L-Ferritin: One Gene, Five Diseases; from Hereditary Hyperferritinemia to Hypoferritinemia—Report of New Cases

 , ,
, ,
Abstract
:1. Introduction
2. Results
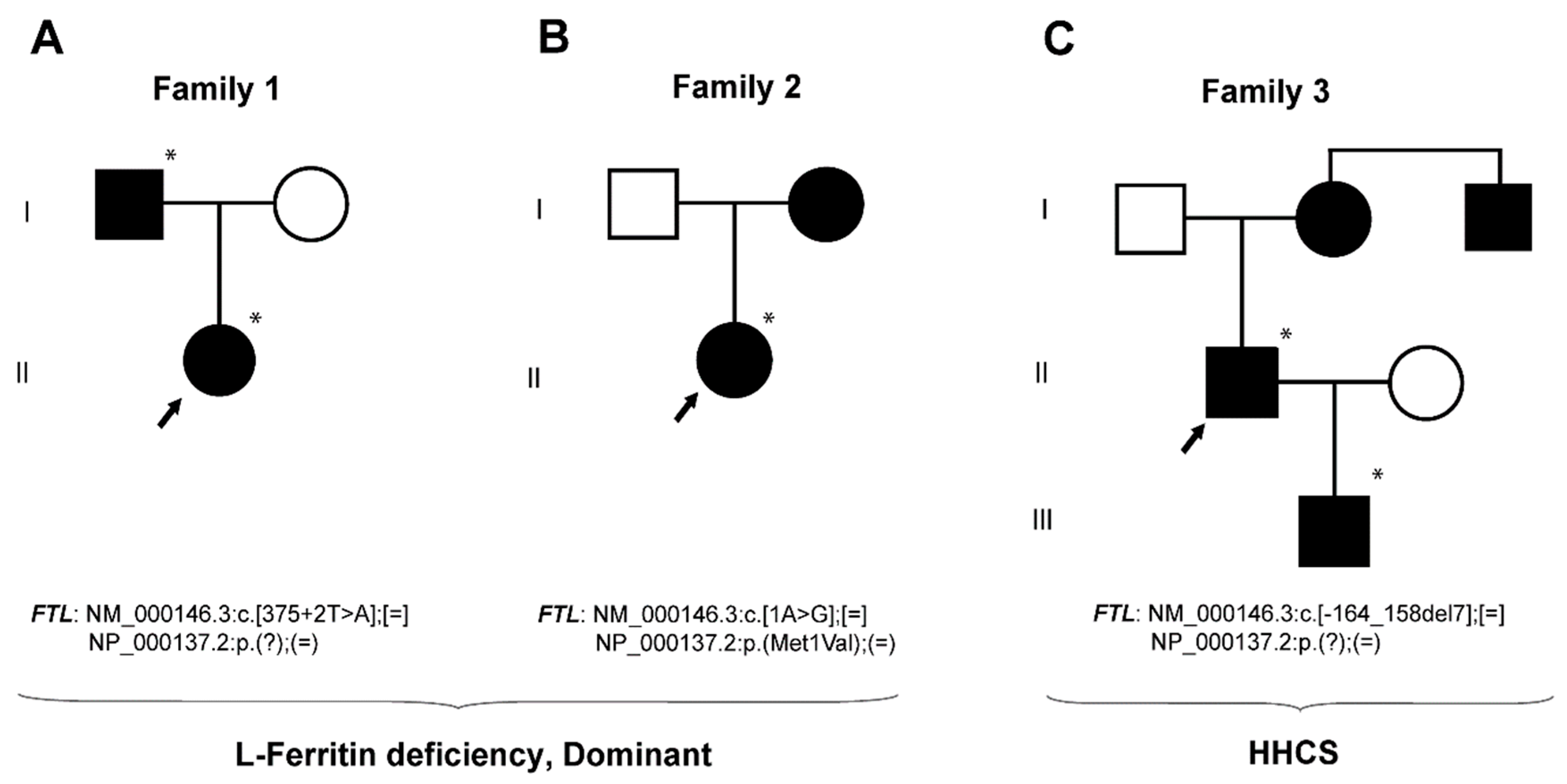
2.1. Case Studies
2.1.1. Family 1—A Case with Autosomal Dominant L-Ferritin Deficiency
2.1.2. Family 2—A Case with Autosomal Dominant L-Ferritin Deficiency
2.1.3. Family 3—A Case with HHCS
2.2. Update on L-Ferritin Mutations and Diseases
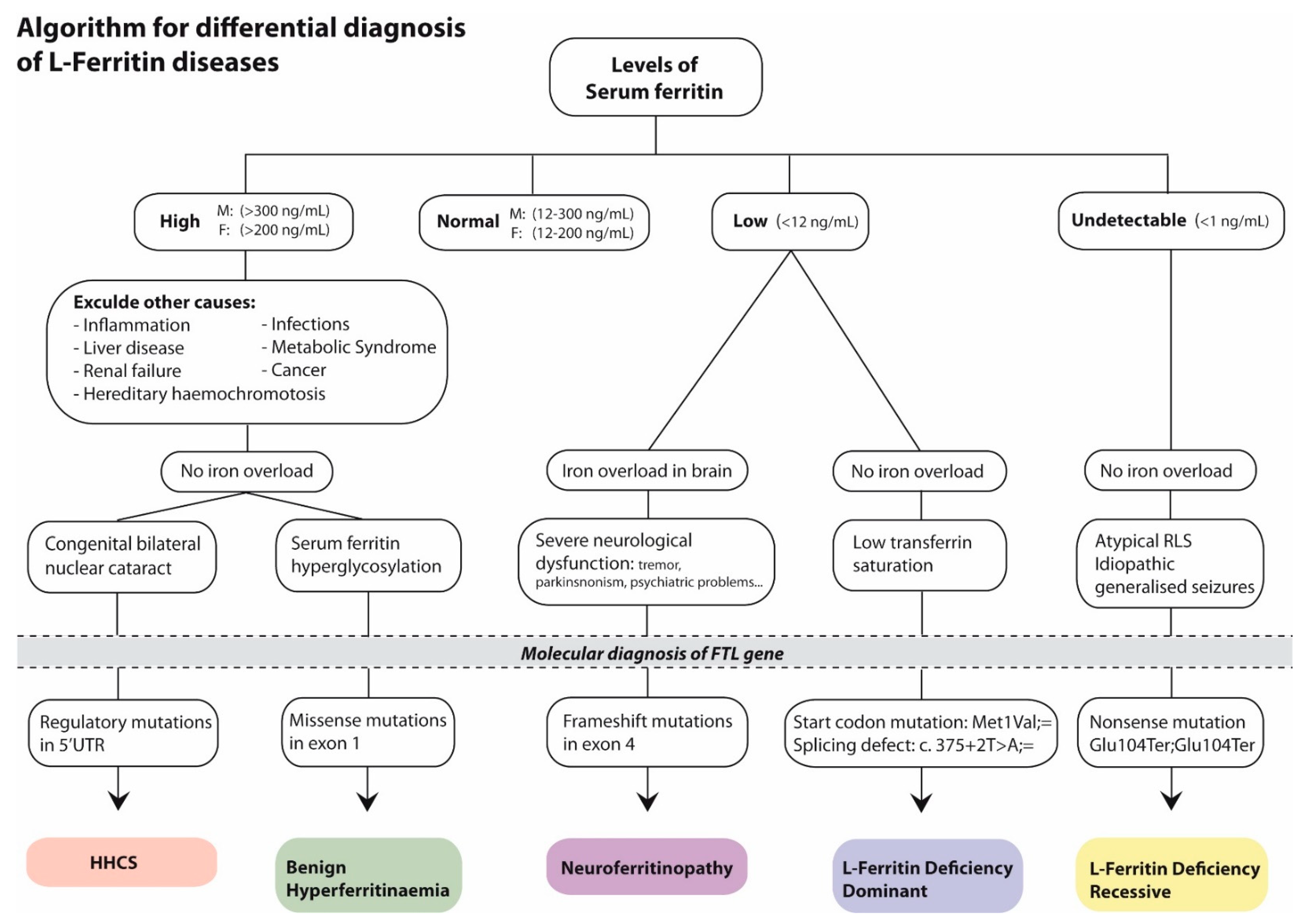
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA Extraction, PCR Amplification, and DNA Sequencing
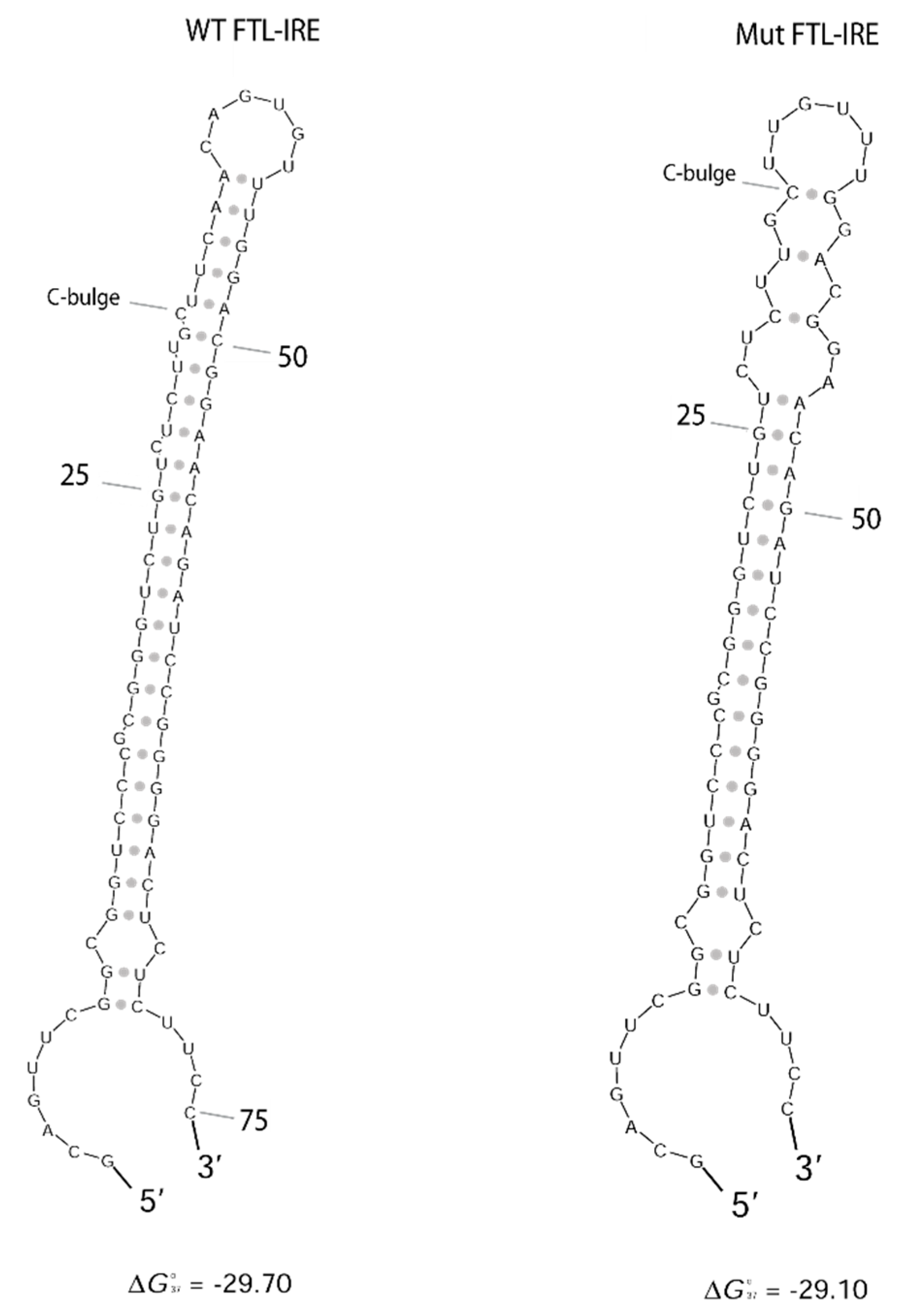
4.3. FTL RNA Fold Predictions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Yokota, M.; Drysdale, J.W. Characterization of serum ferritin in iron overload: Possible identity to natural apoferritin. Br. J. Haematol. 1977, 36, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Fardet, L.; Coppo, P.; Kettaneh, A.; Dehoux, M.; Cabane, J.; Lambotte, O. Low glycosylated ferritin, a good marker for the diagnosis of hemophagocytic syndrome. Arthritis Rheum. 2008, 58, 1521–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [Google Scholar] [CrossRef]
- Santambrogio, P.; Cozzi, A.; Levi, S.; Arosio, P. Human serum ferritin G-peptide is recognized by anti-L ferritin subunit antibodies and concanavalin-A. Br. J. Haematol. 1987, 65, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.A.; Gutierrez, L.; Weiss, A.; Leichtmann-Bardoogo, Y.; Zhang, D.L.; Crooks, D.R.; Sougrat, R.; Morgenstern, A.; Galy, B.; Hentze, M.W.; et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010, 116, 1574–1584. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, C.; Leneuve, P.; Devaux, I.; Scoazec, J.Y.; Berthier, M.; Loiseau, M.N.; Grandchamp, B.; Bonneau, D. Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat. Genet. 1995, 11, 444–446. [Google Scholar] [CrossRef]
