
Characterization and Evaluation of Composite Biomaterial Bioactive Glass–Polylactic Acid for Bone Tissue Engineering Applications

 ,
,  , ,
, ,
Abstract
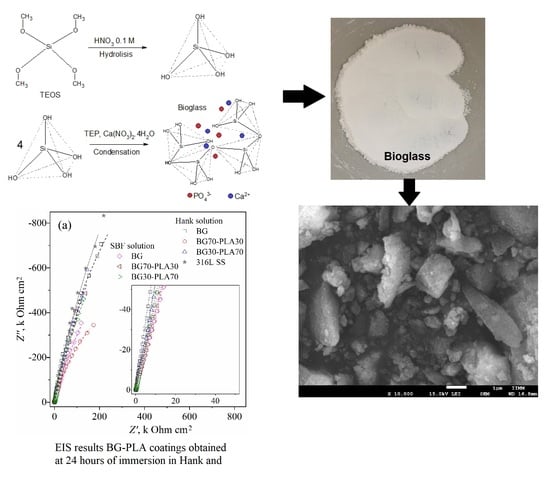
:
1. Introduction
2. Materials and Methods
2.1. PLA Synthesis
2.2. BG-PLA Composite Synthesis
2.3. SBF and PBS Preparation
2.4. BG-PLA Coatings
2.5. BG-PLA Scaffolds Design
2.6. BG-PLA Bioactivity in SBF
2.7. BG-PLA Degradation in PBS
2.8. FTIR and XRD Characterizations
2.9. Electrochemical Tests
3. Results
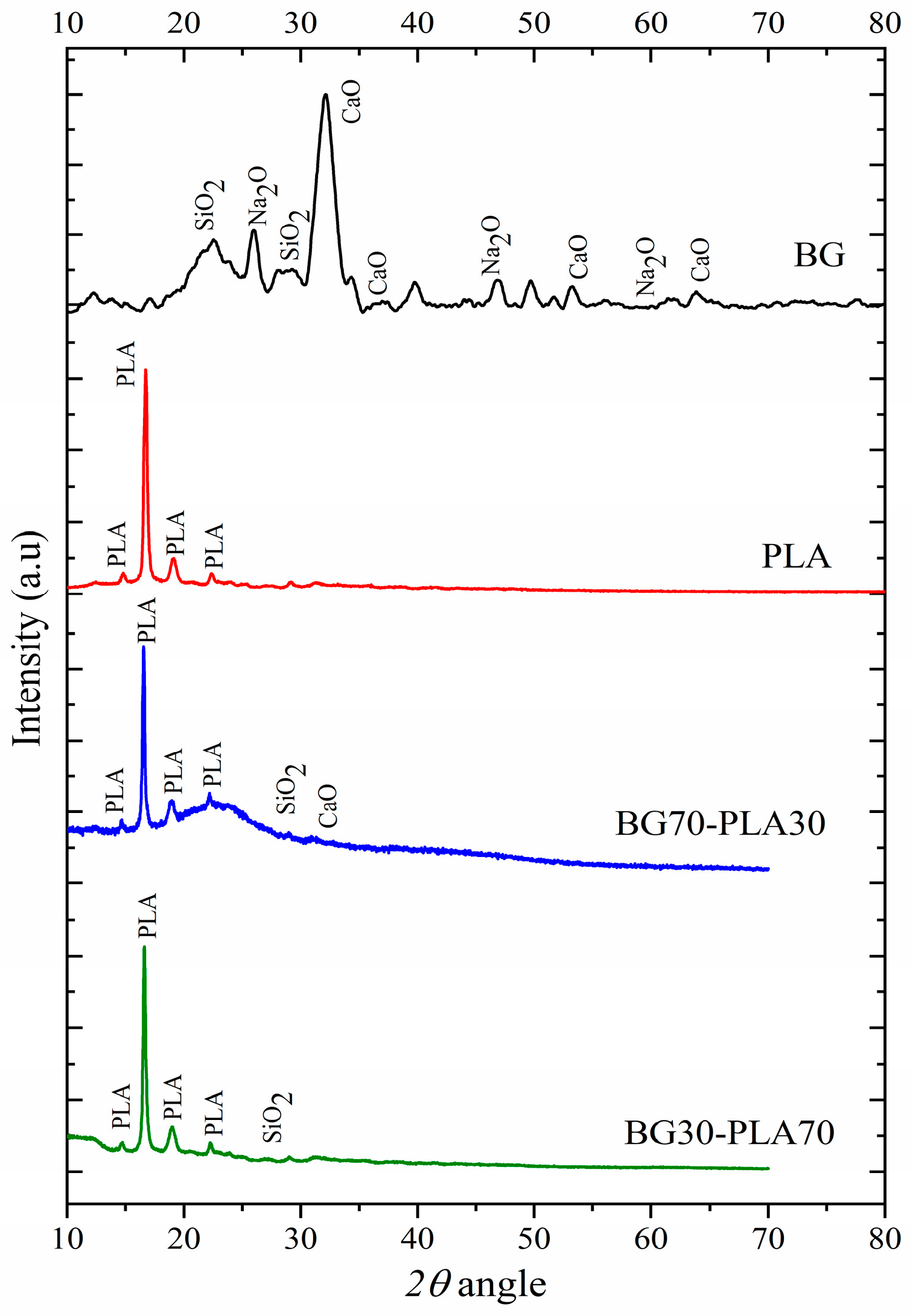
3.1. X-ray Diffraction Analysis
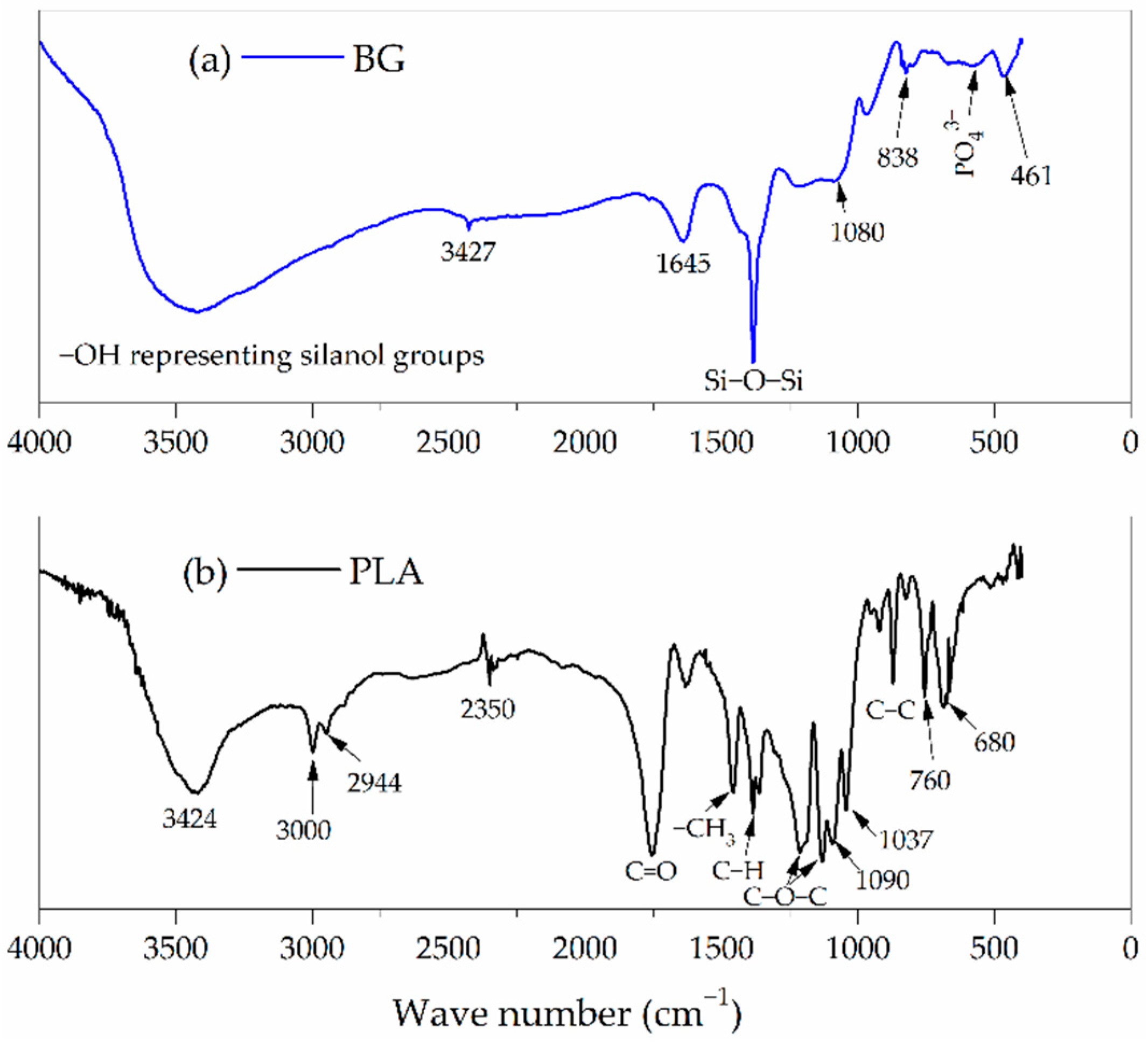
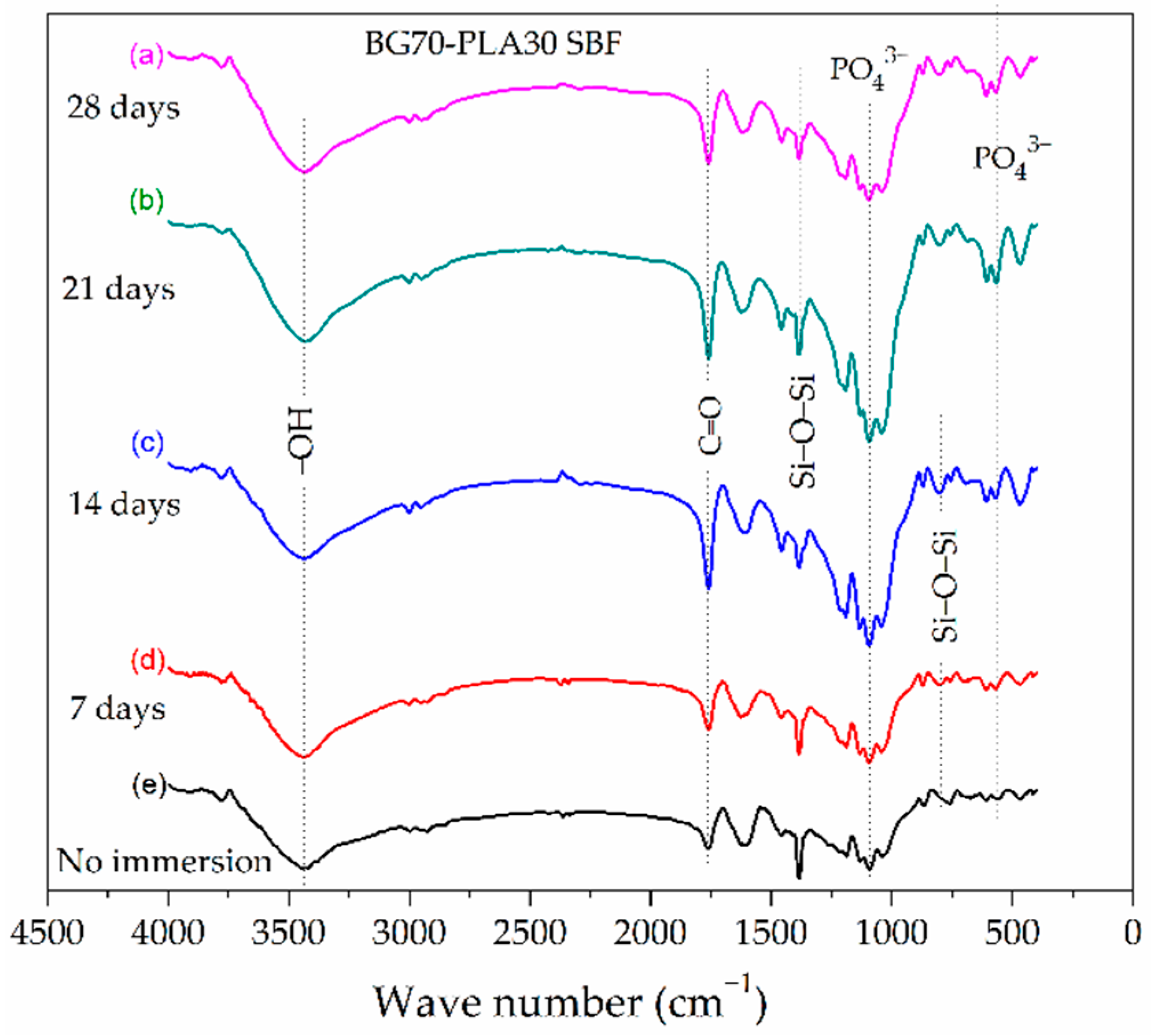
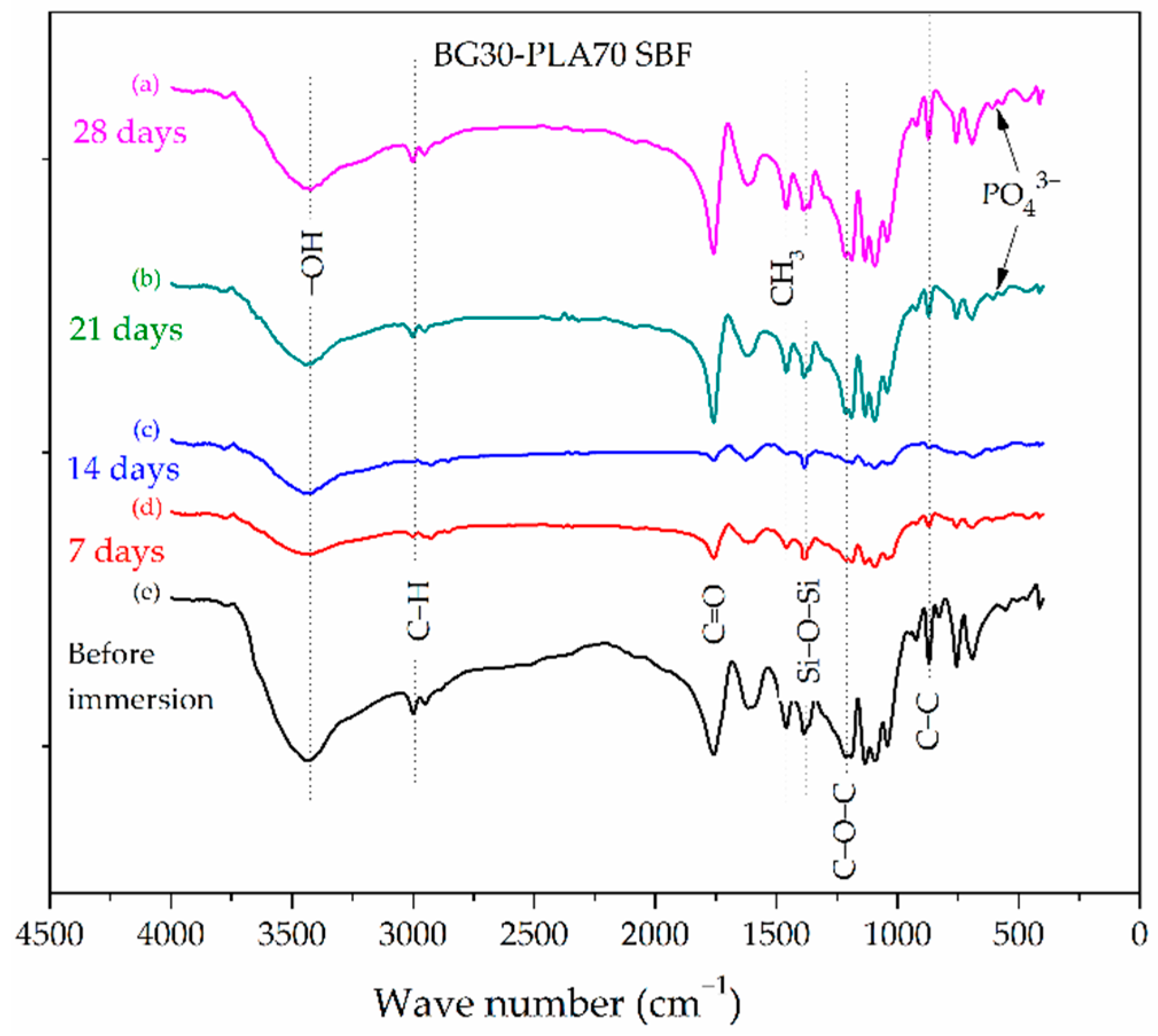
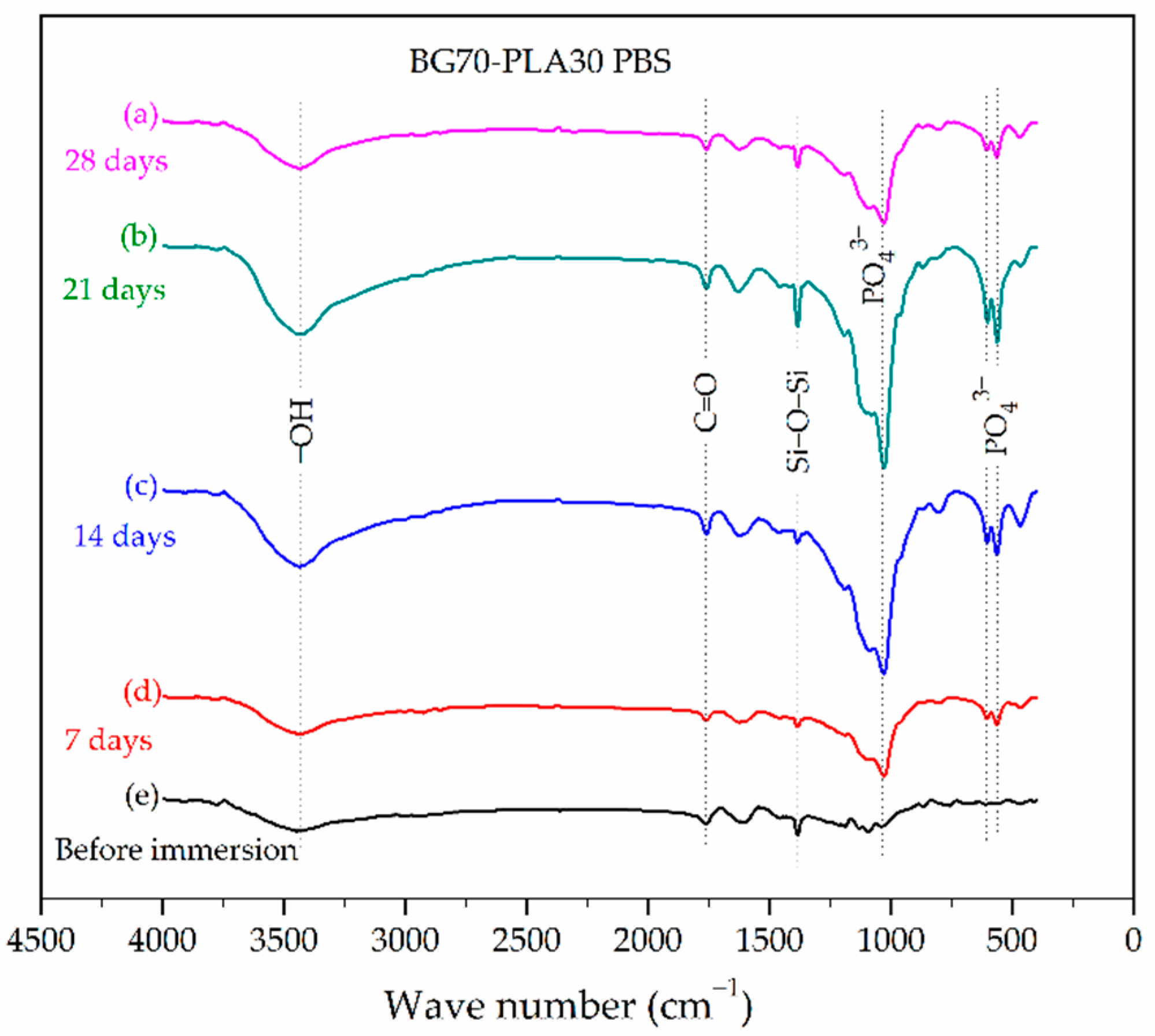
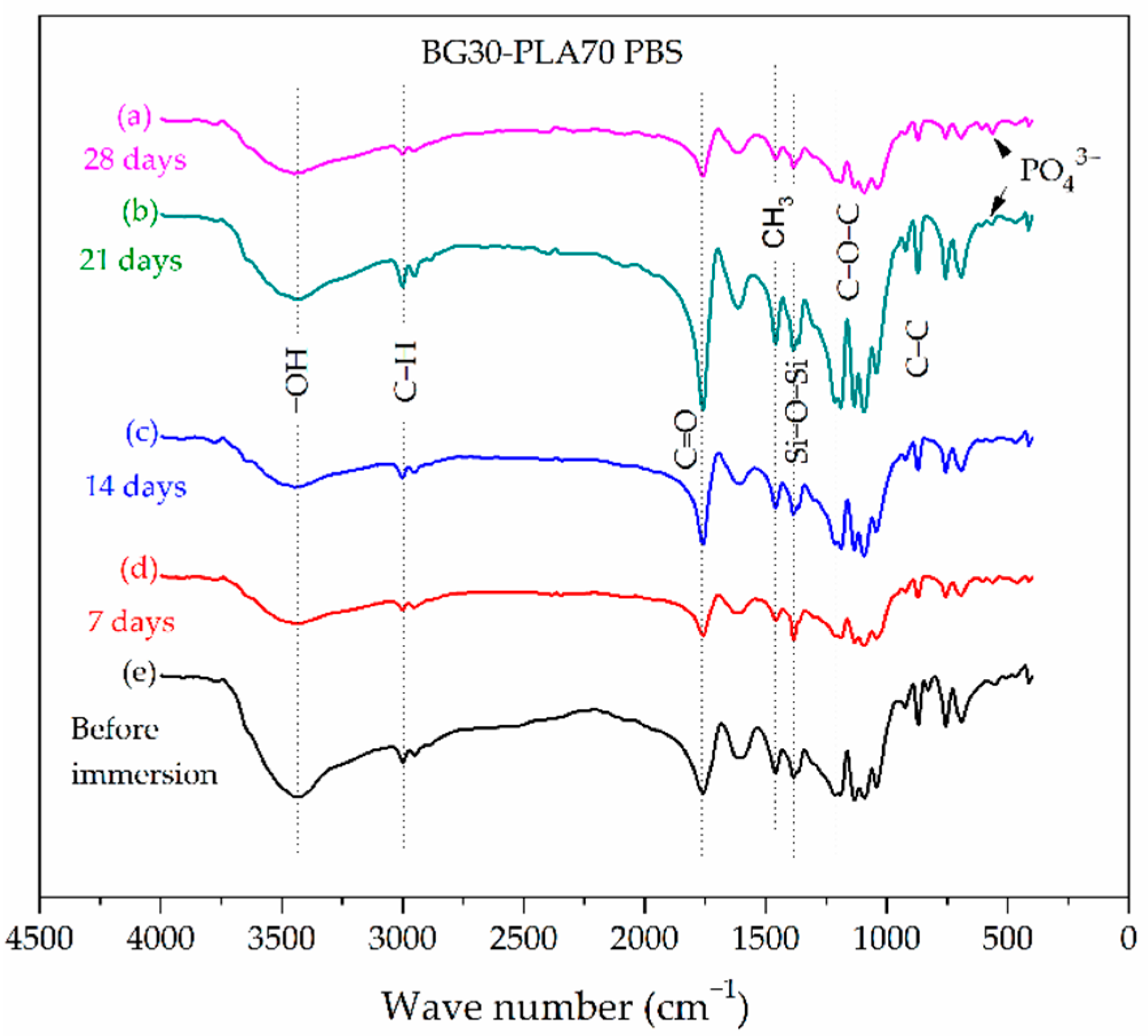
3.2. FTIR Characterization
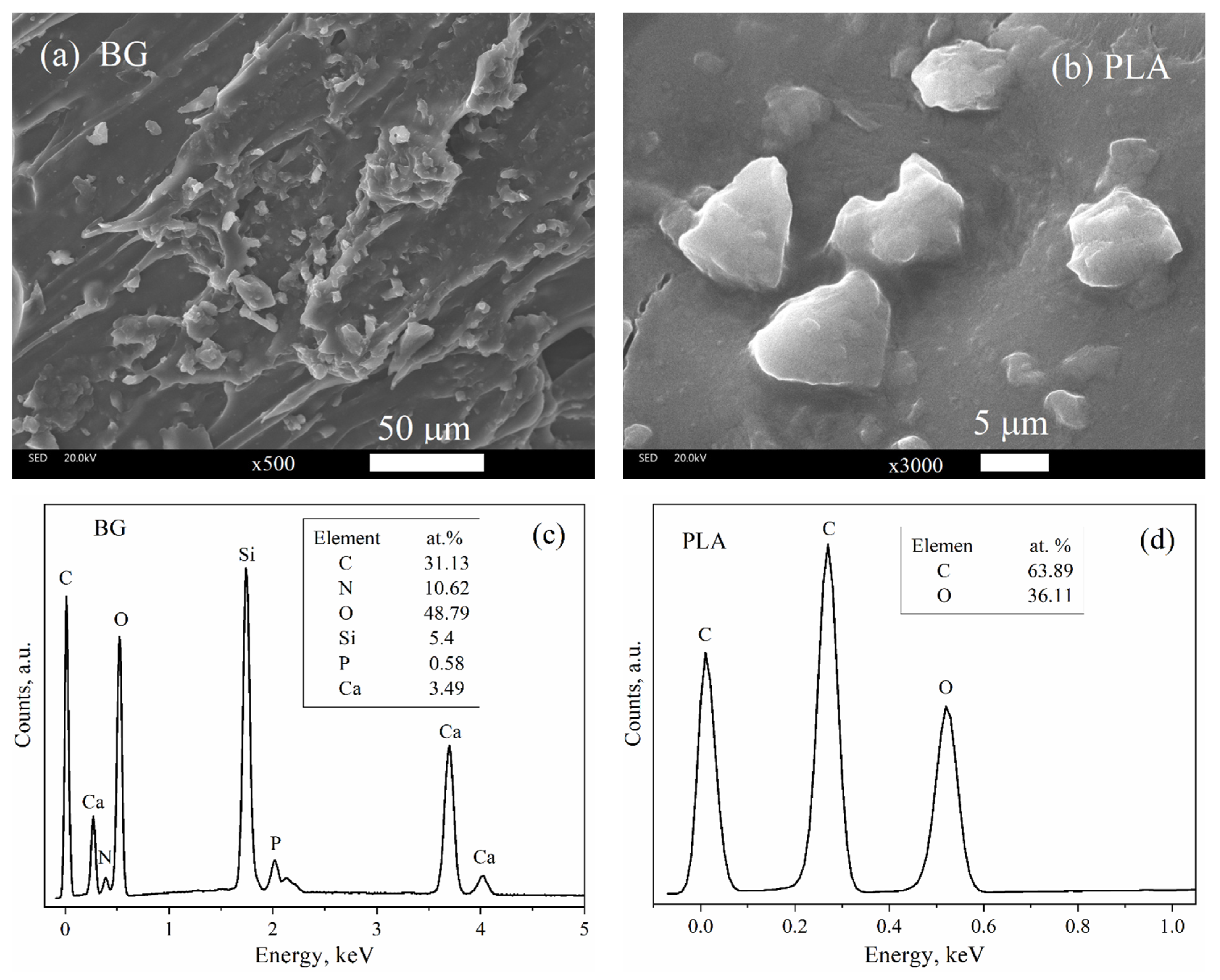
3.3. SEM Characterization
3.4. Evaluation of BG-PLA Bioactivity in SBF
3.5. Evaluation of BG-PLA Degradation in PBS
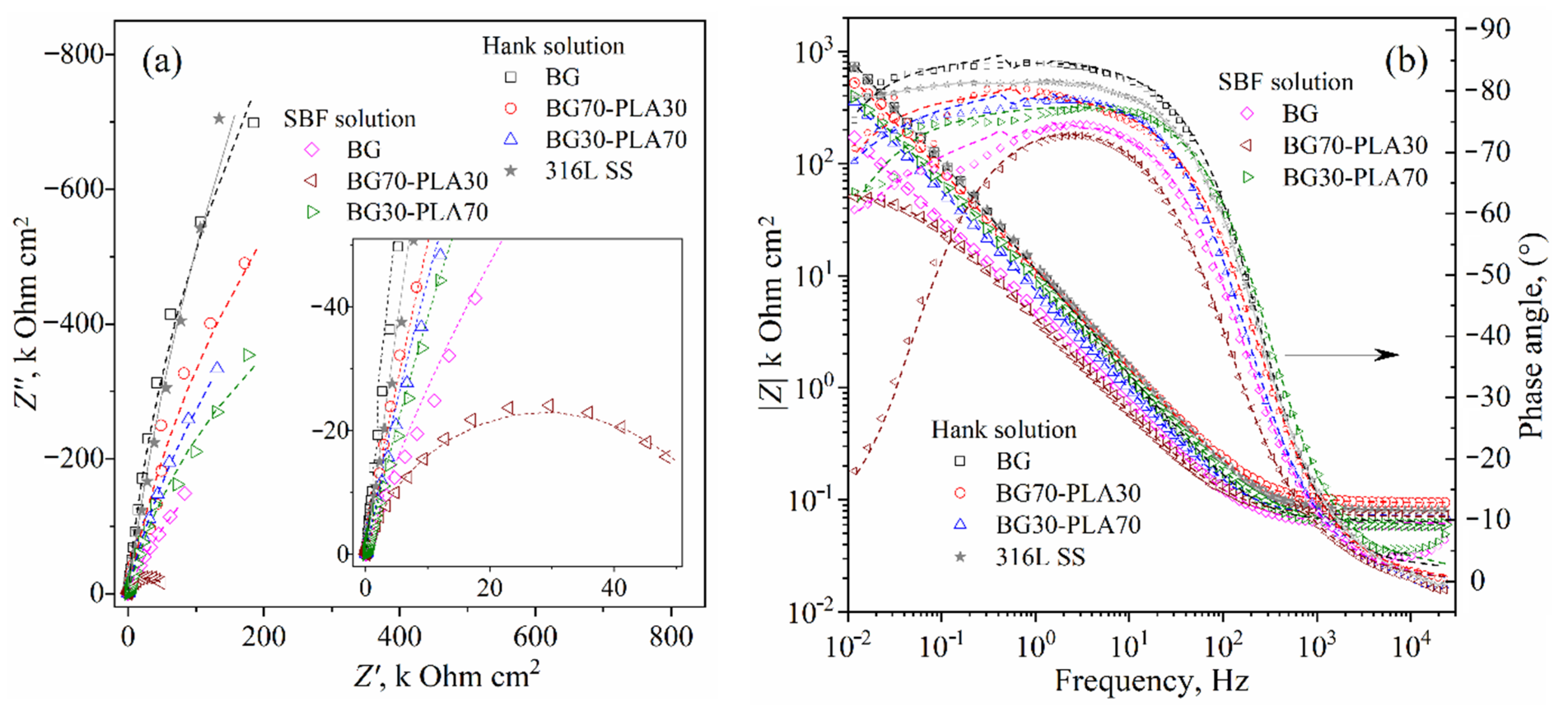
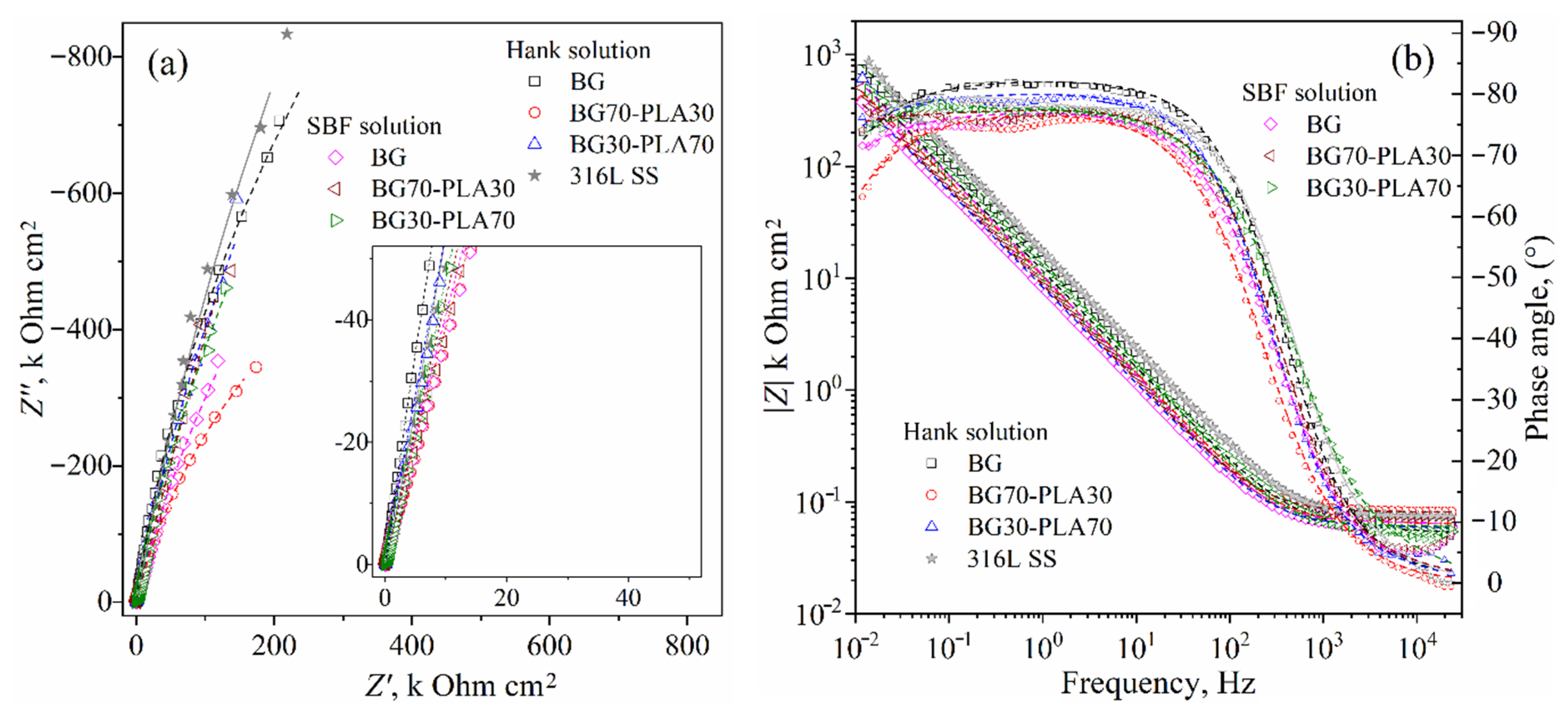
3.6. Electrochemical Evaluation
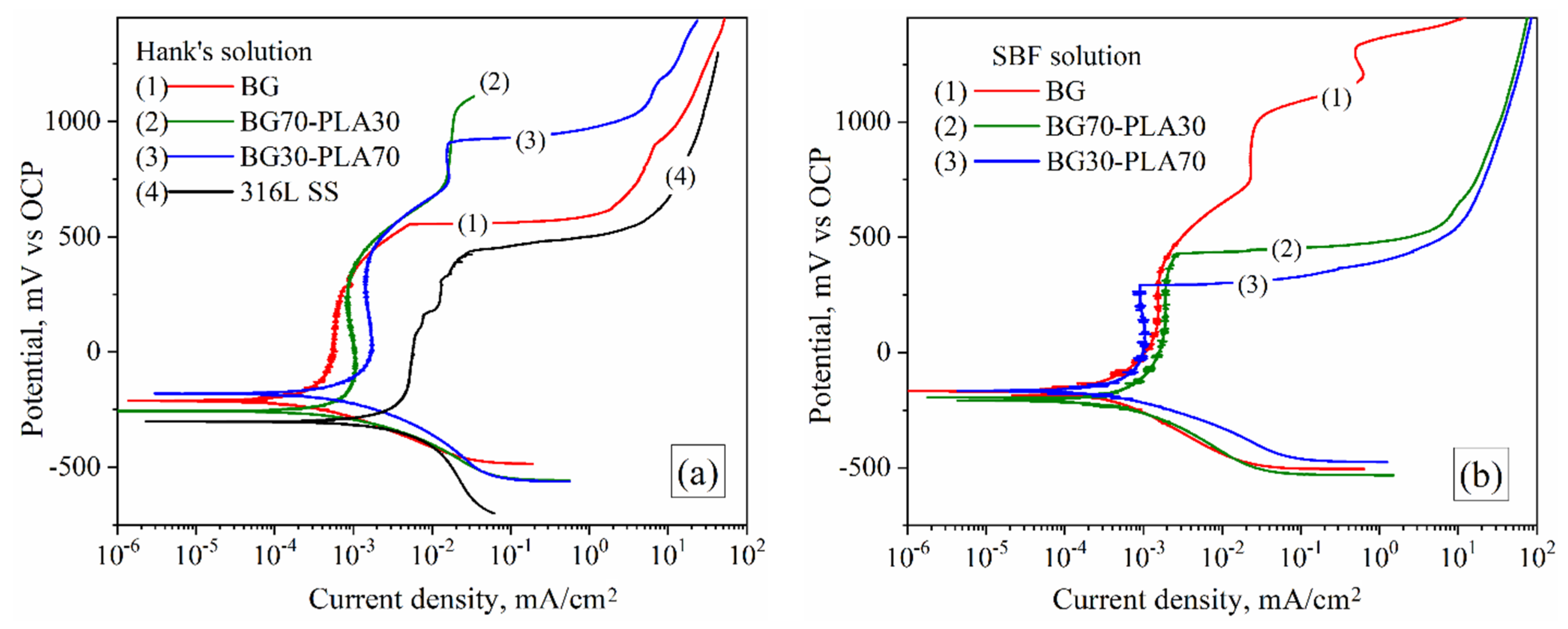
3.6.1. Potentiodynamic Tests
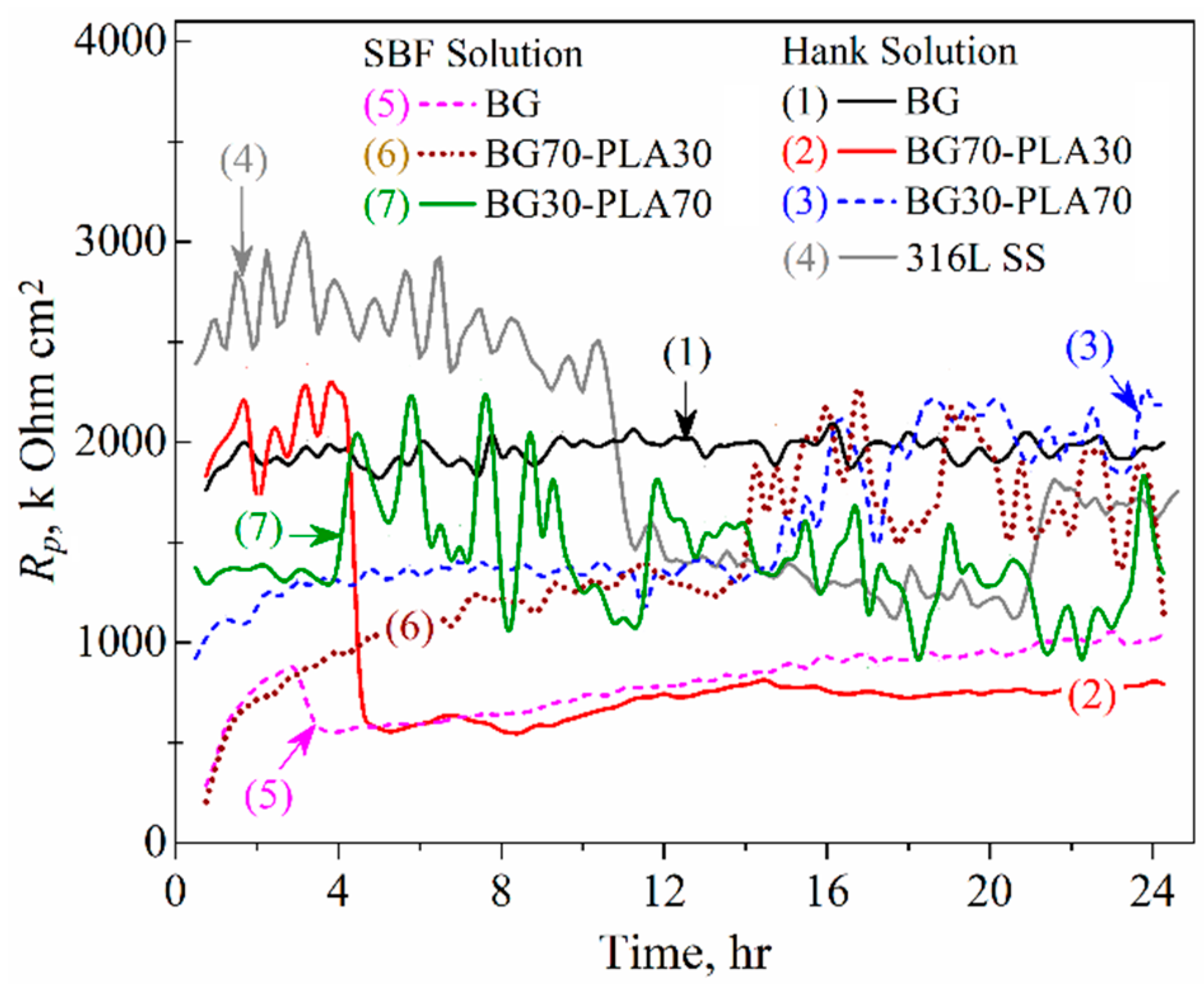
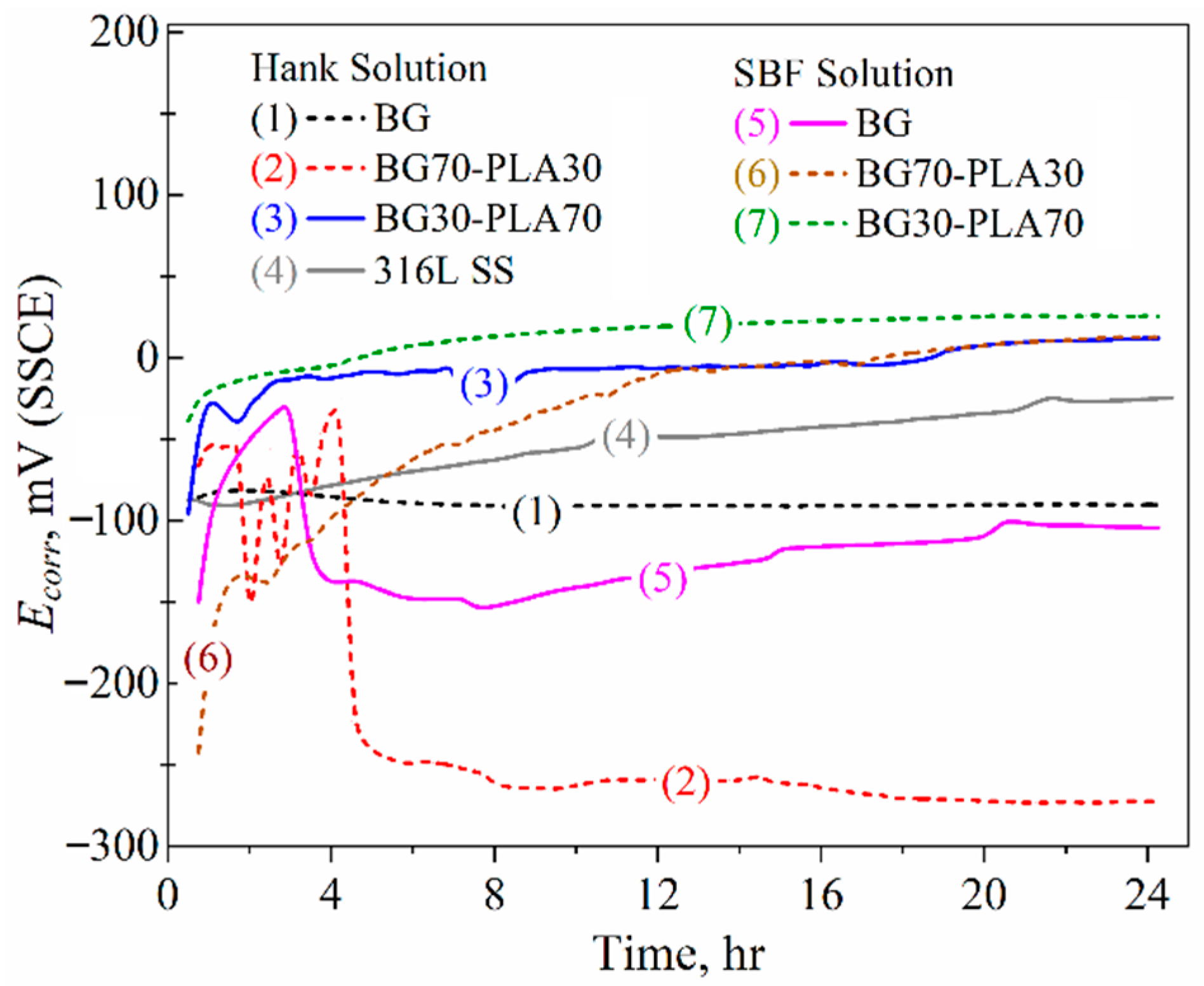
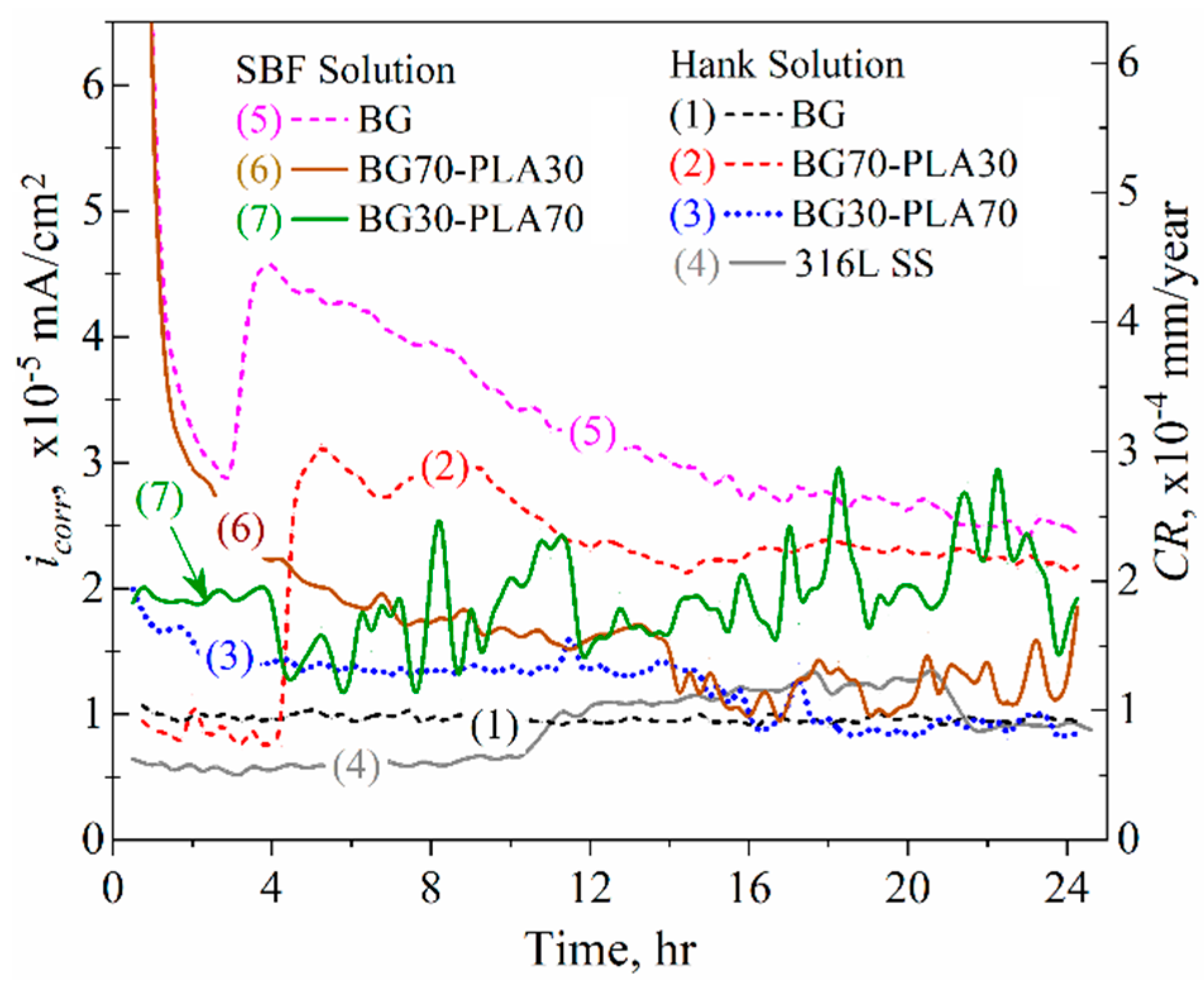
3.6.2. LPR Measurements
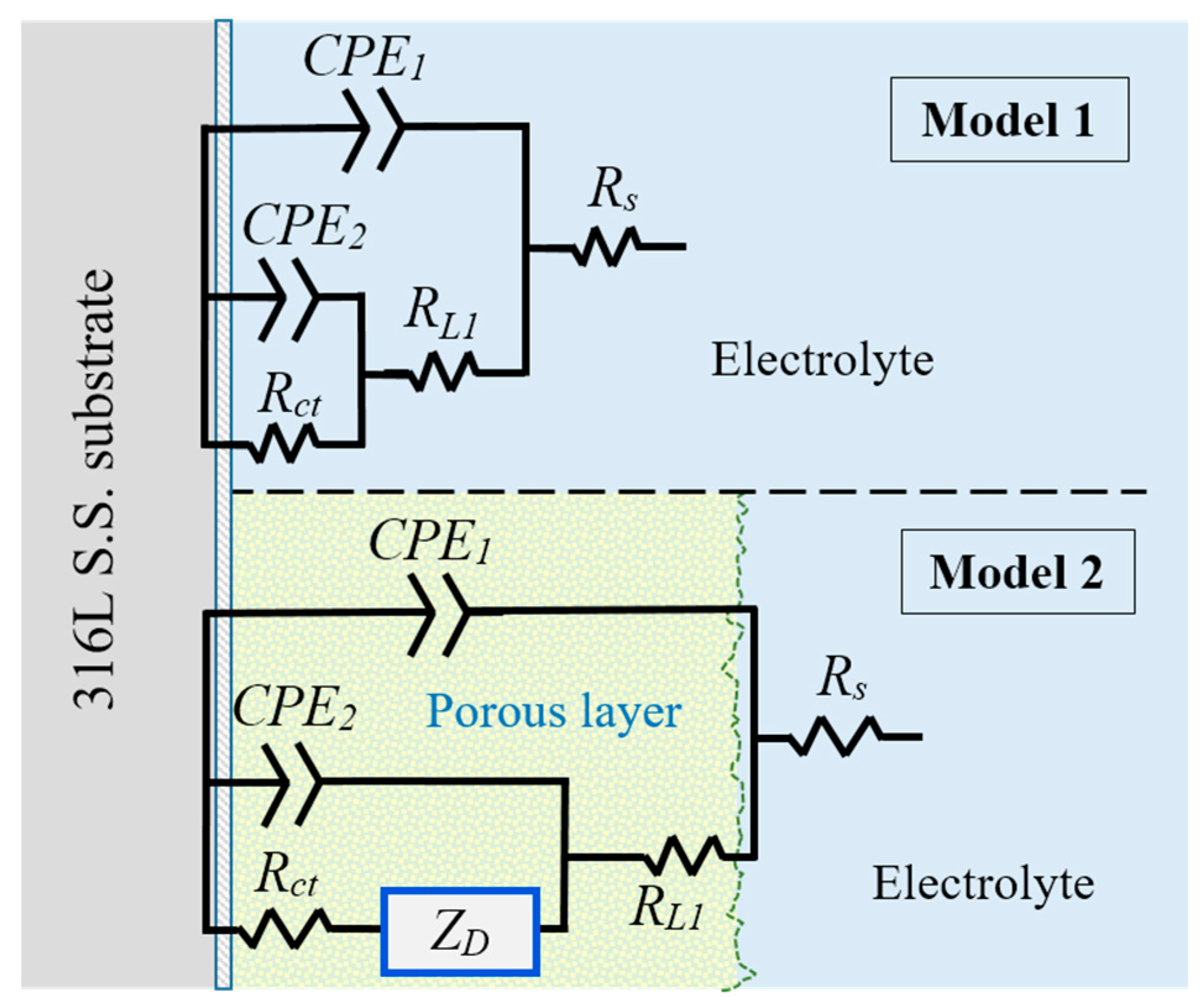
3.6.3. EIS Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernandez, G.; Keller, L. Bone substitutes: A review of their characteristics, clinical use, and perspectives for large bone defects management. J. Tissue Eng. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hasan, A.; Byambaa, B. Advances in osteobiologic materials for bone substitutes. J. Tissue Eng. Regen. Med. 2018, 12, 1448–1468. [Google Scholar] [CrossRef]
