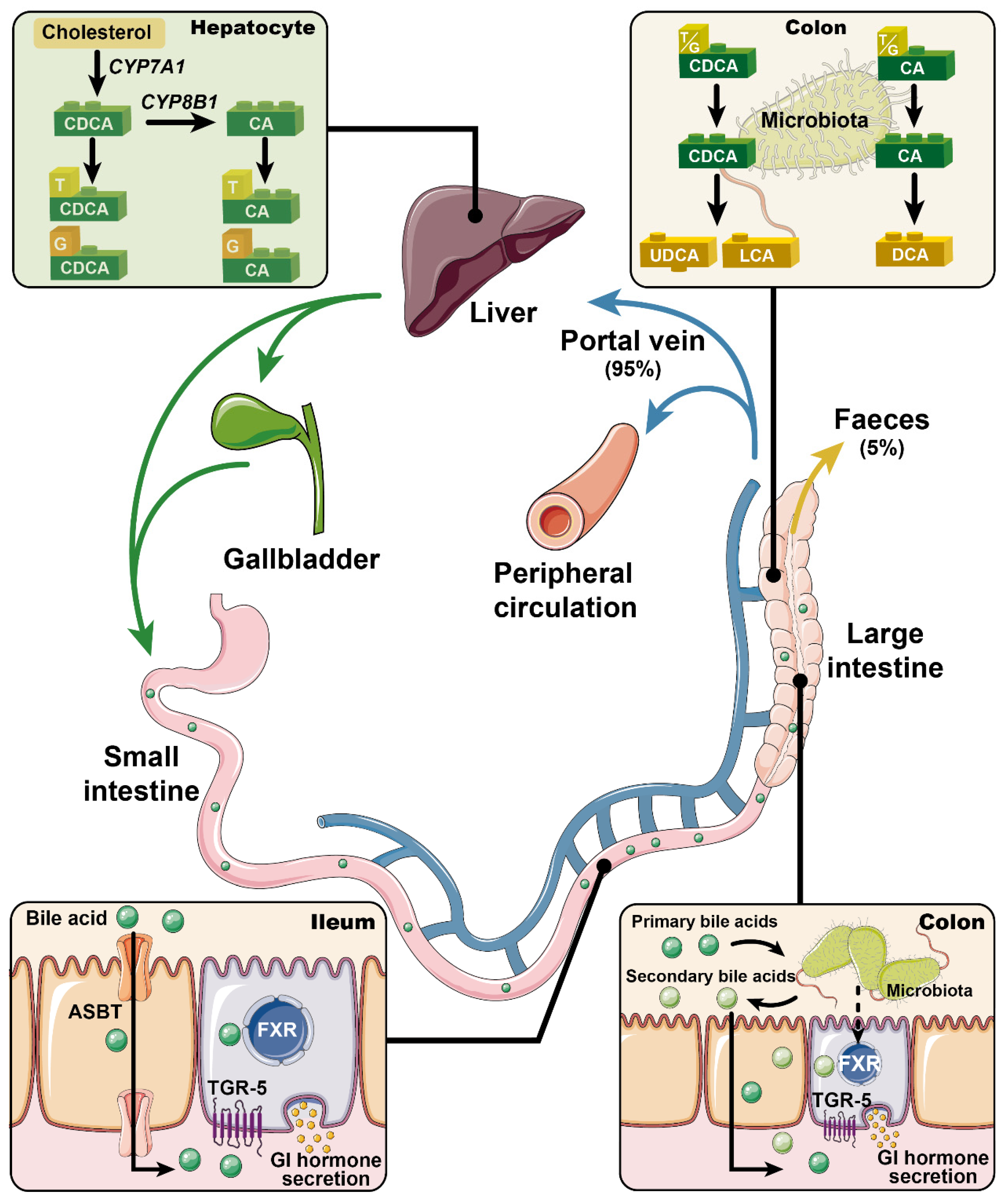{kind=link}
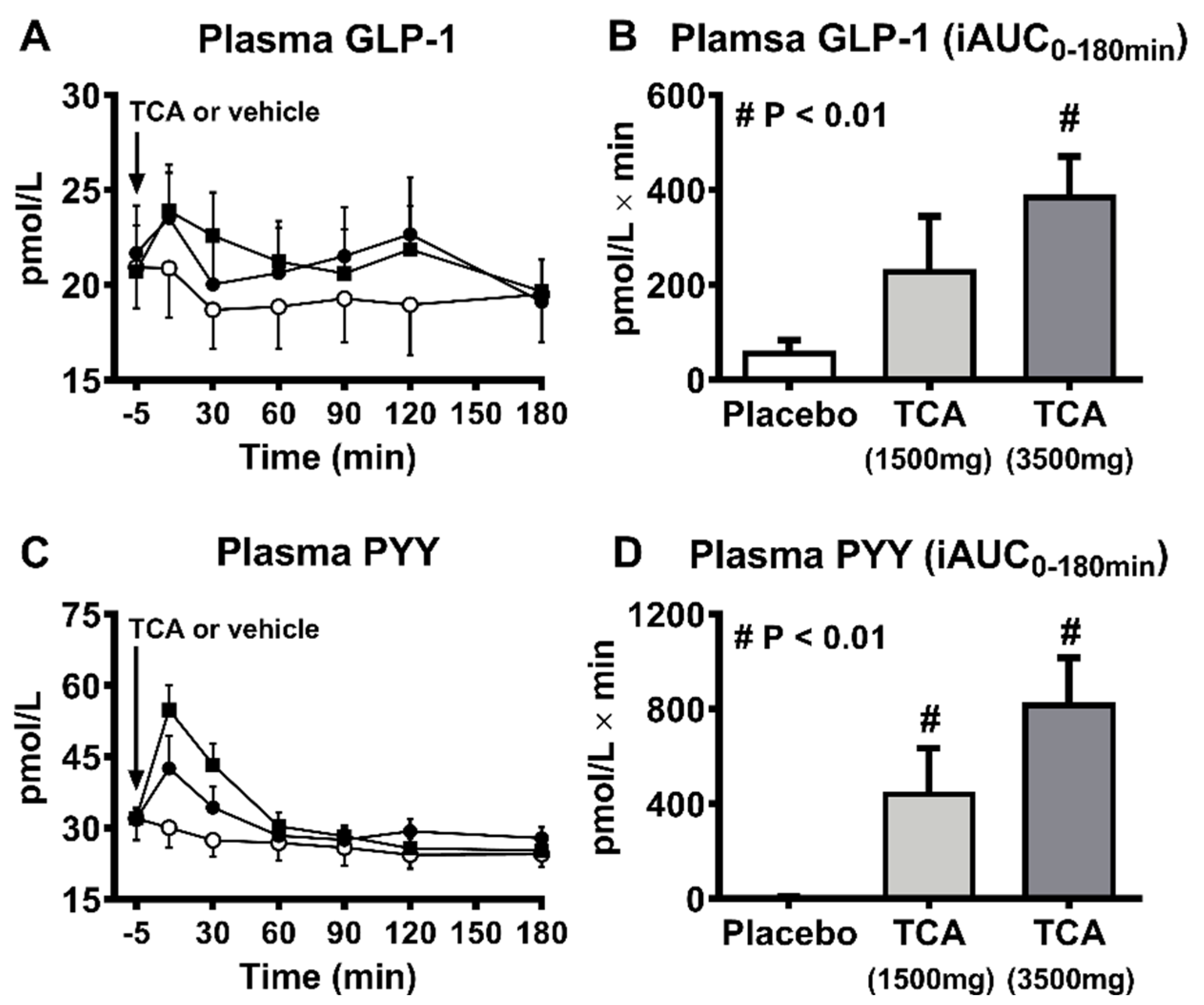{kind=link}
| Bile Acid | TGR5 | FXR | ||||
|---|---|---|---|---|---|---|
| Subjects | Indicator | EC50 | Subjects | Indicator | EC50 | |
| Primary Bile Acids | ||||||
| CA | CHO cells/HEK293 | Intracellular cAMP | 7.72 µM [34]/ >10 µM [10] | CV-1 cells | Reporter gene activation | No effect [45] |
| CDCA | CHO cells/HEK293 | Intracellular cAMP | 4.43 µM [34]/ 4 µM [10] | HepG2 cells /CV-1 cells | Reporter gene activation | 10 µM [9]/ 50 µM [45] |
| CHO cells | Reporter gene activation | 6.71 µM [46] | Cell-free | Ligand-sensing assay | 4.5 µM [47] | |
| Conjugated Primary Bile Acids | ||||||
| TCA/GCA | CHO cells | Reporter gene activation | 4.95 µM/ 13.6 µM [46] | Cell-free | Ligand-sensing assay | No effect [47] |
| TCDCA/ GCDCA | CHO cells | Reporter gene activation | 1.92 µM/ 3.88 µM [46] | Cell-free | Ligand-sensing assay | 10 µM [47] |
| HCA | Cell-free | TR-FRET FXR coactivator assay | 70.06 µM (IC50) [28] | |||
| Secondary bile acids | ||||||
| DCA | CHO cells | Intracellular cAMP | 1.01 µM [34] | HepG2 cells | Reporter gene activation | 100 µM [9] |
| HEK293 | Intracellular cAMP | 575 nM [10] | CV-1 cells | Reporter gene activation | 50 µM [45] | |
| LCA | CHO cells | Intracellular cAMP | 0.53 µM [34] | CV-1 cells | Reporter gene activation | 50 µM [45] |
| HEK293 | Intracellular cAMP | 35 nM [10] | Cell-free | Ligand-sensing assay | 25 µM [6] | |