- Girelli, D.; Olivieri, O.; De Franceschi, L.; Corrocher, R.; Bergamaschi, G.; Cazzola, M. A linkage between hereditary hyperferritinaemia not related to iron overload and autosomal dominant congenital cataract. Br. J. Haematol. 1995, 90, 931–934. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Mumford, A.D.; Cree, I.A.; Arnold, J.D.; Hagan, M.C.; Rixon, K.C.; Harding, J.J. The lens in hereditary hyperferritinaemia cataract syndrome contains crystalline deposits of L-ferritin. Br. J. Ophthalmol. 2000, 84, 697–700. [Google Scholar] [CrossRef] [Green Version]
- Luscieti, S.; Tolle, G.; Aranda, J.; Campos, C.B.; Risse, F.; Morán, É.; Muckenthaler, M.U.; Sánchez, M. Novel mutations in the ferritin-L iron-responsive element that only mildly impair IRP binding cause hereditary hyperferritinaemia cataract syndrome. Orphanet J. Rare Dis. 2013, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, A.R.; Fey, C.; Morris, C.M.; Bindoff, L.A.; Ince, P.G.; Chinnery, P.F.; Coulthard, A.; Jackson, M.J.; Jackson, A.P.; McHale, D.P.; et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat. Genet. 2001, 28, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Maciel, P.; Cruz, V.T.; Constante, M.; Iniesta, I.; Costa, M.C.; Gallati, S.; Sousa, N.; Sequeiros, J.; Coutinho, P.; Santos, M.M. Neuroferritinopathy: Missense mutation in FTL causing early-onset bilateral pallidal involvement. Neurology 2005, 65, 603–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhoberac, B.B.; Vidal, R. Abnormal iron homeostasis and neurodegeneration. Front. Aging Neurosci. 2013, 5, 32. [Google Scholar] [CrossRef]