- Kashirina, A.; Yao, Y. Biopolymers for bone substitutes: A review. Biomater. Sci. 2019, 7, 3961–3983. [Google Scholar] [CrossRef]
- Hasan, M.; Kim, B. In vitro and in vivo evaluation of bioglass microspheres incorporated brushite cement for bone regeneration. Mater. Sci. Eng. C 2019, 103, 1–12. [Google Scholar] [CrossRef]
- Paiva, A.; Risso, J. Bone substitutes and photobiomodulation in bone regeneration: A systematic review in animal experimental studies. J. Biomed. Mater. Res. A 2021, 109, 1765–1775. [Google Scholar]
- Mazzoni, E.; Laquinta, M. Bioactive materials for soft tissue repair. Front. Bioeng. Biotechnol. 2021, 9, 1–17. [Google Scholar] [CrossRef]
- Wajda, A.; Sitarz, M. Structural and microstructural comparison of bioactive melt-derived and gel-derived glasses from CaO-SiO2 binary system. Ceram. Int. 2018, 44, 8727–10020. [Google Scholar] [CrossRef]
- Baino, F.; Fiume, E. Bioactive sol-gel glasses: Processing, properties, and applications. Int. J. Appl. Ceram. Technol. 2018, 15, 841–860. [Google Scholar] [CrossRef]
- Khurshid, Z.; Husain, S. Novel techniques of scaffold fabrication for bioactive glasses. In Biomedical, Therapeutic and Clinical Applications of Bioactive Glasses; Kaur, G., Ed.; Woodhead Publishing: Cambridge, UK, 2019; pp. 497–519. [Google Scholar]
- Fan-Long, J.; Rong-Rong, H. Improvement of thermal behaviour of biodegradable poly(lactic acid) polymer: A review. Compos. B Eng. 2018, 164, 287–296. [Google Scholar]