| UDCA | CHO cells | Reporter gene activation/Intracellular cAMP | 36.4 µM [46]/ No effect [34] | CV-1 cells | Reporter gene activation | No effect [45] |
| HDCA | CHO cells | Reporter gene activation | 31.6 µM [46] | Cell-free | TR-FRET FXR coactivator assay | 62.43 µM [28] (IC50) |
| Conjugated Secondary Bile Acids | ||||||
| TDCA/ GDCA | CHO cells | Reporter gene activation | 0.79 µM /1.18 µM [46] | Cell-free | Ligand-sensing assay | 500 µM [47] (IC50) |
| TLCA/ GLCA | CHO cells | Reporter gene activation | 0.29 µM /0.54 µM [46] | Cell-free | Ligand-sensing assay | 3.8 µM/4.7 µM [47] (IC50) |
| TUDCA/ GUDCA | CHO cells | Reporter gene activation | 30.0 µM /33.9 µM [46] | Cell-free | Ligand-sensing assay | No effect [47] |
| THDCA/GHDCA | CHO cells | Reporter gene activation | 24.2 µM/36.7 µM [46] | |||
| Bile Acid | Model | Dose | Method | Effect | Ref | |
|---|---|---|---|---|---|---|
| Conjugated Bile Acid | ||||||
| Primary | TCA | HFD Sprague-Dawley rat + streptozotocin | 0.05% or 0.3% | Fed with high-fat diet for 12 weeks | Body weight − Energy intake − | [54] |
| Patients with T2DM | 0.66, 2, 6.66, or 20 mmol | Rectal administration | Energy intake ↓ (~47% at 20 mmol) | [41] | ||
| HCA | db/db mice; HFD C57BL/6J mice + streptozotocin; C57BL/6J mice | 100 mg/kg/day | Oral gavage for 28 days | Body weight − Energy intake − | [28] | |
| TUDCA | db/db mice; HFD C57BL/6J mice + streptozotocin; C57BL/6J mice | 100 mg/kg/day | Oral gavage for 28 days | Body weight − Energy intake − | [28] | |