- Kannengiesser, C.; Jouanolle, A.-M.; Hetet, G.; Mosser, A.; Muzeau, F.; Henry, D.; Bardou-Jacquet, E.; Mornet, M.; Brissot, P.; Deugnier, Y.; et al. A new missense mutation in the L ferritin coding sequence associated with elevated levels of glycosylated ferritin in serum and absence of iron overload. Haematologica 2009, 94, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Ravasi, G.; Pelucchi, S.; Mariani, R.; Casati, M.; Greni, F.; Arosio, C.; Pelloni, I.; Majore, S.; Santambrogio, P.; Levi, S.; et al. Unexplained isolated hyperferritinemia without iron overload. Am. J. Hematol. 2017, 92, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Cozzi, A.; Santambrogio, P.; Privitera, D.; Broccoli, V.; Rotundo, L.I.; Garavaglia, B.; Benz, R.; Altamura, S.; Goede, J.S.; Muckenthaler, M.U.; et al. Human L-ferritin deficiency is characterized by idiopathic generalized seizures and atypical restless leg syndrome. J. Exp. Med. 2013, 210, 1779–1791. [Google Scholar] [CrossRef] [Green Version]
- Cremonesi, L.; Cozzi, A.; Girelli, D.; Ferrari, F.; Fermo, I.; Foglieni, B.; Levi, S.; Bozzini, C.; Camparini, M.; Ferrari, M.; et al. Case report: A subject with a mutation in the ATG start codon of L-ferritin has no haematological or neurological symptoms. J. Med. Genet. 2004, 41, e81. [Google Scholar] [CrossRef] [PubMed]
- Campillos, M.; Cases, I.; Hentze, M.W.; Sanchez, M. SIREs: Searching for iron-responsive elements. Nucleic Acids Res. 2010, 38, W360–W367. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chan, C.Y.; Lawrence, C.E. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004, 32, W135–W141. [Google Scholar] [CrossRef]
- Alvarez-Coca-Gonzalez, J.; Moreno-Carralero, M.I.; Martinez-Perez, J.; Mendez, M.; Moran-Jimenez, M.J. The hereditary hyperferritinemia-cataract syndrome: A family study. Eur. J. Pediatr. 2010, 169, 1553–1555. [Google Scholar] [CrossRef] [PubMed]
- Van de Sompele, S.; Pecheux, L.; Couso, J.; Meunier, A.; Sanchez, M.; De Baere, E. Functional characterization of a novel non-coding mutation “Ghent + 49A > G” in the iron-responsive element of L-ferritin causing hereditary hyperferritinaemia-cataract syndrome. Sci. Rep. 2017, 7, 18025. [Google Scholar] [CrossRef] [PubMed]