- Ramírez-Herrera, C.A.; Flores-Vela, A.I. PLA degradation pathway obtained from direct polycondensation of 2-hydroxypropanoic acid using different chain extenders. J. Mater. Sci. 2018, 53, 10846–10871. [Google Scholar] [CrossRef]
- Waizy, H.; Weizbauer, A. In vitro corrosion of ZEK100 plates in Hank’s Balanced Salt Solution. Biomed. Eng. OnLine 2012, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef]
- ISO-10993-13; Biological Evaluation of Medical Devices—Part 13: Identification and Quantification of Degradation Products from Polymeric Medical Devices. 2nd ed. ISO: Geneva, Switzerland, 2010.
- ASTM G 102; Standard Practice for Calculation of Corrosion Rates and Related Information from Electrochemical Measurements. ASTM: West Conshohocken, PA, USA, 1999.
- Cacciotti, I.; Lombardi, M. Sol-gel derived 45S5 bioglass: Synthesis, microstructural evolution and thermal behaviour. J. Mater. Sci. Mater. Med. 2012, 23, 1849–1866. [Google Scholar] [CrossRef]
- Vichery, C.; Nedelec, J.M. Bioactive glass nanoparticles: From synthesis to materials design for biomedical applications. Materials 2016, 9, 288. [Google Scholar] [CrossRef] [Green Version]
- El-Badry, H.; Moustaffa, F. Infrared absorption spectroscopy of some bio-glasses before and after immersion in various solutions. Indian J. Pure App. Phys. 2000, 38, 741–761. [Google Scholar]
- Chen, W.; Long, T. Hydrothermal synthesis of hydroxyapatite coatings with oriented nanorod arrays. RSC Adv. 2014, 4, 185–191. [Google Scholar] [CrossRef]
- Radev, L.; Vladov, D. In vitro Bioactivity of Polycaprolactone/Bioglass Composites. Int. J. Mater. Chem. 2013, 5, 91–98. [Google Scholar]