| Secondary | HDCA | db/db mice; HFD C57BL/6J mice + streptozotocin; C57BL/6J mice | 100 mg/kg/day | Oral gavage for 28 days | Body weight − Energy intake − | [28] |
| Unconjugated Bile Acid | ||||||
| Primary | CA | C57BL/6J mice | 0.5% | High-fat diet fed for 47 days | Body weight ↓ (24%) Energy expenditure ↑ (~50%) Energy intake − | [50] |
| 0.5% | High-fat diet fed for 9 weeks | Body weight ↓ (6g, ~18%) Energy expenditure ↑ (29%) Energy intake ↑ (20%) | [52] | |||
| UDCA | 0.5% | High-fat diet fed for 8 weeks | Body weight ↓ (15%) | [51] | ||
| Secondary | DCA | C57BL/6J mice | 0.1% | High-fat diet fed for 3 weeks | Body weight − Energy intake − | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, C.; Huang, W.; Young, R.L.; Jones, K.L.; Horowitz, M.; Rayner, C.K.; Wu, T. Role of Bile Acids in the Regulation of Food Intake, and Their Dysregulation in Metabolic Disease. Nutrients 2021, 13, 1104. https://doi.org/10.3390/nu13041104
Xie C, Huang W, Young RL, Jones KL, Horowitz M, Rayner CK, Wu T. Role of Bile Acids in the Regulation of Food Intake, and Their Dysregulation in Metabolic Disease. Nutrients. 2021; 13(4):1104. https://doi.org/10.3390/nu13041104
Chicago/Turabian StyleXie, Cong, Weikun Huang, Richard L. Young, Karen L. Jones, Michael Horowitz, Christopher K. Rayner, and Tongzhi Wu. 2021. "Role of Bile Acids in the Regulation of Food Intake, and Their Dysregulation in Metabolic Disease" Nutrients 13, no. 4: 1104. https://doi.org/10.3390/nu13041104