- Kubota, A.; Hida, A.; Ichikawa, Y.; Momose, Y.; Goto, J.; Igeta, Y.; Hashida, H.; Yoshida, K.; Ikeda, S.-I.; Kanazawa, I.; et al. A novel ferritin light chain gene mutation in a Japanese family with neuroferritinopathy: Description of clinical features and implications for genotype-phenotype correlations. Mov. Disord. 2009, 24, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Baraibar, M.A.; Muhoberac, B.B.; Garringer, H.J.; Hurley, T.D.; Vidal, R. Unraveling of the E-helices and Disruption of 4-Fold Pores Are Associated with Iron Mishandling in a Mutant Ferritin Causing Neurodegeneration. J. Biol. Chem. 2010, 285, 1950–1956. [Google Scholar] [CrossRef] [PubMed]
- Bhuva, M.; Sen, S.; Elsey, T.; Atoyebi, W.; Dreau, H.; Bradbury, C.; Johnston, R.; Bignell, P.; Griffiths, W. Sequence analysis of exon 1 of the ferritin light chain (FTL) gene can reveal the rare disorder “hereditary hyperferritinaemia without cataracts”. Br. J. Haematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Thurlow, V.; Vadher, B.; Bomford, A.; DeLord, C.; Kannengiesser, C.; Beaumont, C.; Grandchamp, B. Two novel mutations in the L ferritin coding sequence associated with benign hyperferritinaemia unmasked by glycosylated ferritin assay. Ann. Clin. Biochem. 2012, 49, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Faniello, M.C.; Di Sanzo, M.; Quaresima, B.; Nisticò, A.; Fregola, A.; Grosso, M.; Cuda, G.; Costanzo, F. Bilateral cataract in a subject carrying a C to A transition in the L ferritin promoter region. Clin. Biochem. 2009, 42, 911–914. [Google Scholar] [CrossRef]
- Allerson, C.R.; Cazzola, M.; Rouault, T.A. Clinical severity and thermodynamic effects of iron-responsive element mutations in hereditary hyperferritinemia-cataract syndrome. J. Biol. Chem. 1999, 274, 26439–26447. [Google Scholar] [CrossRef]