- Vaid, C.; Murugavel, S. Alkali oxide containing mesoporous bioactive glasses: Synthesis, characterization and in vitro bioactivity. Mater. Sci. Eng. C 2013, 33, 959–968. [Google Scholar] [CrossRef]
- Doganay, D.; Coskun, S. Electrical, mechanical and thermal properties of aligned silver nanowire/polylactide nanocomposite films. Compos. Part B Eng. 2016, 99, 288–296. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, L. Preparation and characterization of bioactive glass scaffolds and evaluation of bioactivity and cytotoxicity in vitro. Bioact. Mater. 2018, 3, 315–321. [Google Scholar] [CrossRef]
- El-Meliegy, E.; Mabrouk, M. Novel Fe2O3-doped glass/chitosan scaffolds for bone tissue replacement. Ceram. Int. 2018, 44, 8727–10020. [Google Scholar] [CrossRef]
- Sharifianjazi, F.; Parvin, N. Synthesis and characteristics of sol-gel bioactive SiO2-P2O5-CaO-Ag2O glasses. J. Non-Cryst. Solids. 2017, 476, 108–113. [Google Scholar] [CrossRef]
- Xia, W.; Chang, J. Well-ordered mesoporous bioactive glasses (MBG): A promising bioactive drug delivery system. J. Control. Release. 2006, 110, 522–530. [Google Scholar] [CrossRef]
- Felfel, R.; Zakir, K. Accelerated in vitro degradation properties of polylactic acid/phosphate glass fibre composites. J. Mater. Sci. 2015, 50, 3942–3955. [Google Scholar] [CrossRef]
- Zuluaga, F. Algunas aplicaciones del ácido Poli-L-Láctico. Rev. Acad. Colomb. Cienc. 2013, 37, 125–142. [Google Scholar]
- Jokar, M.; Darvishi, S.; Torkaman, R.; Kharaziha, M.; Karbasi, M. Corrosion and bioactivity evaluation of nanocomposite PCL-forsterite coating applied on 316L stainless steel. Surf. Coat. Technol. 2016, 307, 324–331. [Google Scholar] [CrossRef]