- Baraibar, M.A.; Barbeito, A.G.; Muhoberac, B.B.; Vidal, R. Iron-mediated Aggregation and a Localized Structural Change Characterize Ferritin from a Mutant Light Chain Polypeptide That Causes Neurodegeneration. J. Biol. Chem. 2008, 283, 31679–31689. [Google Scholar] [CrossRef] [Green Version]
- Vidal, R.; Ghetti, B.; Takao, M.; Brefel-Courbon, C.; Uro-Coste, E.; Glazier, B.S.; Siani, V.; Benson, M.D.; Calvas, P.; Miravalle, L.; et al. Intracellular ferritin accumulation in neural and extraneural tissue characterizes a neurodegenerative disease associated with a mutation in the ferritin light polypeptide gene. J. Neuropathol. Exp. Neurol. 2004, 63, 363–380. [Google Scholar] [CrossRef]
- Garringer, H.J.; Irimia, J.M.; Li, W.; Goodwin, C.B.; Richine, B.; Acton, A.; Chan, R.J.; Peacock, M.; Muhoberac, B.B.; Ghetti, B.; et al. Effect of Systemic Iron Overload and a Chelation Therapy in a Mouse Model of the Neurodegenerative Disease Hereditary Ferritinopathy. PLoS ONE 2016, 11, e0161341. [Google Scholar] [CrossRef]
- Vidal, R.; Miravalle, L.; Gao, X.; Barbeito, A.G.; Baraibar, M.A.; Hekmatyar, S.K.; Widel, M.; Bansal, N.; Delisle, M.B.; Ghetti, B. Expression of a Mutant Form of the Ferritin Light Chain Gene Induces Neurodegeneration and Iron Overload in Transgenic Mice. J. Neurosci. 2008, 28, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Muhoberac, B.B.; Baraibar, M.A.; Vidal, R. Iron Loading-Induced Aggregation and Reduction of Iron Incorporation in Heteropolymeric Ferritin Containing a Mutant Light Chain that Causes Neurodegeneration. Biochim. Biophys. Acta 2011, 1812, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Luscieti, S.; Santambrogio, P.; Langlois d’Estaintot, B.; Granier, T.; Cozzi, A.; Poli, M.; Gallois, B.; Finazzi, D.; Cattaneo, A.; Levi, S.; et al. Mutant ferritin L-chains that cause neurodegeneration act in a dominant-negative manner to reduce ferritin iron incorporation. J. Biol. Chem. 2010, 285, 11948–11957. [Google Scholar] [CrossRef]
- Baraibar, M.A.; Barbeito, A.G.; Muhoberac, B.B.; Vidal, R. A mutant light chain ferritin that causes neurodegeneration has enhanced propensity toward oxidative damage. Free Radic. Biol. Med. 2012, 52, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Altes, A.; Perez-Lucena, M.J.; Bruguera, M. [Systematic approach to the diagnosis of hyperferritinemia]. Med. Clin. (Barc.) 2014, 142, 412–417. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Family 1 | Family 2 | Family 3 | Reference Values |
|---|---|---|---|---|
| Patient | II.1 | II.1 | II.1 | - |
| Gender | F | F | M | - |
| Age at diagnosis (years) | 4 | 2 | 67 | - |
| Hb (g/dL) | 12.2–13.3 | 13.1–13.7 | 14.0 | 13.5–17.5 (M). 12.1–15.1 (F) |
| MCV (fL) | 78–84 | 80 | 90.2 | 80–95 |
| Ferritin (ng/mL) | 4–9 | 2–7 | 3037 | 12–300 (M), 12–200 (F) |
| Transferrin sat (%) | 12.9 | 17.2–26.2 | 22.0–41.0 | 25–50 |
| Iron (µL/dL) | n/a | 61.95 | 46 | 49–226 |
| Mutation | c.375 + 2T > A | p.Met1Val | c.-164_158del7 | - |
| Novel | Previously reported [18] | Novel | - | |
| Disease | L-ferritin deficiency | L-ferritin deficiency | HHCS | - |
| Inheritance | AD | AD | AD | - |
| Disease | Hereditary Hyperferritinemia Cataract Syndrome | Benign Hyperferritinemia | Neurodegeneration with Brain Iron Accumulation 3 | L-Ferritin Deficiency, Dominant | L-Ferritin Deficiency, Recessive |
|---|---|---|---|---|---|
| First publication | [7,8] | [15] | [12] | [18] | [17] |
| Inheritance | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal recessive |
| Mechanism | LOST OF IRP REGULATION | (DO NOT PROCEED) | DOMINANT NEGATIVE EFFECT | HAPLOINSUFICIENCY | TOTAL LOSS OF FTL |
| Mutation/s | Many in the 5′ IRE | Missense in exon 1 | Frameshift in exon 4 | p.(M1V; =) | p.(E104X; E104X) |