- Zurita-Mendez, N.N.; Carbajal-De la Torre, G.; Estevez, M.; Ballesteros-Almanza, M.L.; Cadenas, E.; Espinosa-Medina, M.A. Evaluation of the electrochemical behavior of TiO2/Al2O3/PCL composite coatings in Hank’s solution. Mater. Chem. Phys. 2019, 235, 1–10. [Google Scholar] [CrossRef]
- Orazem, M.E.; Tribollet, B. Electrochemical Impedance Spectroscopy; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- González-Reyna, M.A.; Espinosa-Medina, M.A.; Esparza, R.; Hernández-Martinez, A.R.; Maya-Cornejo, J.; Estevez, M. Anticorrosive Effect of the Size of Silica Nanoparticles on PMMA-Based Hybrid Coatings. J. Mater. Eng. Perform. 2021, 30, 1054–1065. [Google Scholar] [CrossRef]
- Abdal-hay, A.; Amna, T.; Lim, J.K. Biocorrosion and osteoconductivity of PCL/nHAp composite porous film-based coating of magnesium alloy. Solid State. Sci. 2013, 18, 131–140. [Google Scholar] [CrossRef]
- Abdal-hay, A.; Hwang, M.G.; Lim, J.K. In vitro bioactivity of titanium implants coated with bicomponent hybrid biodegradable polymers. J. Sol-Gel Sci. Technol. 2012, 64, 756–764. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample BG70-PLA30 | SBF Evaluation (Days) | Sample BG30-PLA70 | SBF Evaluation (Days) |
|---|---|---|---|
| BG70-PLA30-SBF-7 | 7 | BG30-PLA70-SBF-7 | 7 |
| BG70-PLA30-SBF-14 | 14 | BG30-PLA70-SBF-14 | 14 |
| BG70-PLA30-SBF-21 | 21 | BG30-PLA70-SBF-21 | 21 |
| BG70-PLA30-SBF-28 | 28 | BG30-PLA70-SBF-28 | 28 |
| Sample BG70-PLA30 | PBS Evaluation (Days) | Sample BG30-PLA70 | PBS Evaluation (Days) |
|---|---|---|---|
| BG70-PLA30-PBS-7 | 7 | BG30-PLA70-PBS-7 | 7 |
| BG70-PLA30-PBS-14 | 14 | BG30-PLA70-PBS-14 | 14 |
| BG70-PLA30-PBS-21 | 21 | BG30-PLA70-PBS-21 | 21 |
| BG70-PLA30-PBS-28 | 28 | BG30-PLA70-PBS-28 | 28 |
| EDS | BG70-PLA30 | BG30-PLA70 |
|---|---|---|
| Element | at.% | at.% |
| C | 38.485 | 50.103 |
| O | 48.356 | 46.811 |
| Si | 9.704 | 0.588 |
| Ca | 3.454 | 2.498 |
| Time, (Days) | SBF | PBS | ||||||
|---|---|---|---|---|---|---|---|---|
| BG30 PLA70 | BG70 PLA30 | BG30 PLA70 | BG70 PLA30 | |||||
| SX | SX | SX | SX | |||||
| 7 | 39.60 | 1.34 | 37.86 | 1.15 | 27.80 | 1.86 | 24.12 | 0.03 |
| 14 | 38.88 | 0.81 | 38.95 | 0.27 | 28.42 | 1.32 | 22.42 | 0.22 |
| 21 | 39.72 | 1.47 | 39.33 | 0.04 | 28.05 | 1.06 | 23.00 | 0.57 |
| 28 | 38.60 | 0.92 | 38.53 | 0.23 | 27.91 | 0.32 | 24.93 | 1.06 |
| Element at.% | BG70-PLA30 | BG30-PLA70 | |||
|---|---|---|---|---|---|
| 14 Days | 28 Days | 14 Days | 28 Days | ||
| S. 1 | S. 2 | ||||
| C | 41.25 | 58.97 | 51.34 | ||
| O | 45.59 | 37.9 | 58.7 | 34.58 | 43.6 |
| Na | 0.88 | 25.1 | 17.3 | 2.35 | 1.26 |
| Si | 4.82 | 13.5 | 8.7 | 1.05 | 2.59 |
| P | 2.91 | 0.24 | |||
| Cl | 0.41 | 19.1 | 9.15 | 2.02 | 0.85 |
| Ca | 4.13 | 4.41 | 6.15 | 0.8 | 0.36 |
| Element at.% | BG70-PLA30 | BG30-PLA70 | |||
|---|---|---|---|---|---|
| 14 Days | 28 Days | 14 Days | 28 Days | ||
| S.1 | S.2 | ||||
| C | 18.96 | 35.73 | 34.87 | 16.31 | |
| O | 62.28 | 72.89 | 51.23 | 44.62 | 57.6 |
| Na | 1.07 | 0.85 | 1.44 | 0.38 | |
| Si | 7.28 | 22.54 | 4.82 | 0.89 | 1.42 |
| P | 0.7 | 1.37 | 3.08 | 6.85 | 3.7 |
| Cl | 0.82 | 0.39 | |||
| Ca | 8.88 | 3.2 | 3.9 | 11.33 | 20.6 |
| Solution | Sample | icorr | Ecorr | βa | βc | Etransp | ipass |
|---|---|---|---|---|---|---|---|
| µA/cm2 | mV | mV | mV | mV | µA/cm2 | ||