| Type | 5′ UTR | Affects the A α-helix near the N-terminus | Predicted to cause loss of the C-terminal secondary structure | Start loss | Nonsense |
| Hematological features | High serum ferritin Normal serum iron Normal transferrin saturation Normal red cell counts Normal hematologic parameters | High serum ferritin Serum ferritin hyperglycosylation Normal hematologic parameters | Low serum ferritin | Low serum ferritin Low transferrin saturation (17%) Normal serum iron Normal hematologic parameters | Undetectable serum ferritin Normal Transferrin saturation Normal hematologic parameters |
| Other features | Congenital bilateral nuclear cataract | Severe neurological dysfunction Gastrointestinal dysphagia | Idiopathic generalized seizures Atypical RLS Progressive hair loss |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cadenas, B.; Fita-Torró, J.; Bermúdez-Cortés, M.; Hernandez-Rodriguez, I.; Fuster, J.L.; Llinares, M.E.; Galera, A.M.; Romero, J.L.; Pérez-Montero, S.; Tornador, C.; et al. L-Ferritin: One Gene, Five Diseases; from Hereditary Hyperferritinemia to Hypoferritinemia—Report of New Cases. Pharmaceuticals 2019, 12, 17. https://doi.org/10.3390/ph12010017
Cadenas B, Fita-Torró J, Bermúdez-Cortés M, Hernandez-Rodriguez I, Fuster JL, Llinares ME, Galera AM, Romero JL, Pérez-Montero S, Tornador C, et al. L-Ferritin: One Gene, Five Diseases; from Hereditary Hyperferritinemia to Hypoferritinemia—Report of New Cases. Pharmaceuticals. 2019; 12(1):17. https://doi.org/10.3390/ph12010017
Chicago/Turabian StyleCadenas, Beatriz, Josep Fita-Torró, Mar Bermúdez-Cortés, Inés Hernandez-Rodriguez, José Luis Fuster, María Esther Llinares, Ana María Galera, Julia Lee Romero, Santiago Pérez-Montero, Cristian Tornador, and et al. 2019. "L-Ferritin: One Gene, Five Diseases; from Hereditary Hyperferritinemia to Hypoferritinemia—Report of New Cases" Pharmaceuticals 12, no. 1: 17. https://doi.org/10.3390/ph12010017