| Hank | BG | 0.108 | −211.7 | 123 | 67 | 545 | 0.593 |
| BG70-PLA30 | 0.241 | −257.5 | 111 | 62 | 394 | 0.817 | |
| BG30-PLA70 | 0.236 | −181.1 | 92 | 79 | 448 | 1.540 | |
| 316L SS | 0.733 | −301.8 | 75 | 67 | 396 | 6.310 | |
| SBF | BG | 0.145 | −166.9 | 134 | 103 | 440 | 1.490 |
| BG70-PLA30 | 0.228 | −210.0 | 135 | 76 | 428 | 0.149 | |
| BG30-PLA70 | 0.190 | −168.0 | 117 | 122 | 289 | 1.040 |
| 316L SS | Time Immersion | |
|---|---|---|
| Model 1 | Beginning | 24 h |
| Rs (Ω cm2) | 78 | 75 |
| TCPE1 (µF cm2) | 11.88 | 10.28 |
| α1 (*) | 0.924 | 0.896 |
| RL1 (Ω cm2) | 167 | 80,654 |
| TCPE1 (µF cm2) | 2.911 | 1.032 |
| α2 (*) | 0.892 | 0.875 |
| Rct (k Ω cm2) | 8250.5 | 8.693 |
| Model 2 | Hank’s Solution | SBF Solution | ||||
| Measurement at the Beginning of Immersion | ||||||
| BG | BG70-PLA30 | BG30-PLA70 | BG | BG70-PLA30 | BG30P-LA70 | |
| Rs (Ω cm2) | 62 | 94 | 71 | 61 | 71 | 57 |
| TCPE1 (µF cm2) | 2.316 | 7.215 | 1.693 | 8.954 | 3.376 | 15.82 |
| α1 (*) | 0.982 | 0.884 | 0.415 | 0.447 | 0.867 | 0.811 |
| RL1 (Ω cm2) | 6.41 | 1.88 | 1.24 | 0.70 | 52.95 | 12.52 |
| TCPE1 (µF cm2) | 12.77 | 10.23 | 25.07 | 38.09 | 15.52 | 6.073 |
| α2 (*) | 0.939 | 0.883 | 0.896 | 0.867 | 0.822 | 0.969 |
| σ (k Ω cm2/s) | 4842.1 | 3122.1 | 4561.9 | 2847.2 | 17.76 | 1025.9 |
| TD (s) | 1.019 | 0.872 | 0.967 | 0.982 | 7.8 × 10−16 | 1.7 × 10−16 |
| P (*) | 0.942 | 0.935 | 0.971 | 0.976 | 0.937 | 0.941 |
| Rct (k Ω cm2) | 15.84 | 22.76 | 5.07 | 3.77 | 40.35 | 476.26 |
| Measurement at the 24 h | ||||||
| BG | BG70-PLA30 | BG30-PLA70 | BG | BG70-PLA30 | BG30-PLA70 | |
| Rs (Ω cm2) | 60 | 81 | 64 | 58 | 63 | 53 |
| TCPE1 (µF cm2) | 10.80 | 22.34 | 15.80 | 19.18 | 17.49 | 8.05 |
| α1 (*) | 0.900 | 0.848 | 0.874 | 0.833 | 0.840 | 0.880 |
| RL1 (Ω cm2) | 12.94 | 3.08 | 20.36 | 14.83 | 20.88 | 20.63 |
| TCPE1 (µF cm2) | 2.285 | 1.426 | 2.456 | 5.729 | 2.471 | 8.751 |
| α2 (*) | 0.979 | 0.992 | 0.992 | 0.934 | 0.992 | 0.852 |
| σ (k Ω cm2/s) | 2070.7 | 825.02 | 5595.5 | 3390.4 | 10,397 | 6207.1 |
| TD (s) | 2.5 × 10−16 | 3.3 × 10−11 | 1.9 × 10−17 | 5.6 × 10−18 | 6.5 × 10−16 | 6.17 |
| P (*) | 0.811 | 0.830 | 0.754 | 0.772 | 0.581 | 0.847 |
| Rct (k Ω cm2) | 2612.7 | 828.52 | 653.23 | 83.42 | 10,252 | 5.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbajal-De la Torre, G.; Zurita-Méndez, N.N.; Ballesteros-Almanza, M.d.L.; Ortiz-Ortiz, J.; Estévez, M.; Espinosa-Medina, M.A. Characterization and Evaluation of Composite Biomaterial Bioactive Glass–Polylactic Acid for Bone Tissue Engineering Applications. Polymers 2022, 14, 3034. https://doi.org/10.3390/polym14153034
Carbajal-De la Torre G, Zurita-Méndez NN, Ballesteros-Almanza MdL, Ortiz-Ortiz J, Estévez M, Espinosa-Medina MA. Characterization and Evaluation of Composite Biomaterial Bioactive Glass–Polylactic Acid for Bone Tissue Engineering Applications. Polymers. 2022; 14(15):3034. https://doi.org/10.3390/polym14153034
Chicago/Turabian StyleCarbajal-De la Torre, Georgina, Nancy N. Zurita-Méndez, María de Lourdes Ballesteros-Almanza, Javier Ortiz-Ortiz, Miriam Estévez, and Marco A. Espinosa-Medina. 2022. "Characterization and Evaluation of Composite Biomaterial Bioactive Glass–Polylactic Acid for Bone Tissue Engineering Applications" Polymers 14, no. 15: 3034. https://doi.org/10.3390/polym14153034